Introduction
Breast cancer is one of the most genetically
heterogeneous cancers. It is also one of the most commonly
diagnosed cancers and the second most common cause of cancer
mortality in women (1). Despite
the improvement of current screening methods which rely on imaging
techniques and the targeted classification of breast cancer based
primarily on hormone receptors (estrogen and progesterone) and the
HER2/NEU status, mortality remains high for advanced breast cancer,
particularly when drug resistance develops.
Since Warburg first hypothesized that cancer cells
had a significantly higher rate of glycolysis than normal cells
(2), researchers have shown that
changes in metabolism can lead to large changes in metabolites that
occur downstream of genomic and proteomic alterations (3,4). How
signal pathways control energy metabolism in cancer cells has
become a high priority in cancer research, reflecting an increased
focus on the interaction of gene expression (i.e. receptors),
microenvironment (i.e. hypoxia), and glucose metabolism, and how
the downstream expression of metabolites can be used for both early
detection of breast cancer as well as targeted drug therapies
(5–7).
Traditional methods of analysis have evolved into
large-scale data gathering through different ‘omics’ approaches:
genomics, transcriptomics, proteomics and more recently,
metabolomics. This latter growing technology permits simultaneous
monitoring of thousands of macro and micro metabolites that serve
as substrates and products of metabolic pathways (8). Integration of these pathways and
their interactions provides insight into the development of
malignant processes and could lead to the discovery of cancer
biomarkers (9).
Several studies have demonstrated the efficacy of
using metabolomics to discriminate between cancer and normal
tissues in different organs including the breast. Sitter et
al (10) analyzed 88 tumor
samples from breast cancer patients and 18 tissue samples from
adjacent non-tumor tissue using high-resolution magic-angle
spinning magnetic resonance spectroscopy (HRMAS). Principal
component analysis (PCA) allowed for correct sample classification
in the majority of the cases with 82% sensitivity and 100%
specificity. Mountford et al (11) performed proton nuclear magnetic
resonance (1HNMR) spectroscopy analysis of breast tumor
extracts. Fine needle aspiration biopsies from 140 patients with
breast lumps (83 malignant and 57 benign) were analyzed by
1HNMR spectroscopy. Using a classification strategy,
they were able to classify samples as malignant or benign with a
sensitivity and specificity of 93 and 92%, respectively. More
recently, using high-throughput gas chromatography with
time-of-flight mass spectrometer (GC-TOFMS)-based metabolomic
analysis, Budczies et al (12) identified significant differences
between metabolites from breast tumors compared to normal tissues,
specifically the cytidine-5-monophosphate/pentadecanoic acid
metabolic ratio. This allowed the discrimination between normal and
cancer tissue samples with high specificity (93.9%) and high
sensitivity (94.8%). Furthermore, a comparison of estrogen receptor
positive and estrogen receptor negative breast cancer revealed
significant changes in glutamine and β-alanine metabolism between
these two breast cancer subtypes (13). Metabolomic profiling was used to
discriminate between localized early breast cancer and advanced
metastatic disease (14), and to
develop a prediction model for the early detection of recurrent
breast cancer from serum samples (15). Of interest, Budhu et al
(16), showed that there was a
specific metabolomic signature of tumors depending on the tissue of
origin and suggested that the metabolites were generally unique for
each tissue and cancer type. Comparing the metabolic changes
between cancer and normal cells could identify the metabolic
reprograming involved in tissue specific tumorigenesis.
To date, metabolomic analysis has been performed on
many different tissue types, including solid tissues, serum, plasma
and urine (17). Originally,
ductal lavage (DL) and nipple aspirate fluid (NAF) were used for
cytological evaluation of breast epithelial cells in the ductal
fluid. They have also been used for different molecular studies.
However, because they contain proteins and metabolites of breast
tissue metabolism in addition to ductal epithelial cells, they are
very useful for metabolomic studies, thus providing a unique
opportunity to evaluate more directly metabolomic changes in the
breast tumor microenvironment itself and avoiding questions of
tissue specificity, which arise when evaluating blood and urine.
The feasibility of performing metabolomic analysis in NAF was
recently demonstrated in a small study of eight subjects (18). The study was conducted on samples
obtained from healthy pre- and post-menopausal individuals and
compared the findings in NAF with matching plasma samples from the
same patients. They showed that NAF is metabolically distinct from
matched plasma samples which supports the theory that the cellular
environment (tumor microenvironment) is more directly mirrored in
breast biofluids (DL and NAF). We have recently identified a panel
of microRNAs that are differentially expressed in ductal fluid from
breasts with tumors compared to paired ductal fluid samples from
the contralateral normal breast (19), further substantiating the
importance of a more direct analysis of the tumor microenvironment
and the potential for biomarker development using ductal fluid
obtained in a non-invasive or minimally invasive approach. Here, we
report the first metabolomic analysis of breast ductal fluid
samples obtained from 43 patients with unilateral breast cancer. We
evaluated paired samples from the breast with cancer compared to
the contralateral non-affected breast (control) and identified
several metabolites with significant changes in levels between
affected and non-affected breasts. This approach provides an
exciting opportunity to detect metabolomic changes in the cellular
microenvironment reflecting tumor evolution, and has the potential
for significantly improving breast cancer screening and
detection
Materials and methods
Patient population
We enrolled 43 patients with unilateral,
biopsy-confirmed, breast tumors [invasive breast cancer (IBC)
and/or ductal carcinoma in situ (DCIS)], who were scheduled
for surgery (mastectomy/lumpectomy) at the MedStar Georgetown
University Hospital. Patients were identified by the surgeon and
offered the opportunity to participate in the study. If they
agreed, they were asked to sign an IRB-approved informed
consent.
Ductal lavage
Prior to starting the operative procedure, for each
subject, the surgeon obtained breast ductal fluid from the affected
breast and the non-affected contralateral breast, using ductal
lavage. Each patient served as her own control. The ductal lavage
procedure was performed as previously described (20), except that the collected fluid was
placed in a sterile tube with no preservative solution, and was
transferred immediately to the laboratory, and divided into
different aliquots which were frozen at −80°C for future studies.
One fresh aliquot was used for cytopathology evaluation to
investigate the presence of benign, atypical or malignant cells by
a certified breast pathologist, using the established criteria for
ductal lavage cytologic analysis (21).
Metabolite extraction from ductal
lavage
Metabolite extraction was performed as per the
protocol described by Sheikh et al (22). Briefly, 150 μl of ductal lavage
fluid was plunged into dry ice for 30 sec followed by heat shock at
37°C for 30 sec. A total of 600 μl of methanol containing
4-nitrobenzoic acid and debrisoquine were then added and the
samples were vortexed, transferred to room temperature and
extracted with chloroform. The tubes were transferred to −20°C for
overnight incubation and subsequently centrifuged at 4°C for 10 min
at 12,000 rpm. The top and bottom phases were transferred to
different tubes carefully avoiding the middle interface (containing
precipitated proteins). An equivalent amount of chilled
acetonitrile (ACN) was then added and the samples cooled on ice for
15 min after vortexing. Samples were centrifuged at 4°C for 10 min
at 12,000 rpm and the supernatant was transferred to a fresh tube
and dried under vacuum. The residual pellet was re-suspended in 200
μl of solvent A (98% water, 2% ACN and 0.1% formic acid) for
UPLC-Q-TOF/MS analysis.
UPLC-QTOF data acquisition
Each sample (5 μl) was injected onto a reverse-phase
50 × 2.1 mm BEH 1.7 μm C18 column using an Acquity UPLC system
(Waters Corp., Milford, MA, USA). The mobile phase comprised of
water containing 0.1% formic acid solution (A) and acetonitrile
containing 0.1% formic acid solution (B). Each sample was resolved
for 10 min at a flow rate of 0.5 ml/min. This approach has been
extensively used for metabolomic profiling of biofluids; UPLC
gradient conditions and the mass spectrometry parameters have been
described in detail (23,24). The column eluent was introduced
directly into the mass spectrometer by electrospray. Mass
spectrometry was performed on a Quadrupole-time-of-flight mass
spectrometer operating in either negative or positive electrospray
ionization mode with a capillary voltage of 3.2 kV and a sampling
cone voltage of 35 V. The desolvation gas flow was 800 l/h and the
temperature was set to 350°C. The cone gas flow was 50 l/h, and the
source temperature was 150°C. Accurate mass was maintained by
infusing sulfadimethoxine (311.0814 m/z) in 50% aqueous
acetonitrile (250 pg/μl) at a rate of 30 μl/min via the lockspray
interface every 10 sec. Data were acquired in centroid mode from 50
to 850 m/z mass range for TOF-MS scanning, in duplicate (technical
replicates) for each sample in positive and negative ionization
mode and checked for chromatographic reproducibility. For all
profiling experiments, the sample queue was staggered by
randomizing samples to eliminate bias. We acquired UPLC-QTOF data
by analysis of DL from 43 subjects. For each subject two samples,
one from the affected and one from the contralateral normal breast,
were generated. Each sample was injected twice.
Data preprocessing
The raw UPLC-QTOF data were converted into Network
Common Data Format (NetCDF) using the MassLynx software (Waters
Corp.). The R-package XCMS (Scripps Center for Metabolomics, La
Jolla, CA, USA) was used to preprocess the datasets acquired in the
electro-spray positive and negative ion modes. The first step in
XCMS is to detect the peaks. The peak detection algorithm first
cuts the data into slices, a fraction of a mass unit wide, and then
applies a model peak matched filter on those individual slices over
the chromatographic time domain. After detecting peaks in
individual samples, the peaks are matched across samples to allow
calculation of retention time (RT) deviations and relative ion
intensity comparison. This is accomplished using a grouping method
that uses kernel density estimation to group peaks in the mass
domain. The peak matching algorithm in XCMS takes into account the
two-dimensional anisotropic nature of data. These groups are then
used to identify and correct drifts in RT from run to run.
Following peak matching, we used the R-package
CAMERA to identify derivative ions originating from the same
compound in the form of adducts, isotopes, and in-source fragments
(25). Adducts of a molecule are
formed during the electrospray ionization (ESI) process, e.g.
sodium and ammonium adduct. Ions of molecular isotopes are detected
with distinct m/z values during MS analysis and the peak with the
lowest m/z is defined as the monoisotopic peak. In-source fragments
are formed during ionization such as ion fragments of
[M+H-H2O]+ or
[M-H-H2O]− through neutral loss of water
molecule. Different adducts/isotopes/water-loss products of the
same compound theoretically share the same retention time in
chromatograms. As long as the scan rate is properly adjusted and
enough scanning points are acquired to define the chromatographic
peaks, the ions from the same compound share similar-shaped elution
profiles which can be represented by their extracted ion
chromatograms (EICs). Thus, clustering of similar elution profiles
was performed by CAMERA prior to statistical analysis. Recognition
of such metabolites, often represented by multiple peaks with
distinct m/z values at similar retention times, can facilitate
metabolite identification in LC-MS based metabolomics.
Statistical analysis
To identify ions with significant changes in
intensity levels, we used parametric statistical methods that we
implemented in-house using MATLAB (MathWorks, Natick, MA, USA) and
R scripts. Before performing the statistical analysis, intensities
from double injections were averaged to achieve a single intensity
for each right or left DL sample. For pre-screening of ions, both
univariate analyses and multivariate analyses were performed. For
each ion, paired t-tests were performed to compare profiles of
metabolites between the tumor samples and the paired normal
samples. P-values obtained from multiple testing were adjusted to
q-values based on Storey's method (26). A robust pairwise fold change (FC)
was calculated based on the median of the relative intensity of
tumor/normal samples for each pair. In addition, we performed
principal component analysis (PCA) and multilevel PLS-DA (MPLS-DA)
that accounts for the paired data (27). The first screening was performed
based on q-values <0.05. To further identify which ions are
associated with the normal/tumor tissues, generalized LASSO
regression adjusting for race, menopausal status, smoking, grade,
and TNM stage and conditional logistic regression with LASSO
penalties were fitted where LASSO penalty parameters were
determined through 10-fold cross-validation (using the ‘glmnet’ and
‘clogitL1’ R package, respectively) (28,29).
Conditional logistic regression with the selected ions was employed
as a predictive model where the diagnostic performance of the
selected ions was assessed using the area under the receiver
operating characteristic (ROC) curves. In addition, one way ANOVA
and linear regression were used to compare cancer metabolomic
profiles of LASSO selected ions across different demographic and
clinical characteristics.
Putative identifications for the resulting ion list
were obtained through a mass-based search using MetaboSearch
(30), which searches for putative
identifications against four databases: the Human Metabolite
DataBase (HMDB) (31), Metlin
(32), Madison Metabolomics
Consortium Database (MMCD) (33),
and LIPID MAPS (34). The mass
tolerance in the database search was set to 10 ppm. The m/z values
of annotated isotopes/adducts/in-source fragments peaks were
converted to the corresponding neutral mono-isotopic masses before
searching them against the databases. Identities of a subset of the
putative metabolite identifications were verified by comparing
their MS/MS fragmentation patterns and RT with those of authentic
standard compounds.
Results
Clinical and tumor characteristics of the patients
are summarized in Table I. Our
patient population was 52 years of age on average, the majority
being post-menopausal (58%). Approximately 60% of our patients were
white, and a quarter of them were African Americans. The great
majority were never or former smokers. Most of the tumors were
invasive ductal carcinoma (IDC) (72%), early stage (I and II)
(81%), with no lymph node involvement (67%), hormone receptor
positive (88% ER+, 70% PR+), and HER2
negative (70%). More than two thirds of our patients had
insufficient cells for analysis in the ductal lavage samples from
the affected breast (70%), emphasizing the limitation of ductal
lavage cytology for detecting breast cancer. Subjects who received
pre-operative chemotherapy were excluded from the study.
 | Table ISubject characteristics. |
Table I
Subject characteristics.
|
Characteristics | N | %a |
|---|
| Age (mean ±
SD) | 52.20 (±12.25) | |
| Menopause |
| Pre | 18 | 41.86 |
| Post | 25 | 58.14 |
| Race/Ethnicity |
| A | 4 | 9.30 |
| AA | 11 | 25.59 |
| CA | 26 | 60.46 |
| H | 2 | 4.65 |
| Family history of
breast cancer |
| Yes | 18 | 41.86 |
| No | 25 | 58.14 |
| Tumor site |
| Right | 16 | 37.21 |
| Left | 27 | 62.79 |
| Smoking
history |
| Current | 4 | 9.30 |
| Former | 7 | 16.28 |
| Never | 32 | 74.42 |
| Histological
type |
| DCIS | 7 | 16.28 |
| IDC | 31 | 72.10 |
| ILC | 4 | 9.30 |
| Mixed | 1 | 2.32 |
| Stage |
| 0 | 7 | 16.28 |
| I | 17 | 39.54 |
| II | 18 | 41.86 |
| III | 1 | 2.32 |
| Grade |
| Low | 3 | 6.98 |
| Intermediate | 14 | 32.56 |
| High | 26 | 60.46 |
| Lymph node
involvement |
| Yes | 13 | 30.23 |
| No | 29 | 67.44 |
| ER |
| Positive | 38 | 88.37 |
| Negative | 5 | 11.63 |
| PR |
| Positive | 30 | 69.77 |
| Negative | 13 | 30.23 |
| HER2 |
| Positive | 8 | 18.60 |
| Negative | 30 | 69.77 |
| Affected breast
cytology |
| Atypical
cells | 4 | 9.30 |
| Benign cells | 9 | 20.93 |
| Insufficient
cells | 30 | 69.77 |
Preprocessing of the UPLC-QTOF data identified 2098
ions, 1560 ions in the positive mode and 538 ions in negative mode
(Table II). These data matrices
were used to select ions with significant differences between
ductal lavage fluid from the affected breast and the non-affected
contralateral control breast. Paired t-tests were applied on these
data matrices and identified 209 ions, out of the 2,098 ions, with
q-values <0.05 (Table II).
Among the 209 significant ions, 66 (31.6%) were assigned putative
compound names by MetaboSearch. When ranking these ions according
to the fold change (FC) in ion intensity levels between the
affected and non-affected breasts, 83 (39.7%) metabolites showed an
FC ≥1.2, 37 of these, being metabolites with a putative compound
name identified through the databases. The great majority of these
compounds were most likely of exogenous origin, being found in
plants, dietary supplements or drugs; however, some were endogenous
metabolites such as amino acid derivatives (N-Acetyl-DL-tryptophan,
FC=−2.02, P=0.006) or products of lipid metabolism such as
N-linoleoyl taurine (FC=−1.24, P=0.001),
trans-2-dodecenoylcarnitine (FC=−3.58, P=0.006),
lysophosphatidylcholine LysoPC(18:2(9Z,12Z)) (FC=−1.20, P=0.018),
and glycerophospholipids PG(18:0/0:0) (FC=−1.45, P=0.018) and
phosphatidylserine PS(20:4(5Z,8Z,11Z,14Z) (FC=−1.49, P<0.001).
Logistic regression with LASSO penalties further selected 21
metabolites, when race, menopausal status, smoking, grade and TNM
stage were adjusted for (Table
III). However, conditional logistic regression with LASSO
penalties yielded five ions where the predictive model does not
provide adequate prediction performance (AUC=0.679,
sensitivity/specificity of 0.744/0.581). Thus, further inferences
focused on the 21 ions.
| Table IINumber of ions detected and those
selected by statistical analysis. |
Table II
Number of ions detected and those
selected by statistical analysis.
| Mode | No. of ions
detected | No. of ions with
adjusted P-value <0.05 | No. of ions
selected by LASSO |
|---|
| Positive | 1560 | 197 | 19 |
| Negative | 538 | 12 | 2 |
| Table IIIMetabolites selected by LASSO. |
Table III
Metabolites selected by LASSO.
| Metabolite ID | Formula | Exact mass | ID (KEGG/HMDB) | Mode | m/z | RT | Cases vs.
control | Fold change | Adjusted
P-value |
|---|
| Putative
identification |
|
3,6,9,12,15,18-Hexaoxaicosaine-1,20-diol | C14H30O8 | 326.1940679 | | + | 327.2019744 | 128.9 | ↓ | −1.29 | 0.0289 |
| Angustifoline | C14H22N2O | 234.1732133 | C10751 | + | 235.1805058 | 143.4 | ↑ | 5.11 | 0.0289 |
| Pentaethylene
glycol | C10H22O6 | 238.1416384 | | + | 239.1485614 | 109.0 | ↓ | −3.58 | 0.0289 |
| Peroxyacetic acid
uroporphyrin III | C39H36N4O17 | 832.2075458 | HMDB03330 | + | 417.1126579 | 338.3 | ↓ | −1.17 | 0.0289 |
| No
identification |
| | | | + | 178.0315745 | 136.0 | ↓ | −1.32 | 0.0013 |
| | | | + | 788.4464449 | 20.4 | ↓ | −1.23 | 0.0021 |
| | | | + | 414.8635376 | 17.2 | ↓ | −1.10 | 0.0046 |
| | | | + | 417.8067865 | 338.1 | ↑ | 1.04 | 0.0046 |
| | | | + | 179.1841366 | 135.9 | ↓ | −1.66 | 0.0110 |
| | | | + | 454.8053723 | 338.2 | ↑ | 1.06 | 0.0110 |
| | | | − | 466.7290136 | 17.9 | ↑ | 1.05 | 0.0171 |
| | | | + | 121.2079108 | 338.4 | ↑ | 1.12 | 0.0196 |
| | | | + | 808.5359146 | 18.4 | ↓ | −1.04 | 0.0196 |
| | | | + | 454.4746506 | 337.7 | ↑ | 1.05 | 0.0289 |
| | | | + | 636.3472375 | 174.4 | ↑ | 1.02 | 0.0289 |
| | | | + | 664.7500890 | 17.7 | ↑ | 1.06 | 0.0289 |
| | | | + | 702.8673044 | 17.1 | ↑ | 1.05 | 0.0289 |
| | | | + | 760.6118168 | 18.3 | ↑ | 1.02 | 0.0289 |
| | | | − | 336.7892724 | 17.9 | ↑ | 1.02 | 0.0350 |
| | | | + | 622.6270105 | 18.4 | ↓ | −1.11 | 0.0491 |
| | | | + | 724.4910051 | 20.4 | ↓ | −1.04 | 0.0491 |
We performed principal component analysis (PCA) and
multilevel PLS-DA (MPLS-DA) that accounts for the cross-over data
and examined the performance of selected biomarkers. The PCA plot
based on the 21 ions (identified using the LASSO regression
analysis; Fig. 1B) shows visually
better clustering performances of both sets of ions than the PCA
plot based on 209 ions (q-values <0.05; Fig. 1A).
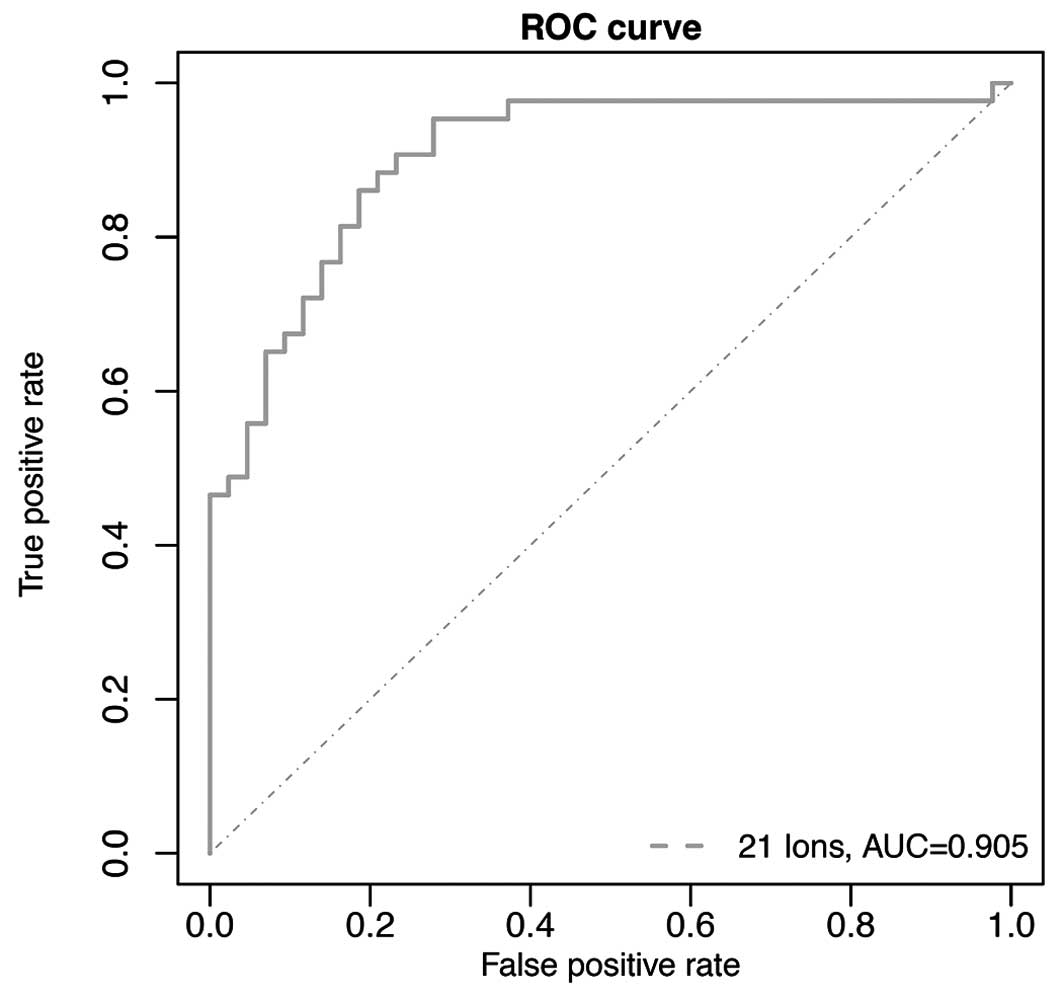
We evaluated whether the LASSO-selected 21 ions can
be used as potential biomarkers for breast cancer detection.
Conditional logistic regression with the 21 ions was employed as a
predictive model where the diagnostic performance of the 21 ions
was assessed using the area under the receiver operating
characteristic (ROC) curves. ROC based on the conditional logistic
regression model gave highly accurate diagnostic performance with
an area under the curve (AUC) of 0.956 along with a sensitivity of
90.7% and specificity of 88.4% (Fig.
2). In addition, one way ANOVA and linear regression were used
to compare cancer metabolomic profiles of the LASSO-selected ions
across different demographic and clinical characteristics. Among
the 21 LASSO-identified ions, we found ten metabolites that show
statistically significant differences based on menopausal status
(pre/post), ER (+/−), and HER2 (+/−). Table IV shows these metabolites.
P-values are in bold for the metabolites with P<0.05.
| Table IVMetabolites, among the LASSO
identified ions, that show statistically significant differences
based on menopausal status (pre/post), ER (+/−) and HER2 (+/−). |
Table IV
Metabolites, among the LASSO
identified ions, that show statistically significant differences
based on menopausal status (pre/post), ER (+/−) and HER2 (+/−).
| M/z | RT | Menopause | ER | Her2 |
|---|
| 239.1486 | 109.0287 | 0.849 | 0.045 | 0.437 |
| 179.1841 | 135.8769 | 0.026 | 0.432 | 0.226 |
| 121.2079 | 338.3861 | 0.049 | 0.788 | 0.000 |
| 178.0316 | 136.0036 | 0.124 | 0.3 | 0.036 |
| 417.1127 | 338.331 | 0.238 | 0.997 | 0.022 |
| 454.4747 | 337.7336 | 0.538 | 0.388 | 0.003 |
| 622.627 | 18.4193 | 0.04 | 0.777 | 0.280 |
| 724.491 | 20.4252 | 0.269 | 0.407 | 0.004 |
| 788.4464 | 20.4252 | 0.649 | 0.673 | 0.001 |
| 466.729 | 17.86 | 0.026 | 0.774 | 0.133 |
We found that menopausal status can affect the level
of metabolite expression among our patients. Three metabolites
(M179.1841, M121.2079 and M466.729) are higher in post-menopausal
patients (P<0.05; Table IV).
On the other hand, one metabolite (M622.627) is higher in
pre-menopausal patients (P=0.040; Table IV). HER2 positive receptor status
also shows an association with increased levels of metabolites
(M724.491 and M788.4464) in patients (P<0.005; Table IV), while four metabolites
(M121.2079, M178.0316, M417.1127 and M454.4747) are lower in HER2
positive patients (P<0.05; Table
IV).
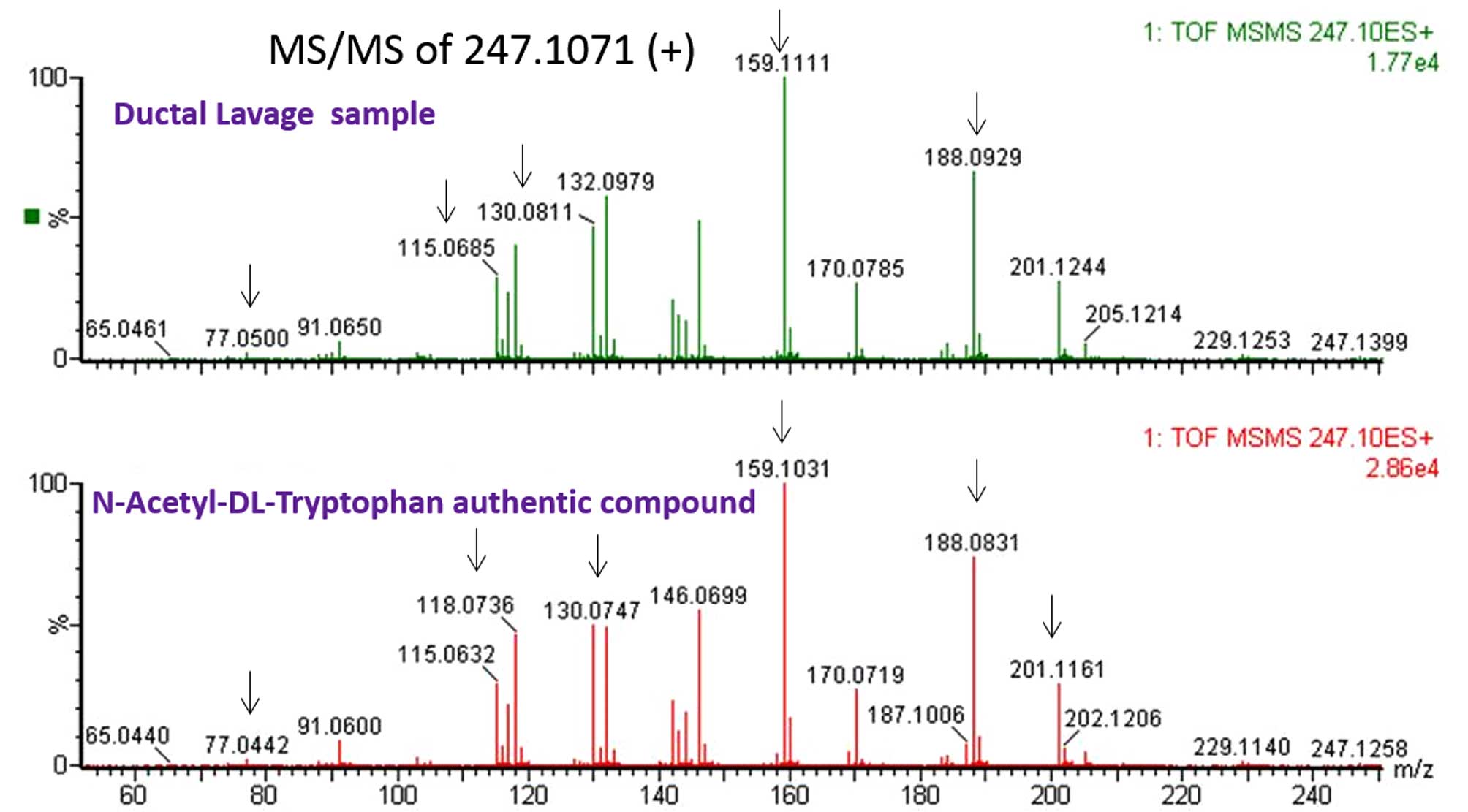
From the list of 209 significant monoisotopic ions
with putative identifications, we selected eight metabolites with
putative IDs and standard compounds for verification by comparing
their TOF-MS/MS spectra against their corresponding synthetic
compounds. Specifically, we ran an authentic compound for each
candidate and a patient sample side by side and compared the
fragmentation patterns and their LC retention time. We were able to
verify six metabolites against their standards, experimentally.
Table V shows a list of the six
verified and two unverified metabolites, and Fig. 3 shows an example of the
verification result for N-acetyl-DL-tryptophan, comparing the
fragmentation pattern of the analyte in ductal lavage against the
pattern obtained from an authentic compound.
| Table VThe verified and unverified
metabolites. |
Table V
The verified and unverified
metabolites.
| M/Z | RT(sec) | RT(min) | Monoisotopic
mass | Putative name of
the compound | FC | q-value |
|---|
| Verified
metabolites |
| 247.1071 | 187 | 3.12 | 247.1070665 |
N-Acetyl-Dtryptophan | −2.02309 | 0.049087 |
| 134.0966 | 136 | 2.27 | 134.0965659 |
1,2,3,4-tetrahydroisoquinoline | −1.12989 | 0.049087 |
| 179.0568 | 136 | 2.27 | 179.0567725 | Gluconolactone | −1.26445 | 0.049087 |
| 513.3184 | 351 | 5.85 | 513.3184402 | Phosphatidyl
Glycerol (18:0/0:0) | −1.45152 | 0.049087 |
| 174.1487 | 234 | 3.90 | 174.1487168 | 9-amino-nonanoic
acid | −1.15122 | 0.028858 |
| 433.2574 | 440 | 7.33 | 433.2574371 | Hydrocortisone
butyrate | −1.08314 | 0.028858 |
| Unverified
metabolites |
| 620.5977 | 19 | 0.32 | 620.5977263 | Ceramide
(d18:2/22:0) | −1.08974 | 0.028858 |
| 429.2271 | 365 | 6.08 | 429.2270941 | Phosphatidyl
glycerol (12:0/0:0) | 1.03441 | 0.019619 |
Discussion
The emerging technology of metabolomics has provided
insights into cancer metabolic pathways in different organs,
including the mammary gland, which can lead to the discovery of new
cancer biomarkers and therapies. To the best of our knowledge, this
is the first study that compares the metabolomic profiles in the
ductal fluid from cancer affected breasts vs. non-affected
contralateral control breasts in the same subjects, allowing us to
view the metabolites in the tumor microenvironment where breast
cancer arises, in a unique well-controlled setting where each
patient serves as her own control.
We analyzed ductal fluid samples obtained by ductal
lavage from 43 subjects with unilateral breast cancer from both the
affected breast and the contralateral non-affected control breast
and identified 209 ions that showed a significant difference in
intensity levels (q-values <0.05) between the fluid from the
affected breast vs. the fluid from the non-affected control breast.
We observed significant differences between the fluid from the
affected breast vs. the fluid from the non-affected control breast
in the levels of tryptophan, products of lipid metabolism,
derivatives of amino acid metabolism and phospholipids.
Interestingly, metabolomic profiles are not only
different between cancer and normal tissue as we and others have
shown, but also are cancer tissue type specific (16,35).
In addition, specific metabolite changes occur at earlier stages
for each tissue and cancer type. For instance, amino acid related
metabolites, such as N-Acetyl-amino acids, and lipid related
metabolites, such as carnitine and glycerophopsholipids, were shown
to be significantly altered in breast tumors (14,16);
in serum, amino acid changes were mainly observed in early stage
breast cancer (stages I and II) compared to metastatic disease
(14). A supervised class
comparison analysis of tumor vs. non-tumor tissues revealed
specific metabolites that could discriminate between liver,
pancreas and breast, with the most evident differences occurring in
amino acid and lipid pathways; glycerol and linolenate were
upregulated only in breast tumors (16). In individuals with early stage
disease, 8 metabolites were significantly and uniquely altered in
breast tumors vs. normal tissues; in a similar analysis, 81
metabolites in liver and 18 in pancreatic tumors were significantly
and differentially altered (16).
Comparing the metabolic changes between tissue specific cancer and
normal cells could pinpoint the metabolic reprograming involved in
tissue specific tumorigenesis.
In the present study we identified lower levels of
N-Acetyl-DL-tryptophan in the fluid from the affected breasts
compared to the control breast fluid, consistent with previous
reports showing decreased amino acid metabolites in breast cancer
(36–38). Cancer metabolism requires energy
derived from both anaerobic and aerobic glycolysis, which means
lower substrate levels like glucose and glutamine being siphoned
off to the tricarboxylic acid (TCA) cycle, and higher levels of
bioenergic substrates, like lactate, which stimulates continuous
growth (36). In a recent study,
Willmann et al (39), used
metabolomic profiling to sub-classify different breast cancer cell
lines and differentiate not only between them, but also between the
cancer cell lines and a non-cancer breast epithelial cell line as
well. Research has shown that elevated glycine and lactate indicate
increased metabolic changes due to rapid growth of cancer cells,
and hence poorer prognosis (5).
Levels of metabolites were also reported to differ
by race (38). However, in this
study, we were not able to detect a statistically significant
difference in the metabolite levels based on the subjects' race
possibly due to the small sample size per each race. None of the
LASSO-identified 21 ions showed statistically significant
difference among patients based on race (minimum P-values >0.50;
data not shown).
When we stratified our patients by receptor status,
we observed that the M239.1486 metabolite tends to be less
expressed in ER+ patients (P=0.045; Table IV). Fan et al (35) analyzed plasma metabolite profiles
of 96 breast cancer patients compared to 79 normal controls by
UPLC-Q/TOF-MS and GC-Q/MS, and identified several metabolites that
could discriminate between breast cancer and controls, and also
between breast cancer subtypes according to HER2 and estrogen
receptor status. ER positive (ER+) patients showed
elevated alanine, aspartate and glutamate metabolism, decreased
glycerolipid catabolism and enhanced purine metabolism compared to
the ER negative (ER−) group (also described in plasma
metabolites from breast cancer patients vs. normal controls
(38). ER− and
triple-negative receptor status is indicative of more aggressive
tumors and a poorer prognosis. In breast cancer, the substrate
glutamine and end product lactate enhance cancer aggressiveness;
additionally ER− breast cancers are dependent upon
serine synthesis for continued growth (36).
In the present study, metabolites involved in lipid
signaling were decreased in the fluid from the breasts with tumors,
similar to previously published study (36). Lower levels of
lysophosphatidylcholines and higher levels of sphingomyelins and
acylcarnitines were detected in the plasma of cancer patients
compared to controls (37). The
decrease in glycerophopsholipid levels in cancer patients may be
due to a higher expression of phospholipase A2 (PLA2), a gene
encoding for the enzymes responsible for their metabolism (37), and whose expression was found to be
upregulated in breast cancer cell lines and tissues (40,41).
Higher levels of lipid metabolites, including fatty acids and
carnitine metabolites, have been found in breast cancer patients
compared to normal controls (35,38,42).
Wang et al (9) reported
that several lipids including phosphatidylglycerol (PG) were
upregulated in highly invasive breast cancer cells. Breast tumors
are described as developing a ‘lipogenic phenotype’, and while cell
growth is dependent on lipogenesis, lipolysis is upregulated, and
is associated with tumor aggressiveness (36,37).
Although our findings do not show exactly the same
metabolites being altered as reported in other studies, the
metabolites that we identified belong to the same class of
compounds and same metabolic pathways, especially in the case of
lipid metabolites. Cancer cells have been shown to have an
increased glucose uptake (Warburg effect), conversion to lactate
(glycolysis) and changes in protein and lipid metabolism (43). Some differences between studies can
be attributed to the biological specimens analyzed (ductal lavage)
vs. tissue and blood, or to technical or methodological
differences. Moreover, a large part of the differentially expressed
metabolites from the present study are still unknown, emphasizing
the complexity of metabolic alterations in cancer.
Because the ductal lavage samples were collected
under anesthesia immediately before surgery when the patient was
already medicated, we identified many drug-related metabolites in
our samples. However, because many of these drug-related
metabolites were different between the breasts with tumors and
normal breasts, this altered drug metabolism in the affected breast
could be of possible therapeutic benefit and requires additional
analysis. Identification of the specific metabolites could be used
to target specific enzymes. Enzyme expression levels and metabolic
pathways could be used to target specific breast cancer phenotypes,
or cancer cell survival within tumors (7).
A metabolomics based profile for the early detection
of breast cancer recurrence has been developed by Asiago et
al (15). They used 11
metabolites in their prediction model, several of which we have
reported as being significantly differentially expressed in the
ductal lavage samples. A follow-up analysis of our patients might
yield comparable results and contribute to improving earlier
detection and better treatment in the cases of breast cancer
recurrence.
Although our study design was unique in that we used
ductal fluid obtained from the contralateral normal breast for
comparison, which allowed us to control for several variables that
could not have been controlled for otherwise, a drawback of such a
design is the possibility that the contra-lateral breast cells may
already harbor metabolomic changes. We, and others, have shown that
genomic aberrations occur very early in tumorigenesis and may
precede morphologic changes (20,44–46). Therefore, we cannot rule
out the possibility of similar changes affecting some metabolites
in the contralateral clinically normal breast. A future study may
include an additional control group of normal subjects (e.g.,
patients undergoing reduction mammoplasty) to account for this
possibility.
In summary, this is the first study to assess
metabolomic profiles in ductal fluid samples from breast cancer
patients. The differences we saw were directly related to the tumor
microenvironment and provide a snapshot of ongoing breast cancer
metabolism. However, the identification of several metabolites with
altered levels in the cancer affected breast, especially lipid
related metabolites, previously reported in breast cancer tissue,
cell lines and the circulation, provide confidence that these could
constitute the basis for metabolomics based markers for breast
cancer detection. This study shows the feasibility of conducting a
comprehensive metabolomic profiling of breast tumors using breast
ductal fluid to detect changes in the cellular microenvironment of
the tumors and shows the potential for this approach to be used to
improve detection of breast cancer.
Acknowledgements
The present study was supported by a grant from the
Avon Foundation for Women.
References
|
1
|
American Cancer Society. Global Cancer
Facts and Figures. 3rd edition. American Cancer Society; Atlanta,
GA: 2015
|
|
2
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Denkert C, Bucher E, Hilvo M, Salek R,
Orešič M, Griffin J, Brockmöller S, Klauschen F, Loibl S, Barupal
DK, et al: Metabolomics of human breast cancer: New approaches for
tumor typing and biomarker discovery. Genome Med.
4:372012.PubMed/NCBI
|
|
4
|
Sengupta D and Pratx G: Imaging metabolic
heterogeneity in cancer. Mol Cancer. 15:42016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Long JP, Li XN and Zhang F: Targeting
metabolism in breast cancer: How far we can go? World J Clin Oncol.
7:122–130. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mannello F and Ligi D: Resolving breast
cancer heterogeneity by searching reliable protein cancer
biomarkers in the breast fluid secretome. BMC Cancer. 13:3442013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shajahan-Haq AN, Cheema MS and Clarke R:
Application of metabolomics in drug resistant breast cancer
research. Metabolites. 5:100–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Claudino WM, Quattrone A, Biganzoli L,
Pestrin M, Bertini I and Di Leo A: Metabolomics: Available results,
current research projects in breast cancer, and future
applications. J Clin Oncol. 25:2840–2846. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang J, Zuo Y, Man YG, Avital I,
Stojadinovic A, Liu M, Yang X, Varghese RS, Tadesse MG and Ressom
HW: Pathway and network approaches for identification of cancer
signature markers from omics data. J Cancer. 6:54–65. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sitter B, Lundgren S, Bathen TF, Halgunset
J, Fjosne HE and Gribbestad IS: Comparison of HR MAS MR
spectroscopic profiles of breast cancer tissue with clinical
parameters. NMR Biomed. 19:30–40. 2006. View Article : Google Scholar
|
|
11
|
Mountford CE, Somorjai RL, Malycha P,
Gluch L, Lean C, Russell P, Barraclough B, Gillett D, Himmelreich
U, Dolenko B, et al: Diagnosis and prognosis of breast cancer by
magnetic resonance spectroscopy of fine-needle aspirates analysed
using a statistical classification strategy. Br J Surg.
88:1234–1240. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Budczies J, Denkert C, Müller BM,
Brockmöller SF, Klauschen F, Györffy B, Dietel M,
Richter-Ehrenstein C, Marten U, Salek RM, et al: Remodeling of
central metabolism in invasive breast cancer compared to normal
breast tissue - a GC-TOFMS based metabolomics study. BMC Genomics.
13:3342012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Budczies J, Brockmöller SF, Müller BM,
Barupal DK, Richter-Ehrenstein C, Kleine-Tebbe A, Griffin JL,
Orešič M, Dietel M, Denkert C, et al: Comparative metabolomics of
estrogen receptor positive and estrogen receptor negative breast
cancer: Alterations in glutamine and beta-alanine metabolism. J
Proteomics. 94:279–288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jobard E, Pontoizeau C, Blaise BJ,
Bachelot T, Elena-Herrmann B and Trédan O: A serum nuclear magnetic
resonance-based metabolomic signature of advanced metastatic human
breast cancer. Cancer Lett. 343:33–41. 2014. View Article : Google Scholar
|
|
15
|
Asiago VM, Alvarado LZ, Shanaiah N, Gowda
GA, Owusu-Sarfo K, Ballas RA and Raftery D: Early detection of
recurrent breast cancer using metabolite profiling. Cancer Res.
70:8309–8318. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Budhu A, Terunuma A, Zhang G, Hussain SP,
Ambs S and Wang XW: Metabolic profiles are principally different
between cancers of the liver, pancreas and breast. Int J Biol Sci.
10:966–972. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Armitage EG and Barbas C: Metabolomics in
cancer biomarker discovery: Current trends and future perspectives.
J Pharm Biomed Anal. 87:1–11. 2014. View Article : Google Scholar
|
|
18
|
Tredwell GD, Miller JA, Chow HH, Thompson
PA and Keun HC: Metabolomic characterization of nipple aspirate
fluid by 1H NMR spectroscopy and GC-MS. J Proteome Res.
13:883–889. 2014. View Article : Google Scholar
|
|
19
|
Do Canto LM, Marian C, Willey S, Sidawy M,
Da Cunha PA, Rone JD, Li X, Gusev Y and Haddad BR: MicroRNA
analysis of breast ductal fluid in breast cancer patients. Int J
Oncol. 48:2071–2078. 2016.PubMed/NCBI
|
|
20
|
Isaacs C, Cavalli LR, Cohen Y, Pennanen M,
Shankar LK, Freedman M, Singh B, Liu M, Gallagher A, Rone JD, et
al: Detection of LOH and mitochondrial DNA alterations in ductal
lavage and nipple aspirate fluids from high-risk patients. Breast
Cancer Res Treat. 84:99–105. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Masood S: Development of a novel approach
for breast cancer prediction and early detection using minimally
invasive procedures and molecular analysis: How cytomorphology
became a breast cancer risk predictor. Breast J. 21:82–96. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sheikh KD, Khanna S, Byers SW, Fornace A
Jr and Cheema AK: Small molecule metabolite extraction strategy for
improving LC/MS detection of cancer cell metabolome. J Biomol Tech.
22:1–4. 2011.PubMed/NCBI
|
|
23
|
Kaur P, Rizk N, Ibrahim S, Luo Y, Younes
N, Perry B, Dennis K, Zirie M, Luta G and Cheema AK: Quantitative
metabolomic and lipidomic profiling reveals aberrant amino acid
metabolism in type 2 diabetes. Mol Biosyst. 9:307–317. 2013.
View Article : Google Scholar
|
|
24
|
Patterson AD, Li H, Eichler GS, Krausz KW,
Weinstein JN, Fornace AJ Jr, Gonzalez FJ and Idle JR:
UPLC-ESI-TOFMS-based metabolomics and gene expression dynamics
inspector self-organizing metabolomic maps as tools for
understanding the cellular response to ionizing radiation. Anal
Chem. 80:665–674. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kuhl C, Tautenhahn R, Böttcher C, Larson
TR and Neumann S: CAMERA: An integrated strategy for compound
spectra extraction and annotation of liquid chromatography/mass
spectrometry data sets. Anal Chem. 84:283–289. 2012. View Article : Google Scholar
|
|
26
|
Storey J: False discovery rates.
International Encyclopedia of Statistical Science. 1st edition.
Lovric M: Springer; pp. 16732011
|
|
27
|
Westerhuis JA, van Velzen EJ, Hoefsloot HC
and Smilde AK: Multivariate paired data analysis: Multilevel PLSDA
versus OPLSDA. Metabolomics. 6:119–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Friedman J, Hastie T and Tibshirani R:
Regularization paths for generalized linear models via coordinate
descent. J Stat Softw. 33:1–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reid S and Tibshirani R: Regularization
paths for conditional logistic regression: The clogitL1 package. J
Stat Softw. 58:122014. View Article : Google Scholar
|
|
30
|
Zhou B, Wang J and Ressom HW:
MetaboSearch: Tool for mass-based metabolite identification using
multiple databases. PLoS One. 7:e400962012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wishart DS, Knox C, Guo AC, Eisner R,
Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S, et al:
HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res.
37(Database): D603–D610. 2009. View Article : Google Scholar :
|
|
32
|
Smith CA, O'Maille G, Want EJ, Qin C,
Trauger SA, Brandon TR, Custodio DE, Abagyan R and Siuzdak G:
METLIN: A metabolite mass spectral database. Ther Drug Monit.
27:747–751. 2005. View Article : Google Scholar
|
|
33
|
Cui Q, Lewis IA, Hegeman AD, Anderson ME,
Li J, Schulte CF, Westler WM, Eghbalnia HR, Sussman MR and Markley
JL: Metabolite identification via the Madison Metabolomics
Consortium Database. Nat Biotechnol. 26:162–164. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sud M, Fahy E, Cotter D, Brown A, Dennis
EA, Glass CK, Merrill AH Jr, Murphy RC, Raetz CR, Russell DW, et
al: LMSD: LIPID MAPS structure database. Nucleic Acids Res.
35(Database): D527–D532. 2007. View Article : Google Scholar
|
|
35
|
Fan Y, Zhou X, Xia TS, Chen Z, Li J, Liu
Q, Alolga RN, Chen Y, Lai MD, Li P, et al: Human plasma
metabolomics for identifying differential metabolites and
predicting molecular subtypes of breast cancer. Oncotarget.
7:9925–9938. 2016.PubMed/NCBI
|
|
36
|
Mishra P and Ambs S: Metabolic signatures
of human breast cancer. Mol Cell Oncol. 2:22015.
|
|
37
|
Qiu Y, Zhou B, Su M, Baxter S, Zheng X,
Zhao X, Yen Y and Jia W: Mass spectrometry-based quantitative
metabolomics revealed a distinct lipid profile in breast cancer
patients. Int J Mol Sci. 14:8047–8061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shen J, Yan L, Liu S, Ambrosone CB and
Zhao H: Plasma metabolomic profiles in breast cancer patients and
healthy controls: By race and tumor receptor subtypes. Transl
Oncol. 6:757–765. 2013. View Article : Google Scholar
|
|
39
|
Willmann L, Schlimpert M, Halbach S, Erbes
T, Stickeler E and Kammerer B: Metabolic profiling of breast
cancer: Differences in central metabolism between subtypes of
breast cancer cell lines. J Chromatogr B Analyt Technol Biomed Life
Sci. 1000:95–104. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yamashita J, Ogawa M and Sakai K:
Prognostic significance of three novel biologic factors in a
clinical trial of adjuvant therapy for node-negative breast cancer.
Surgery. 117:601–608. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yamashita S, Yamashita J, Sakamoto K,
Inada K, Nakashima Y, Murata K, Saishoji T, Nomura K and Ogawa M:
Increased expression of membrane-associated phospholipase A2 shows
malignant potential of human breast cancer cells. Cancer.
71:3058–3064. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Takayama T, Tsutsui H, Shimizu I, Toyama
T, Yoshimoto N, Endo Y, Inoue K, Todoroki K, Min JZ, Mizuno H, et
al: Diagnostic approach to breast cancer patients based on target
metabolomics in saliva by liquid chromatography with tandem mass
spectrometry. Clin Chim Acta. 452:18–26. 2016. View Article : Google Scholar
|
|
43
|
Hadi NI and Jamal Q: ‘OMIC’ tumor markers
for breast cancer: A review. Pak J Med Sci. 31:1256–1262. 2015.
View Article : Google Scholar : PubMed/NCBI
|