Introduction
Pancreatic cancer (PC) is the fourth most common
cause of cancer death in Western societies with a 5-year survival
of <5% (1–6). PC often presents asymptomatically,
and as a consequence is advanced in the majority of cases at
diagnosis (5,7). Surgical resection currently offers
the only option for long-term survival, however, only 20% of
patients are suitable for surgical intervention (8,9).
Current chemotherapeutic and radiation treatments have also met
with limited success (5,9). Novel diagnostic and therapeutic
strategies are urgently needed to improve outcomes for this
disease.
Epigenetic therapeutic agents, such as histone
deacetylase inhibitors (HDACi) and DNA methyltransferase inhibitors
(DNMTi), have demonstrated efficacy in the treatment of cutaneous
T-cell lymphoma (CTCL) (10–12)
and myelodysplastic syndrome (MDS) (13,14).
Preclinical studies of epigenetic modulating drugs in other cancers
have also demonstrated antitumour activity with tolerable toxicity,
suggesting a potential role for these drugs in cancer treatment
(15–19). HDACi and DNMTi are thought to act
by regulating gene expression and remodelling the chromosome
structure, which may lead to cell cycle arrest and cell death
(20,21).
HDACi induce accumulation of acetylated histones,
resulting in the relaxation of chromatin structure to promote
access of transcriptional machinery (22). Induction of p21WAF1
expression after treatment with HDACi is common and appears to play
a major role in arresting the growth of transformed cells (23–26).
DNMTi reduce genomic DNA methylation by binding to DNA
methyltransferases (DNMT) after being incorporated into newly
synthesized DNA (16,27). DNA methylation, a major epigenetic
mechanism of gene regulation, is usually associated with gene
silencing (28). Treatment with
DNMTi reverses aberrant DNA methylation and thus reactivates the
transcription of many genes, including putative tumour suppressor
genes (16,28,29).
The re-expression of tumour suppressor genes is thought to
contribute, at least in part, to the DNMTi anticancer effect
(29,30). In acute myeloid leukemia cells,
DNMTi alter cell cycle progression, reduce cell proliferation and
induce apoptosis (16). Treatment
combining DNMTi with HDACi results in synergistic cell death, which
may reflect re-expression of silenced genes as well as the
potentiation of cell death through acetylation of non-histone
proteins (10,31).
Previous studies have identified several genes with
tumour suppressor properties that are epigenetically regulated in
PC (32–35), including the regulation of mucin
expression [MUC1 (36), MUC2
(37) and MUC4 (38)], which is associated with
carcinogenesis and tumour invasion (36–39).
The mechanism of epigenetic alterations in PC is poorly understood
(40). Treatment of PC cells with
HDACi induces cell death and enhances the apoptotic effects of
gemcitabine (24). In the
gemcitabine-resistant PC cell line, PANC1, treatment with
suberoylanilide hydroxamic acid (SAHA) restores sensitivity to
gemcitabine (24). Treatment with
5-AZA-2′ deoxycytidine (5-AZA-dc) restores the expression of BNIP3
and induces hypoxia-mediated cell death (35). Together, these data suggest
potential for epigenetic modulating drugs in the treatment and
management of PC.
Materials and methods
Cell culture
MiaPaCa2 cells were cultured in DMEM supplemented
with 10% FBS and 2.5% horse serum according to American Type
Culture Collection (ATCC) protocols. Human pancreatic ductal
epithelial (HPDE) cells, a gift from Dr Ming-Sound Tsao, were used
as a normal control and cultured in keratinocyte serum-free medium
(KSF) supplemented with 50 mg/ml bovine pituitary extract and 5
ng/ml epidermal growth factor (41). These cell lines were maintained in
a humidified atmosphere of 5% CO2 at 37°C.
5-AZA-dc and SAHA treatment
MiaPaCa2 cells were plated at 1.5×105
cells/well in 6-well plates for protein/nucleic acid harvest; or at
3.9×105 cells in T25 flasks for flow cytometry analysis
(Day 0). The cells were treated as follows: i) treated on Day 1
with 300 μM of 5-AZA-dc (Sigma-Aldrich) and harvested on Day 6; ii)
treated on Days 1–3 and Day 5 with 5 μM of SAHA (Cayman Chemical)
and harvested on Day 6; iii) treated on Day 1 with 5-AZA-dc, Day 2,
3 and 5 with SAHA and harvested on Day 6. Untreated MiaPaCa2 and
HPDE cells were used as controls. During treatment, the media was
changed daily, and cells were washed twice with cold PBS prior to
nucleic acid and/or protein extraction.
MTS assay
An MTT assay was performed using CellTiter
96® AQueous Non-Radioactive Cell Proliferation Assay
according to the manufacturer’s protocol (Promega Corporation).
MiaPaCa2 and HPDE cells were plated at a density of
0.5×104 cells/well in 96-well plates to measure cell
proliferation. Cells were treated on Days 1–5 with SAHA (Cayman
Chemical) at a concentration of 1, 3 or 5 μM. Untreated MiaPaCa2
and HPDE cells were included in each plate as controls. Each
treatment group was plated in triplicate and repeated at least
three times. Media was changed daily during treatment. An
additional untreated plate was prepared as a baseline. Absorbance
was measured at 490 nM.
BrdU and PI staining
MiaPaCa2 cells were pulsed with bromodeoxyuridine
(BrdU) (Sigma-Aldrich) for 1 h prior to harvesting. Cells were
harvested and fixed in 70% ethanol overnight at 4°C before BrdU and
propidium iodide (PI) (Sigma-Aldrich) staining. The next day,
ethanol was removed and cells resuspended in PBS/1% Tween-20 (PBST)
followed by DNA denaturation using 1.5 M HCl for 20 min. The cells
were in PBST (3X), resuspended in 100 μl PBST, followed by the
addition of 5 μl of 250 μg/ml FITC-anti-BrdU (MAB3262F; Chemicon)
for 1 h at 37°C in the dark. After incubation, cells were washed
with 1 ml cold PBST and resuspended in 470 μl PBST. PI (5 μl of 1
mg/ml) and RNase A (25 μl of 10 mg/ml) (Sigma-Aldrich) were added
and mixed gently by pipetting. The cells were incubated between 1–4
h in the dark at room temperature. Prior to running on the BD
FACSCalibur™ or BD FACSCanto™ flow cytometer (BD Biosciences),
cells were syringed gently to avoid cell clumping. Flow cytometry
data were analysed using either BD CellQuest™ (BD Biosciences), or
FlowJo 8.7.3 or 8.8.2 (Tree Star, Inc.).
Protein extraction
Cell lysis and protein extraction from cell lines
were performed on ice. Media was removed and cell monolayers washed
twice with cold PBS. Lysis buffer (50 μl) containing protease
inhibitors (0.5% deoxycholate, 150 mM NaCl, 1% sodium
pyrophosphate, 50 mM Tris pH 8.0, 0.1% SDS, 10% glycerol, 5 mM
EDTA, 20 mM NaF, 10 μg/ml apoprotinin, 10 μg/ml leupeptin, 1 mM
phenylmethylsufonyl fluoride (PMSF), 200 μM sodium orthovanadate;
Sigma-Aldrich) was added to each well of a 6-well plate. The cells
were scraped and transferred to microcentrifuge tubes, vortexed and
centrifuged at 13,000 rpm for 5 min at 4°C. The supernatant was
collected and 2–5 μl of the supernatant was aliquoted for protein
quantitation. The remaining supernatant was stored at −80°C.
Protein quantification was performed using the Bio-Rad Protein
Assay kit according to the manufacturer’s instructions.
Western blot analysis
Following normalization for protein concentration,
SDS sample buffer was added and lysates denatured at 70°C for 10
min, protein was separated using 4–12% Bis-Tris NuPAGE®
precast gels (Invitrogen Life Technologies) and transferred to
polyvinylidene fluoride (PVDF) membranes according to the
manufacturer’s protocols. Non-specific binding was blocked in 10%
(w/v) skim milk powder in TBS/Tween-20 [10 mM Tris pH 7.4, 150 mM
NaCl, 0.1% (v/v) Tween-20; TBST]. Membranes were incubated with
primary antibodies diluted in TBS/BSA solution (10 mM Tris pH 7.4,
150 mM NaCl, 5% w/v BSA, 0.02% azide). Table I contains the list of primary
antibodies and their incubation conditions. Membranes were washed
in TBST for 30 min, then incubated for 1 h at room temperature with
HRP-conjugated anti-rabbit or anti-mouse secondary antibody
(1:2,000; Amersham Pharmacia Biotech) in 5% (w/v) skim milk powder
in TBST. Membranes were washed for 30 min in TBST before proteins
were visualized using the Enhanced Chemiluminescence (ECL)
detection system (Perkin Elmer) on X-ray film (Fujifilm Medical
Systems USA). Cell cycle marker (cyclin E, A, D1, B1 and
p21WAF1) protein levels were determined relative to
β-actin by densitometry using ImageJ software [National Institutes
of Health (NIH)] and normalized to DMSO-treated control
samples.
 | Table IPrimary antibodies for western blot
analysis. |
Table I
Primary antibodies for western blot
analysis.
| Primary | Dilution | Manufacturer | Incubation |
|---|
| β-actin (AC-15;
A5441) | 1:40,000 | Sigma-Aldrich | 1 h RT |
| Cyclin |
| E (He12;
SC-247) | 1:500 | Santa
Cruz.
Biotechnology, Inc. | O/N 4°C |
| A (C19;
SC-596) | 1:500 | Santa
Cruz
Biotechnology, Inc. | O/N 4°C |
| D1 (DCS-6) | 1:100 | Novocastra | O/N 4°C |
| B1 (V152;
4135) | 1:2,000 | Cell
Signaling
Technology, Inc. | O/N 4°C |
| p21WAF1
(610234) | 1:1,000 | BD Biosciences | O/N 4°C |
In vivo study of the efficacy of
epigenetic therapeutic agents in a xenograft model of PC
Ethics approval was obtained from Garvan Institute
and St. Vincent’s Hospital Animal Ethics Committee to examine the
effect of epigenetic therapeutic agents in a mouse xenograft model
of PC (Protocol no. 07/06).
MiaPaCa2 PC cells were injected subcutaneously into
female athymic nude mice (BALB/c nu/nu) (6–8 weeks old, mean weight
of 16 g). Prior to injection, MiaPaCa2 cells were harvested,
counted and diluted to the appropriate concentration in cold media.
For each mouse, 1.0×106 MiaPaCa2 cells were diluted in
1:1 mixture of cold media and BD Matrigel™ Basement Membrane Matrix
(BD Biosciences) to a total volume of 100 μl and kept on ice until
injection.
The mice were divided into three treatment groups:
i) vehicle (control) (n=5), ii) SAHA alone (n=5); and iii) 5-AZA-dc
and SAHA (n=5). 5-AZA-dc was administered in a single dose at a
concentration of 0.25 mg/kg, followed by daily intraperitoneal
administration of SAHA for 21 days at a dose of 50 mg/kg (in a
maximum volume of 0.1 ml). Treatment began 1 week after the
injection of MiaPaca2 cells to examine the ability of the drugs to
prevent tumour growth (Prevention Studies); or once a solid tumour
was present to test the ability of the drugs to reduce tumour
growth (Therapeutic Studies). The mice were euthanased upon
cessation of drug treatments and tumour weight was measured. The
pancreas, abdominal cavity, mesentery, spleen and liver were
assessed for the presence of metastases. Tumour size was measured,
and tumour volume was calculated using the formula 1/2 length ×
breadth × width (42). The in
vivo experiments were performed in duplicate, with tumour
measurements from each experiment combined for statistical
analysis.
Statistical analysis
All in vitro experiments were carried out at
least in triplicate, and the in vivo studies in duplicate.
Mean, standard deviation and Student’s t-test were calculated using
Microsoft Excel. Statistical analyses of univariate analysis of
variance (ANOVA), Tukey’s post hoc tests and calculation of 95%
confidence interval were performed using SPSS16.0 (SPSS Inc.)
and/or R v.2.10.1 (R Foundation for Statistical Computing).
Results
Treatment with SAHA and 5-AZA-dc
decreases cell proliferation in MiaPaCa2 cells
Treatment with SAHA alone, or in combination with
5-AZA-dc, resulted in higher cell death and lower DNA synthesis
(Fig. 1A) compared to 5-AZA-dc
alone or DMSO-treated cells. In MiaPaCa2 cells, treatment with SAHA
was more effective in inducing cell death than treatment with
5-AZA-dc (p=0.002 and p=0.008 respectively), while SAHA treatment
alone was not significantly different to combination therapy
(p=0.019; Fig. 1A). The dramatic
effect of SAHA may potentially be due to a global effect of
chromatin remodelling in regulation of cellular function, as
defective histone dynamics during S phase, DNA repair and mitosis
result in cell cycle arrest and cell death (43).
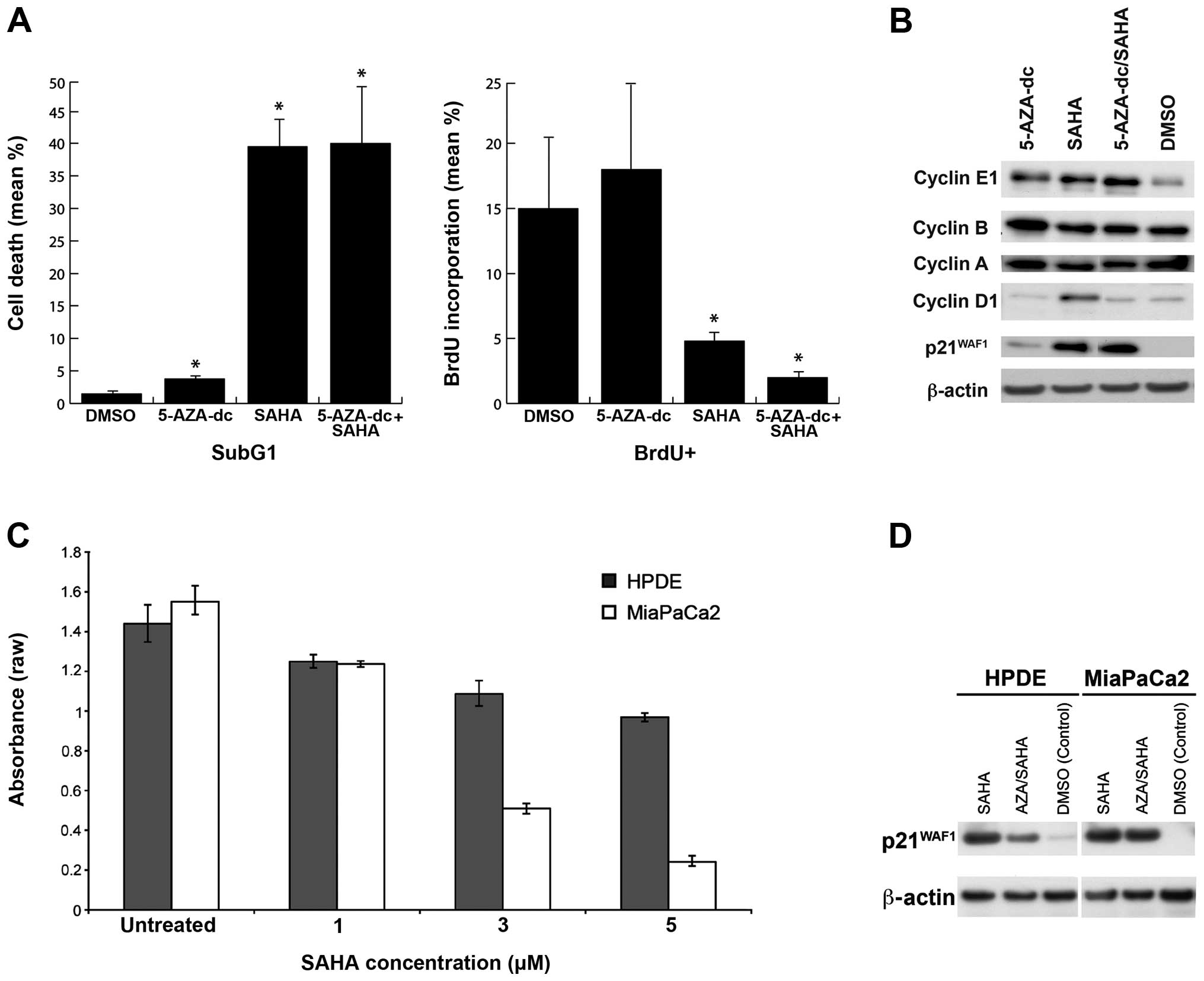 | Figure 1(A) Effect of 5-AZA-2′ deoxycytidine
(5-AZA-dc) and/or suberoylanilide hydroxamic acid (SAHA) treatment
on cell proliferation and DNA synthesis in MiaPaCa2 cells. Cell
proliferation: MiaPaCa2 cells treated with 5-AZA-dc, SAHA, or
combination SAHA/5-AZA-dc significantly increased cell death, as
determined by the SubG1 population compared to control cells. DNA
synthesis: MiaPaCa2 cells treated with SAHA, with or without
5-AZA-dc reduced DNA synthesis compared to treatment with 5-AZA-dc
alone and control (DMSO), as determined by bromodeoxyuridine (BrdU)
incorporation. *P<0.05 compared to untreated control;
Student’s t-test. (B) Cell cycle markers 3 days post-treatment with
SAHA and/or 5-AZA-dc. Treatment with 5-AZA-dc alone increased the
expression of cyclin E and p21WAF1 compared with
controls, while treatment with SAHA alone also led to higher
expression of p21WAF1. The combination treatment with
5-AZA-dc and SAHA increased p21WAF1 expression and
increased the expression of cyclin E1, but did not alter expression
of cyclin B, A and D1. (C) Efficacy of SAHA treatment on MiaPaCa2
vs. human pancreatic ductal epithelial (HPDE) cell proliferation.
Treatment with SAHA reduced MiaPaCa2 and HPDE cell proliferation.
MiaPaCa2 cells were more sensitive to SAHA than were HPDE cells.
Data are expressed as raw absorbance values ± 95% CI. (D)
p21WAF1 expression in HPDE and MiaPaCa2 cells treated
with 5-AZA-dc and/or SAHA. Treatment with SAHA alone, and in
combination with 5-AZA-dc, increased expression of
p21WAF1 compared to control cells (DMSO-treated
cells). |
DNA synthesis levels, as measured by BrdU
incorporation, were significantly reduced upon treatment with SAHA,
either alone (p=0.001) or in combination with 5-AZA-dc (p=0.005),
while treatment with 5-AZA-dc alone had no significant effect
compared to DMSO-treated cells (Fig.
1A). These data correlate with higher levels of
p21WAF1 observed in SAHA and SAHA/5-AZA-dc
combination-treated samples relative to control and
5-AZA-dc-treated cells (Fig. 1B),
which is consistent with findings in other cancer types (44).
Western blot analysis of cell cycle markers cyclin
E1, B, A, D1 and p21WAF1 revealed that MiaPaCa2 cells
treated with 5-AZA-dc alone expressed higher cyclin E protein
compared to control (DMSO) cells (Fig.
1B). Cells treated with SAHA, alone or in combination with
5-AZA-dc, expressed higher levels of cyclin E1 and significantly
higher p21WAF1 protein levels that could be contributing
to G1/S-phase cell cycle arrest (45).
MiaPaCa2 cells are more sensitive than
HPDE cells to treatment with SAHA
Previous studies have reported that normal cells are
more tolerant to epigenetic drug therapy compared to cancer cells
(46–49). We investigated this premise using
the ‘normal’ pancreatic ductal cell line, HPDE, and MiaPaCa2
cells.
Increasing SAHA concentration significantly
decreased cell proliferation in both MiaPaCa2 (p<0.0001) and
HPDE cells (p<0.0001; Fig. 1C).
However, the cell lines demonstrated different cell proliferation
rates in response to drug treatment, suggesting that MiaPaCa2 cells
are more sensitive to epigenetic modifying agents than HPDE cells
(p=0.034).
Treatment with SAHA and/or 5-AZA-dc
increases expression of p21WAF1
Induction of p21WAF1 expression upon
treatment with epigenetic drugs reportedly induces cell cycle
arrest in some cancer types, including MiaPaCa2 cells. The
induction of p21WAF1 expression in HPDE and MiaPaCa2
cells after treatment with SAHA, with or without 5-AZA-dc (Fig. 1D), was associated with decreased
proliferation (Fig. 1C),
suggesting that the induction of p21WAF1 expression
following treatment with epigenetic drugs may contribute to
decreased cell proliferation.
The efficacy of 5-AZA-dc and SAHA in a
pancreatic xeno-graft model
Prevention studies
Using a subcutaneous xenograft model of PC we
investigated the efficacy of pharmacological epigenetic modulation
in the prevention of tumour growth. Treatment with SAHA and/or
5-AZA-dc significantly increased the tumour lag period compared to
control (p=0.001 and p<0.001 respectively; Fig. 2A), while the lag period between the
two treatment groups (5-AZA-dc/SAHA and SAHA alone) was similar
(p=0.882; Fig. 2A). The lag period
was defined as the time required for the tumour volume to reach 100
mm3 (indicated by the shaded area in Fig. 2A). Tumour growth rate measured
after the lag cut-off point (100 mm3), was significantly
different between groups (p=0.001) (Fig. 2A), indicating an effect following
treatment. However, overall tumour growth rates between the two
treatment groups (5-AZA-dc/SAHA and SAHA alone) were similar. No
significant difference was observed in the mean tumour weights
between all groups upon completion of treatment (p=0.994). This
suggests that after the lag period, tumours in the treated groups
grew at an increased rate when compared to the control group.
Therapeutic studies
As PC is usually diagnosed at an advanced stage, we
investigated the ability of epigenetic therapies to reduce tumour
growth using an established subcutaneous xenograft model of PC.
Similar tumour growth rates were observed between treatment groups
and control (p=1.000; Fig. 2B),
with no significant difference in tumour weight observed (p=0.448).
These results indicate that in this model, treatment with SAHA,
alone or in combination with 5-AZA-dc, was not effective in the
treatment of established pancreatic tumours in vivo.
Discussion
Epigenetic therapies have shown promising
antitumourigenic effects in some malignancies (15–19,50).
These include enzyme inhibitors, specifically DNMTi and HDACi,
which induce epigenetic modifications. In this study, we
demonstrated that treatment of PC cells with HDACi (SAHA) in
combination with DNMTi (5-AZA-dc) decreased cell proliferation and
induced cell death. This may be mediated through upregulation of
p21WAF1, and is associated with cell cycle arrest,
apoptosis and decreased cell proliferation (23–26,44).
This study also demonstrated that MiaPaCa2 cells were more
sensitive to epigenetic drugs compared to the ‘normal’ HPDE
pancreatic cells.
Treatment with SAHA induced significant cell death
and reduced DNA synthesis, potentially as a result of cell cycle
arrest. Dysregulated histone modification, which can be promoted by
HDACi, may lead to aberrant chromatin remodelling during DNA
replication and repair, and mitosis (43,51,52).
Treatment with SAHA also induces a more open chromatin structure,
increasing the susceptibility of DNA to damage (53,54).
These events correlate with cell cycle arrest leading to cell death
(43,51,52).
Our study of MiaPaCa2 cells demonstrate that treatment with SAHA
(HDACi) was more effective than treatment with 5-AZA-dc (DNMTi)
alone. Consequently, aberrant chromatin modification following
treatment with HDACi, such as SAHA, may play an important role in
an anticancer effect. While the effect of HDACi in chromatin
remodelling would also apply to normal cells, these are likely to
be more resistant to epigenetic treatment (46–49).
Therefore, this mechanism does not fully explain the anticancer
effects of SAHA in MiaPaCa2 cells, or the higher tolerance of HPDE
cells to epigenetic treatment.
Our data show that decreased cell proliferation and
induction of cell death following treatment with SAHA and 5-AZA-dc
may be mediated via upregulation of p21WAF1.
p21WAF1 is tightly regulated by p53, however, as the
MiaPaCa2 cell line used in this study is p53 defective,
p21WAF1 must be regulated by pathways independent of p53
[26, 55, reviewed in (56)]. Previous studies have indicated
that treatment with epigenetic drugs is likely to increase the
expression of p21WAF1 by regulating the chromatin
structure and increasing the acetylation of histone 3 on the
p21WAF1 promoter (12,23,26,57,58).
More recently, Vijayaraghavalu et al demonstrated that
sequential treatment of resistant breast cancer cells with 5-AZA-dc
and doxorubicin induces a highly synergistic effect and caused the
resistant cells to undergo G2/M cell cycle arrest, which was due to
upregulation of p21WAF1 expression. Induction of
p21WAF1 was correlated with depletion of DNA
methyltransferase 1 (DNMT1), which promotes DNA methylation,
suggesting that p21WAF1 may be a methylation-suppressed
gene in specific cell types (59).
A further explanation for the reduction of MiaPaCa2
cell proliferation observed in our study may be via the process of
autophagy. Recent studies provide a strong link between HDAC
inhibition and cell death by the process of autophagy (60,61).
In particular, Robert et al (61)showed that using valproic acid, a
class I and II HDAC inhibitor, triggers Sae2 (CtIP in human)
degradation by promoting autophagy that affects the DNA damage
sensitivity of hda1 and rpd3 mutants. While beyond the scope of
this study, further experiments are necessary to address the
difference in cellular response in cancer and normal cells after
treatment with epigenetic drugs, such as SAHA, particularly in the
way these cells regulate chromatin remodelling (61).
Since the effectiveness of epigenetic therapy is
cell and context-dependent (10,22,45,62),
the anticancer activities in vitro, may not be translated
into in vivo settings. Thus the efficacy of epigenetic drugs
in vivo was assessed using a subcutaneous xenograft model of
PC. The prevention studies demonstrated that treatment with SAHA,
alone or in combination with 5-AZA-dc, delayed tumour progression
during the early stage (lag period) of tumour development. However,
the therapeutic studies demonstrated that treatment with SAHA, with
or without 5-AZA-dc, did not reduce tumour growth rate nor tumour
weight, indicating that treatment with epigenetic drugs alone is
unlikely to be effective for the treatment of established
pancreatic tumours. These data suggest that treatment with
epigenetic drugs during early pancreatic carcinogenesis may provide
an opportunity for the use of combination treatment with other
chemotherapeutic drugs, thereby increasing the susceptibility of
tumour cells to cytotoxic agents, with many studies demonstrating
the synergistic effect of epigenetic drugs with existing
therapeutic agents (10,19,53,63–65).
In particular, SAHA increases the sensitivity of PC cells to the
chemotherapeutic agent gemcitabine (24). This may be particularly useful in
an adjuvant setting, where a systemic adjuvant approach for
resected pancreatic adenocarcinoma is the most effective means for
improving overall survival (66–69).
This approach has been investigated by Mohammed et al
(70), who demonstrated the
chemopreventative efficacy of the EGFR inhibitor, gefitinib, in
delaying the progression of pancreatic intraepithelial neoplasia
(PanIN) lesions to pancreatic adenocarcinoma, while not showing
efficacy in advanced disease.
The exact mechanisms of the anticancer activity of
combination treatments have yet to be fully elucidated, but may
include regulation of chromatic structures and the induction of
pro-apoptotic genes (19,24,64,65,71).
Our data show that the epigenetic agents modulate the cell cycle
and inhibit cell growth, however, one important question arises.
How effective are combination treatments likely to be if many
anticancer agents need an efficient cell cycle to exert their
effect? Venturelli et al recently showed that the epigenetic
agent 5-AZA-cytidine sensitises cancer cells to tumour necrosis
factor-related apoptosis-inducing ligand (TRAIL) by: i) inhibiting
protein biosynthesis of tumour-protecting factors, thus enabling
TRAIL-induced apoptosis; and ii) reversing the
malignancy-associated methylation phenotype. The ability of
5-AZA-cytidine to inhibit protein biosynthesis was associated with
the ability of the drug to be incorporated into cellular RNA and
disrupt cellular protein biosynthesis (72). This study suggested that epigenetic
drugs could exert their anticancer mechanisms via non-epigenetic
modes of action, which may provide a more complete picture of the
anticancer activities of combination treatments with epigenetic
agents. Further, in a recent study by Shakya et al, 5-AZA-dc
was administered in a mouse model of aggressive stromal-rich
pancreatic adenocarcinoma, and demonstrate that 5-AZA-dc
significantly reduced DNA methylation and slowed progression of
pancreatic adenocarcinoma. 5-AZA-dc upregulated
interferon-inducible genes (e.g., STAT1), and that treatment with
5-AZA-dc and interferon γ had an antiproliferative effect (73). These studies support the rationale
for future studies combining epigenetic agents with other
anticancer agents, as well as with cytokines and immunotherapy.
In conclusion, this study illustrated that treatment
with epigenetic agents decreased cell proliferation and induced
cell death in PC cells, while in vivo studies demonstrated a
delay in tumour progression following treatment. These data suggest
that epigenetic therapy has the potential to delay early pancreatic
carcinogenesis, and may have potential application in an adjuvant
setting for the management of resected PC.
Abbreviations:
|
PC
|
pancreatic cancer
|
|
HDACi
|
histone deacetylase inhibitors
|
|
DNMTi
|
DNA methyltransferase inhibitors
|
References
|
1
|
Cancer Institute NSW. Cancer in New South
Wales: Incidence, Mortality and Prevalence 2005. Cancer Institute
NSW; Sydney: 2007
|
|
2
|
National Cancer Institute: Cancer Trends
Progress Report - 2007 Update. National Cancer Institute; Bethesda,
MD: 2007
|
|
3
|
National Cancer Institute. A Snapshot of
Pancreatic Cancer. National Cancer Institute; Bethesda, MD:
2007
|
|
4
|
Australian Institute of Health and Welfare
(AIHW) and Australasian Association of Cancer Registries (AACR).
Cancer in Australia: an overview, 2008. AIHW and AACR; Canberra:
2008
|
|
5
|
American Cancer Society. Cancer Facts and
Figures 2008. American Cancer Society; Atlanta, GA: 2008
|
|
6
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
American Cancer Society. Cancer Facts and
Figures 2007. American Cancer Society; Atlanta, GA: 2007
|
|
8
|
Von Hoff DD, Evans DB and Hruban RH:
Pancreatic Cancer. 1st edition. Jones and Bartlett Publishers;
Sudbury, MA: 2005
|
|
9
|
Chang DK, Merrett ND and Biankin AV: NSW
Pancreatic Cancer Network: Improving outcomes for operable
pancreatic cancer: is access to safer surgery the problem? J
Gastroenterol Hepatol. 23:1036–1045. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Glaser KB: HDAC inhibitors: clinical
update and mechanism-based potential. Biochem Pharmacol.
74:659–671. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Duvic M, Talpur R, Ni X, et al: Phase 2
trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA)
for refractory cutaneous T-cell lymphoma (CTCL). Blood. 109:31–39.
2007. View Article : Google Scholar
|
|
12
|
Zhang C, Richon V, Ni X, Talpur R and
Duvic M: Selective induction of apoptosis by histone deacetylase
inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to
mechanism of therapeutic action. J Invest Dermatol. 125:1045–1052.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gore SD: Combination therapy with DNA
methyltransferase inhibitors in hematologic malignancies. Nat Clin
Pract Oncol. 2(Suppl 1): S30–S35. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Issa JP and Byrd JC: Decitabine in chronic
leukemias. Semin Hematol. 42(Suppl 2): S43–S49. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Batty N, Malouf GG and Issa JP: Histone
deacetylase inhibitors as anti-neoplastic agents. Cancer Lett.
280:192–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Flotho C, Claus R, Batz C, et al: The DNA
methyltransferase inhibitors azacitidine, decitabine and zebularine
exert differential effects on cancer gene expression in acute
myeloid leukemia cells. Leukemia. 23:1019–1028. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Laurenzana A, Petruccelli LA, Pettersson
F, et al: Inhibition of DNA methyltransferase activates tumor
necrosis factor alpha-induced monocytic differentiation in acute
myeloid leukemia cells. Cancer Res. 69:55–64. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Munster PN, Troso-Sandoval T, Rosen N,
Rifkind R, Marks PA and Richon VM: The histone deacetylase
inhibitor suberoylanilide hydroxamic acid induces differentiation
of human breast cancer cells. Cancer Res. 61:8492–8497.
2001.PubMed/NCBI
|
|
19
|
Shiozawa K, Nakanishi T, Tan M, et al:
Preclinical studies of vorinostat (suberoylanilide hydroxamic acid)
combined with cytosine arabinoside and etoposide for treatment of
acute leukemias. Clin Cancer Res. 15:1698–1707. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu WS, Parmigiani RB and Marks PA: Histone
deacetylase inhibitors: molecular mechanisms of action. Oncogene.
26:5541–5552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Buchwald M, Krämer OH and Heinzel T: HDACi
- targets beyond chromatin. Cancer Lett. 280:160–167. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nolan L, Johnson PW, Ganesan A, Packham G
and Crabb SJ: Will histone deacetylase inhibitors require
combination with other agents to fulfil their therapeutic
potential? Br J Cancer. 99:689–694. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gui CY, Ngo L, Xu WS, Richon VM and Marks
PA: Histone deacetylase (HDAC) inhibitor activation of
p21WAF1 involves changes in promoter-associated
proteins, including HDAC1. Proc Natl Acad Sci USA. 101:1241–1246.
2004. View Article : Google Scholar
|
|
24
|
Arnold NB, Arkus N, Gunn J and Korc M: The
histone deacetylase inhibitor suberoylanilide hydroxamic acid
induces growth inhibition and enhances gemcitabine-induced cell
death in pancreatic cancer. Clin Cancer Res. 13:18–26. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eyüpoglu IY, Hahnen E, Buslei R, et al:
Suberoylanilide hydroxamic acid (SAHA) has potent anti-glioma
properties in vitro, ex vivo and in vivo. J Neurochem. 93:992–999.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kumagai T, Wakimoto N, Yin D, et al:
Histone deacetylase inhibitor, suberoylanilide hydroxamic acid
(Vorinostat, SAHA) profoundly inhibits the growth of human
pancreatic cancer cells. Int J Cancer. 121:656–665. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hurd PJ, Whitmarsh AJ, Baldwin GS, et al:
Mechanism-based inhibition of C5-cytosine DNA methyltransferases by
2-H pyrimidinone. J Mol Biol. 286:389–401. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
29
|
Jackson-Grusby L, Beard C, Possemato R, et
al: Loss of genomic methylation causes p53-dependent apoptosis and
epigenetic deregulation. Nat Genet. 27:31–39. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Scott SA, Lakshimikuttysamma A, Sheridan
DP, Sanche SE, Geyer CR and DeCoteau JF: Zebularine inhibits human
acute myeloid leukemia cell growth in vitro in association with
p15INK4B demethylation and reexpression. Exp Hematol. 35:263–273.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gilbert J, Gore SD, Herman JG and Carducci
MA: The clinical application of targeting cancer through histone
acetylation and hypomethylation. Clin Cancer Res. 10:4589–4596.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sato N, Fukushima N, Maehara N, et al:
SPARC/osteonectin is a frequent target for aberrant methylation in
pancreatic adenocarcinoma and a mediator of tumor-stromal
interactions. Oncogene. 22:5021–5030. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sato N, Parker AR, Fukushima N, et al:
Epigenetic inactivation of TFPI-2 as a common mechanism associated
with growth and invasion of pancreatic ductal adenocarcinoma.
Oncogene. 24:850–858. 2005. View Article : Google Scholar
|
|
34
|
Fu B, Guo M, Wang S, et al: Evaluation of
GATA-4 and GATA-5 methylation profiles in human pancreatic cancers
indicate promoter methylation patterns distinct from other human
tumor types. Cancer Biol Ther. 6:1546–1552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Abe T, Toyota M, Suzuki H, et al:
Upregulation of BNIP3 by 5-aza-2′-deoxycytidine sensitizes
pancreatic cancer cells to hypoxia-mediated cell death. J
Gastroenterol. 40:504–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamada N, Nishida Y, Tsutsumida H, et al:
MUC1 expression is regulated by DNA methylation and histone H3
lysine 9 modification in cancer cells. Cancer Res. 68:2708–2716.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yamada N, Hamada T, Goto M, et al: MUC2
expression is regulated by histone H3 modification and DNA
methylation in pancreatic cancer. Int J Cancer. 119:1850–1857.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vincent A, Ducourouble MP and Van
Seuningen I: Epigenetic regulation of the human mucin gene MUC4 in
epithelial cancer cell lines involves both DNA methylation and
histone modifications mediated by DNA methyltransferases and
histone deacetylases. FASEB J. 22:3035–3045. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yonezawa S, Goto M, Yamada N, Higashi M
and Nomoto M: Expression profiles of MUC1, MUC2, and MUC4 mucins in
human neoplasms and their relationship with biological behavior.
Proteomics. 8:3329–3341. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Omura N and Goggins M: Epigenetics and
epigenetic alterations in pancreatic cancer. Int J Clin Exp Pathol.
2:310–326. 2009.PubMed/NCBI
|
|
41
|
Furukawa T, Duguid WP, Rosenberg L,
Viallet J, Galloway DA and Tsao MS: Long-term culture and
immortalization of epithelial cells from normal adult human
pancreatic ducts transfected by the E6E7 gene of human papilloma
virus 16. Am J Pathol. 148:1763–1770. 1996.PubMed/NCBI
|
|
42
|
Vonlaufen A, Joshi S, Qu C, et al:
Pancreatic stellate cells: partners in crime with pancreatic cancer
cells. Cancer Res. 68:2085–2093. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Probst AV, Dunleavy E and Almouzni G:
Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell
Biol. 10:192–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Missiaglia E, Donadelli M, Palmieri M,
Crnogorac-Jurcevic T, Scarpa A and Lemoine NR: Growth delay of
human pancreatic cancer cells by methylase inhibitor
5-aza-2′-deoxycytidine treatment is associated with activation of
the interferon signalling pathway. Oncogene. 24:199–211. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Butler LM, Agus DB, Scher HI, et al:
Suberoylanilide hydroxamic acid, an inhibitor of histone
deacetylase, suppresses the growth of prostate cancer cells in
vitro and in vivo. Cancer Res. 60:5165–5170. 2000.PubMed/NCBI
|
|
47
|
Butler LM, Zhou X, Xu WS, et al: The
histone deacetylase inhibitor SAHA arrests cancer cell growth,
up-regulates thioredoxin-binding protein-2, and down-regulates
thioredoxin. Proc Natl Acad Sci USA. 99:11700–11705. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Marks PA and Breslow R: Dimethyl sulfoxide
to vorinostat: development of this histone deacetylase inhibitor as
an anticancer drug. Nat Biotechnol. 25:84–90. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Henderson C, Mizzau M, Paroni G, Maestro
R, Schneider C and Brancolini C: Role of caspases, Bid, and p53 in
the apoptotic response triggered by histone deacetylase inhibitors
trichostatin-A (TSA) and suberoylanilide hydroxamic acid (SAHA). J
Biol Chem. 278:12579–12589. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Egger G, Liang G, Aparicio A and Jones PA:
Epigenetics in human disease and prospects for epigenetic therapy.
Nature. 429:457–463. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Polo SE and Almouzni G: Histone metabolic
pathways and chromatin assembly factors as proliferation markers.
Cancer Lett. 220:1–9. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Koundrioukoff S, Polo S and Almouzni G:
Interplay between chromatin and cell cycle checkpoints in the
context of ATR/ATM-dependent checkpoints. DNA Repair (Amst).
3:969–978. 2004. View Article : Google Scholar
|
|
53
|
Graham JS, Kaye SB and Brown R: The
promises and pitfalls of epigenetic therapies in solid tumours. Eur
J Cancer. 45:1129–1136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Polo SE and Almouzni G: DNA damage leaves
its mark on chromatin. Cell Cycle. 6:2355–2359. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Vrana JA, Decker RH, Johnson CR, et al:
Induction of apoptosis in U937 human leukemia cells by
suberoylanilide hydroxamic acid (SAHA) proceeds through pathways
that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but
independent of p53. Oncogene. 18:7016–7025. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Abbas T and Dutta A: p21 in cancer:
intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mills J, Hricik T, Siddiqi S and
Matushansky I: Chromatin structure predicts epigenetic therapy
responsiveness in sarcoma. Mol Cancer Ther. 10:313–324. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yin D, Ong JM, Hu J, et al:
Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor:
effects on gene expression and growth of glioma cells in vitro and
in vivo. Clin Cancer Res. 13:1045–1052. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Vijayaraghavalu S, Dermawan JK, Cheriyath
V and Labhasetwar V: Highly synergistic effect of sequential
treatment with epigenetic and anticancer drugs to overcome drug
resistance in breast cancer cells is mediated via activation of p21
gene expression leading to G2/M cycle arrest. Mol Pharm.
10:337–352. 2013. View Article : Google Scholar :
|
|
60
|
Carew JS, Nawrocki ST and Cleveland JL:
Modulating autophagy for therapeutic benefit. Autophagy. 3:464–467.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Robert T, Vanoli F, Chiolo I, et al: HDACs
link the DNA damage response, processing of double-strand breaks
and autophagy. Nature. 471:74–79. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Dokmanovic M, Perez G, Xu W, et al:
Histone deacetylase inhibitors selectively suppress expression of
HDAC7. Mol Cancer Ther. 6:2525–2534. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Mitsiades CS, Mitsiades NS, McMullan CJ,
et al: Transcriptional signature of histone deacetylase inhibition
in multiple myeloma: biological and clinical implications. Proc
Natl Acad Sci USA. 101:540–545. 2004. View Article : Google Scholar :
|
|
64
|
Cooper AL, Greenberg VL, Lancaster PS, van
Nagell JR Jr, Zimmer SG and Modesitt SC: In vitro and in vivo
histone deacetylase inhibitor therapy with suberoylanilide
hydroxamic acid (SAHA) and paclitaxel in ovarian cancer. Gynecol
Oncol. 104:596–601. 2007. View Article : Google Scholar
|
|
65
|
Nimmanapalli R, Fuino L, Stobaugh C,
Richon V and Bhalla K: Cotreatment with the histone deacetylase
inhibitor suberoylanilide hydroxamic acid (SAHA) enhances
imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia
cells. Blood. 101:3236–3239. 2003. View Article : Google Scholar
|
|
66
|
Holzman DC: Pancreatic cancer: will
incremental advances begin to make a difference? J Natl Cancer
Inst. 102:1821–1823. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Neoptolemos JP, Dunn JA, Stocken DD, et
al: Adjuvant chemoradiotherapy and chemotherapy in resectable
pancreatic cancer: a randomised controlled trial. Lancet.
358:1576–1585. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Neoptolemos JP, Stocken DD, Bassi C, et
al: Adjuvant chemotherapy with fluorouracil plus folinic acid vs
gemcitabine following pancreatic cancer resection: a randomized
controlled trial. JAMA. 304:1073–1081. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
O’Reilly EM: Refinement of adjuvant
therapy for pancreatic cancer. JAMA. 304:1124–1125. 2010.
View Article : Google Scholar
|
|
70
|
Mohammed A, Janakiram NB, Li Q, et al: The
epidermal growth factor receptor inhibitor gefitinib prevents the
progression of pancreatic lesions to carcinoma in a conditional
LSL-KrasG12D/+ transgenic mouse model. Cancer Prev Res (Phila).
3:1417–1426. 2010. View Article : Google Scholar
|
|
71
|
Appleton K, Mackay HJ, Judson I, et al:
Phase I and pharmacodynamic trial of the DNA methyltransferase
inhibitor decitabine and carboplatin in solid tumors. J Clin Oncol.
25:4603–4609. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Venturelli S, Berger A, Weiland T, et al:
Dual antitumour effect of 5-azacytidine by inducing a breakdown of
resistance-mediating factors and epigenetic modulation. Gut.
60:156–165. 2011. View Article : Google Scholar
|
|
73
|
Shakya R, Gonda T, Quante M, et al:
Hypomethylating therapy in an aggressive stroma-rich model of
pancreatic carcinoma. Cancer Res. 73:885–896. 2013. View Article : Google Scholar :
|















