Contents
Introduction
EGFR and autophagy regulation
Autophagy inhibition may be a promising therapeutic
strategy for enhancing the effects of EGFR-targeted therapy in
NSCLC
Issues and perspectives
Introduction
Non-small-cell lung cancer (NSCLC) remains the
leading cause of cancer-related mortality worldwide (1). NSCLC is closely associated with
mutations of the epidermal growth factor receptor (EGFR), which are
associated with adenocarcinoma histology, female gender, Asian
ethnicity and a non-smoker status (2,3).
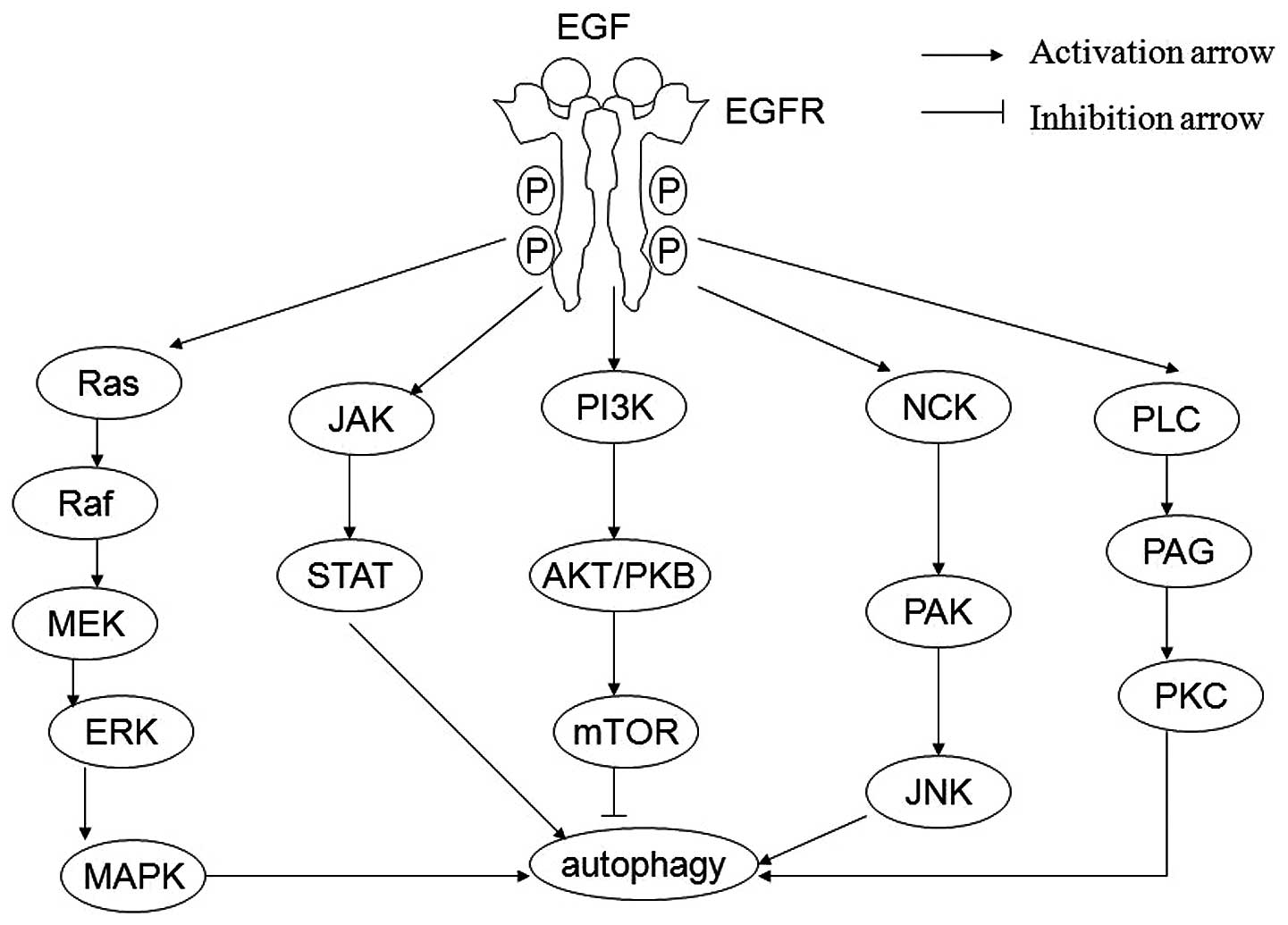
Subsequent to ligand binding, EGFR becomes activated via
phosphorylation. Phosphorylated EGFR subsequently activates
pro-survival and antiapoptotic signals through its downstream
targets, such as Ras/Raf/MEK/MAPK, JAK/STAT, PI3K/AKT/mTOR,
NCK/PAK/JNK and PLC/PAG/PKC (Fig.
1) (4,5). Consequently, inhibition of the EGFR
network may activate the intrinsic mitochondrial apoptotic pathway
and exhibit antitumor activity against NSCLC. Two specific
EGFR-tyrosine kinase inhibitors (TKIs), gefitinib and erlotinib,
have been approved by the Food and Drug Administration (FDA) thus
far for the treatment of patients with advanced NSCLC.
Notwithstanding the success of these drugs in cases of NSCLC with
classical EGFR mutations, it appears that all the cases ultimately
relapse, resulting in acquired resistance to EGFR-TKIs (6). These unsatisfactory outcomes are, in
part, due to macroautophagy (hereafter autophagy), which is
considered to be crucial in the resistance to EGFR-TKIs.
Autophagy is a catabolic process in which portions
of cytosol and organelles are sequestered into a double-membrane
vesicle and delivered to the lysosome for bulk degradation to
maintain cellular homeostasis (7,8). The
role of autophagy in regulating cancer cell death or survival has
not been fully elucidated. When cancer cells are subjected to
unfavorable conditions, such as nutrient-deficient environment or
treatment with anticancer drugs, autophagy is rapidly upregulated
to function as a cytoprotective mechanism which enhances cancer
cell survival, and may contribute to the resistance to anticancer
therapies (9). Excessive autophagy
may also lead to autophagic cell death, which is also referred to
as type II programmed cell death to distinguish it from type I
programmed cell death (apoptosis) (10). Various stress signals may induce
autophagy, including p53, phosphatidylinositol 3-kinase
(PI3K)/mammalian target of rapamycin (mTOR), AMP-activated protein
kinase (AMPK) and other signaling pathways (11,12).
Among these signals, the AMPK/mTOR axis is critical for the
regulation of autophagy. Since PI3K/AKT/mTOR signaling is involved
in the regulation of EGFR as well as in autophagic flux, there is
likely an association between autophagy and EGFR. Our laboratory
recently reported that EGFR-TKIs may activate cell autophagy to
impair the sensitivity of lung cancer cells to targeted therapy by
inhibiting the AKT/mTOR/p70S6K signaling pathway (13). In this review, we aimed to
summarize recent studies on the ability of EGFR-TKIs to induce
autophagy, assess whether this ability is correlated with
EGFR-targeted therapy resistance and investigate the prospect of
manipulating this ability as a novel method of sensitizing cancer
cells to these type of drugs.
EGFR and autophagy regulation
The EGFR signaling pathway is frequently
dysregulated in a variety of human malignancies, which has
attracted interest in EGFR as a target of anticancer therapies.
Several small-molecule TKIs, such as erlotinib or gefitinib, as
well as anti-EGFR antibodies, such as cetuximab, have been approved
by the FDA and the European Medicines Evaluation Agency for the
treatment of patients with breast and colorectal cancer, NSCLC,
squamous cell carcinoma of the head and neck and pancreatic cancer
(14–16).
Although generally known as a plasma membrane
protein, EGFR has also been identified in the nucleus and
mitochondria (17,18). Increasing evidence has indicated
that the EGFR functionality may be dependent on its subcellular
location. In the plasma, EGFR is best known for its classical
function as a membrane-bound receptor tyrosine kinase, which is
phosphorylated following ligand binding to activate its downstream
targets, which are involved in the regulation of autophagy.
However, in the nucleus, EGFR behaves as a transcriptional
regulator and mediator of other physiological processes. There is
currrently no evidence to elucidate whether EGFR is associated with
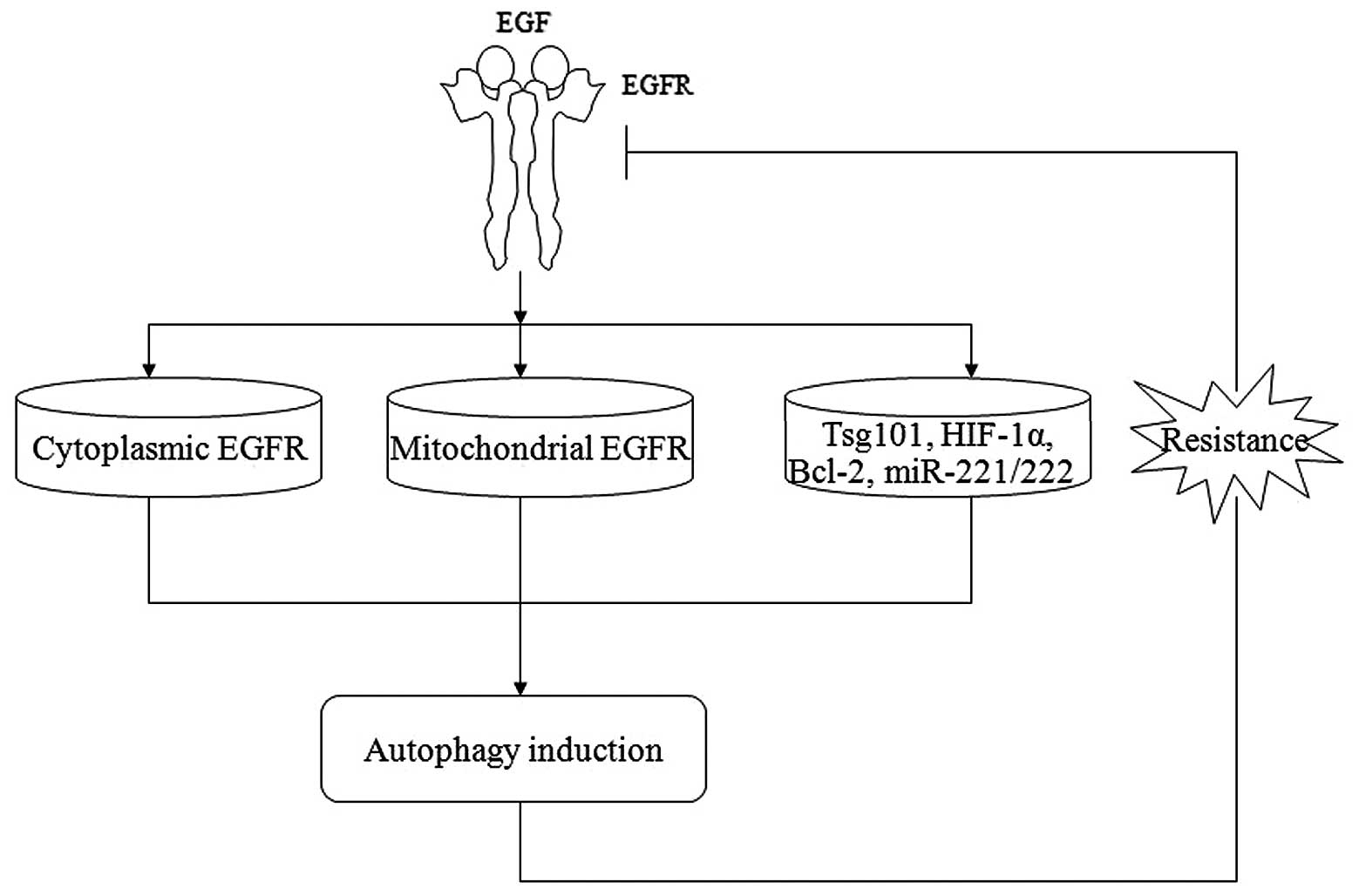
autophagy. Recently, the mitochondrial localization of EGFR was
also reported, although the role of mitochondrially localized EGFR
is unknown (19). A novel role of
mitochondrial EGFR, that appears to regulate apoptosis and
autophagy, was recently identified (Fig. 2) (19,20).
Cytoplasmic EGFR is a member of the human epidermal
growth factor receptor (HER)/ErbB family of receptor tyrosine
kinases. Upon binding to its ligand, EGFR undergoes homo- or
heterodimerization with its family members ErbB2/neu, ErbB3/HER3
and ErbB4/HER4, which leads to the phosphorylation of EGFR and
other intracellular signaling proteins and subsequently initiates a
number of downstream signaling molecules that are critical for
cancer cell growth, survival and proliferation (21,22).
The main pathways triggered by EGFR activation include MAPK,
JAK/STAT, PI3K/AKT/mTOR, NCK/PAK/JNK and PLC/PAG/PKC, all of which
exert a potent stimulatory effect on autophagy (Fig. 1) (4,5).
Although autophagy enhanced by troglitazone is considered to be
independent of EGFR transactivation, it was more recently
demonstrated that EGFR may mediate autophagy through its downstream
signals, such as the AMPK/mTOR pathway (23). Mammary epithelial cells expressing
the delE746-A750EGFR oncogene have a functional
AMP/LKB1/AMPKα sensor circuitry. Moreover, EGFR activation protects
against ultraviolet-induced apoptosis in cultured mouse dendritic
cells through the induction of the LKB1/AMPK pathway (24,25).
The EGFR inhibitor BIBU may induce apoptosis and prevent execution
of autophagy in glioma cells via the inhibition of the pro-survival
pathways AKT/mTOR and gp130/JAK/STAT3 (26). The MAPK pathway may transmit
signals from the receptor to the nucleus through a series of
intermediate proteins, including Ras, Raf, MEK and ERK. Akhtar
et al (27) reported that
activation of EGFR via pathways involving ERK1/2, p38 MAPK and
AKT/FOXO promoted cardiac cell survival during ischemia-reperfusion
injury. Chen et al (28)
demonstrated that the surface receptor EGFR and its downstream
ERK/MAPK signaling pathway are crucial in the activation of
autophagy in cathepsin S-targeted cells. Furthermore, a recent
study indicated that EGFR downstream signal JNK exerts a major
protective effect by increasing autophagy, leading to antagonising
apoptosis and ROS generation in A431 cells (29). These data support the fact that
cytoplasmic EGFR contributes to autophagic regulation by its
downstream signalings, although the association between EGFR and
autophagy remains unelucidated.
Mitochondrial detection of EGFR was first reported
by Boerner et al (18).
That study demonstrated that EGFR may translocate to the
mitochondria through interaction with cytochrome c oxidase
subunit II, which may positively regulate survival pathways that
contribute to the development of breast cancer. Recent studies
indicated that mitochondrial EGFR and EGFRvIII, a constitutively
activated variant of EGFR, may contribute to tumor resistance to
anticancer therapy through the regulation of autophagy (20,30).
EGFR mitochondrial translocation may be enhanced in A431 cells by
rapamycin, an autophagy inducer. Conversely, autophagy inhibition
by 3-methyladenine (an inhibitor of autophagy) or Beclin 1
knockdown by small interfering RNA (siRNA) leads to a reduction of
the rapamycin-induced mitochondrial import of EGFR (20). Cao et al (31) demonstrated that tumor cells
exhibiting mitochondrial EGFRvIII accumulation were highly
resistant to Iressa-mediated growth inhibition and mitochondrial
accumulation of EGFR as well as EGFRvIII, contributes to cancer
drug resistance. Thus, under certain unfavorable and harsh
microenvironment conditions, EGFR mitochondrial translocation
modulated by autophagy may be critical for cell survival in certain
cancer cell types.
Several target genes and microRNAs (miRNAs) are also
involved in the regulation of EGFR and autophagy (Fig. 2). A knockout of the tumor
susceptibility gene 101 (Tsg101), an essential requirement for cell
cycle progression and cell viability, leads to decreased expression
of EGFR and induction of autophagy prior to cell death, which
suggests that Tsg101 knockout cells may utilize autophagy as an
ultimate survival mechanism prior to their death (32). The EGFR antibody cetuximab has been
proven to induce autophagy to protect cancer cells against
apoptosis by downregulating HIF-1α and Bcl-2 and activating the
Beclin 1/hVps34 complex (33). In
addition, two miRNAs, miR-221 and miR-222, were recently identified
as downstream targets of the EGFR/Ras/Raf/MEK pathway, which are
involved in the acquisition of resistance to antineoplastic
therapies (34). However, the
molecular mechanisms underlying the antitumor activity of these
genes and miRNAs have not been fully elucidated.
Autophagy inhibition may be a promising
therapeutic strategy for enhancing the effects of EGFR-targeted
therapy in NSCLC
Recent studies have demonstrated that tumor
resistance to anticancer therapies, including radiation therapy,
chemotherapy and targeted therapies, may be increased through the
upregulation of autophagy in different tumor cell lines (35,36).
The resistance to gefitinib and erlotinib is often mediated by
acquisition of the T790M mutation, which accounts for 50% of NSCLCs
(37,38). Notably, EGFR T790M expression did
not affect the activity of the EGFR downstream pro-survival
signaling pathways. Moreover, the inhibition of signaling molecules
downstream of EGFR did not enhance the sensitivity of NSCLC cells
with EGFR T790M mutation to EGFR-TKIs, suggesting that there may be
another route mediating EGFR-TKI resistance (39). A recent study demonstrated that
autophagy inhibition acted synergistically with erlotinib to induce
glioblastoma cell death (40). Guo
et al (41) investigated
the utility of autophagy-related proteins Beclin-1 and LC3 in
predicting cetuximab efficacy in advanced colorectal cancer (ACRC)
and observed that the patients with low Beclin-1/LC3 expression
exhibited a longer progression-free survival and objective response
rate compared to those with high Beclin-1 expression, indicating
that autophagy may reduce cetuximab efficacy in patients with ACRC.
Our laboratory recently reported that autophagy, as a
cytoprotective response, may be activated by EGFR-TKIs in lung
cancer cells and autophagy inhibition by chloroquine (CQ) and
siRNAs targeting ATG5 and ATG7 enhanced the cytotoxicity effect of
the EGFR-TKIs gefitinib and erlotinib (13). These results indicate that the
combination of autophagy inhibitors and EGFR-TKIs may represent
novel clinical strategies for enhancing the cytotoxic effect of
EGFR-TKIs.
Several autophagy inhibitors, such as CQ and
hydroxychloroquine (HCQ) have been investigated in preclinical
studies or clinical trials. CQ and HCQ are antimalarial drugs that
recently attracted attention as potential anticancer agents and
chemosensitizers, when used in combination with anticancer drugs
(42–45). In order to assess the efficacy of
the combination of HCQ with conventional chemotherapeutics, three
clinical trials (NCT00809237, NCT01649947, NCT00977470) are
currently being launched. A phase I study on advanced NSCL patients
with prior clinical benefit from EGFR-TKIs investigated the safety,
maximum tolerated dose, clinical response and pharmacokinetics of
HCQ, with and without erlotinib. As a result, HCQ, with or without
erlotinib, was shown to be safe and the recommended phase II dose
for HCQ with 150 mg erlotinib is 1,000 mg daily (46). Although this study did not
elaborate on the difference in survival, the safety of adding HCQ
to erlotinib was established. However, the efficacy of the
combination of HCQ with EGFR-TKIs requires further
investigation.
Issues and perspectives
NSCLC cells with EGFR mutations are sensitive to the
small-molecule EGFR-TKIs gefitinib and erlotinib, although acquired
resistance eventually develops. EGFR/EGFRvIII on the cell surface
and mitochondria may transmit pro-survival and antiapoptotic
signals, such as PI3K/AKT/mTOR. All these signals exert a strong
stimulatory effect on autophagy. The cytoplasmic EGFR may modulate
autophagy, whereas EGFR mitochondrial translocation may be
increased by autophagy.
Autophagy, as a lysosomal degradation process, is
involved in the development of cancer cell resistance to certain
treatments (37). In addition to
the mutations in the EGFR exons, recent studies (24,38)
have demonstrated that autophagy may be another probable route for
the mediation of EGFR-TKI resistance. Although it has been
previously indicated that combining EGFR-TKIs with
autophagy-inducing drugs enhances the cytotoxicity of gefitinib or
erlotinib through stimulating autophagic cell death, more evidence
supports the role of autophagy as a survival pathway to protect
tumor cells against anticancer therapy (47,48).
Furthermore, the inhibition of autophagy may enhance the
sensitivity of cancer cells to various cancer therapies, including
chemotherapy, radiotherapy or certain targeted therapies (49).
Our laboratory reported that autophagy, as a
cyto-protective response, may be activated by EGFR-TKIs in lung
cancer cells, which may be associated with acquired EGFR-TKIs
resistance (13). Consistent with
this hypothesis, Eimer et al (40) reported that inhibition of autophagy
induced a marked increase in cell death induced by erlotinib. Based
on that finding, the combination of autophagy inhibitors with
EGFR-TKIs is attracting increasing attention in cancer therapy.
However, the mechanism through which EGFR-TKIs induce autophagy has
not been elucidated. One or more pathways should be focused on to
elucidate the reason for autophagy in EGFR-TKI-resistant cells. The
understanding of the functional association of autophagy with
EGFR-TKI resistance may provide a promising therapeutic strategy to
circumvent resistance and enhance the effects of EGFR-TKIs in
patients with NSCLC.
Acknowledgements
This study was supported by grants
from National Natural Science Foundation of China (Grant Nos.
81301891, 81272593, 81071651 and 81071963) and Zhejiang Provincial
Natural Science Foundation of China (Grant No. LQ13H160008).
References
|
1.
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
2.
|
Shigematsu H, Lin L, Takahashi T, Nomura
M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, et
al: Clinical and biological features associated with epidermal
growth factor receptor gene mutations in lung cancers. J Natl
Cancer Inst. 97:339–346. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Riely GJ, Politi KA, Miller VA and Pao W:
Update on epidermal growth factor receptor mutations in non-small
cell lung cancer. Clin Cancer Res. 12:7232–7241. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Henson ES and Gibson SB: Surviving cell
death through epidermal growth factor (EGF) signal transduction
pathways: implications for cancer therapy. Cell Signal.
18:2089–2097. 2006. View Article : Google Scholar
|
|
5.
|
Yarden Y and Shilo BZ: SnapShot: EGFR
signaling pathway. Cell. 131:10182007. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kobayashi S, Boggon TJ, Dayaram T, Janne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Klionsky DJ, Abdalla FC, Abeliovich H, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy. 8:445–544. 2012. View Article : Google Scholar
|
|
8.
|
Yang Z and Klionsky DJ: Eaten alive: a
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Maiuri MC, Galluzzi L, Morselli E, Kepp O,
Malik SA and Kroemer G: Autophagy regulation by p53. Curr Opin Cell
Biol. 22:181–185. 2010. View Article : Google Scholar
|
|
12.
|
Glick D, Barth S and Macleod KF:
Autophagy: cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Han W, Pan H, Chen Y, et al: EGFR tyrosine
kinase inhibitors activate autophagy as a cytoprotective response
in human lung cancer cells. PLoS One. 6:e186912011. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Sridhar SS, Seymour L and Shepherd FA:
Inhibitors of epidermal-growth-factor receptors: a review of
clinical research with a focus on non-small-cell lung cancer.
Lancet Oncol. 4:397–406. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Galizia G, Lieto E, De Vita F, Orditura M,
Castellano P, Troiani T, Imperatore V and Ciardiello F: Cetuximab,
a chimeric human mouse anti-epidermal growth factor receptor
monoclonal antibody, in the treatment of human colorectal cancer.
Oncogene. 26:3654–3660. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Vecchione L, Jacobs B, Normanno N,
Ciardiello F and Tejpar S: EGFR-targeted therapy. Exp Cell Res.
317:2765–2771. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Lin SY, Makino K, Xia W, Matin A, Wen Y,
Kwong KY, Bourguignon L and Hung MC: Nuclear localization of EGF
receptor and its potential new role as a transcription factor. Nat
Cell Biol. 3:802–808. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Boerner JL, Demory ML, Silva C and Parsons
SJ: Phosphorylation of Y845 on the epidermal growth factor receptor
mediates binding to the mitochondrial protein cytochrome c
oxidase subunit II. Mol Cell Biol. 24:7059–7071. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Han W and Lo HW: Landscape of EGFR
signaling network in human cancers: biology and therapeutic
response in relation to receptor subcellular locations. Cancer
Lett. 318:124–134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Yue X, Song W, Zhang W, Chen L, Xi Z, Xin
Z and Jiang X: Mitochondrially localized EGFR is subjected to
autophagic regulation and implicated in cell survival. Autophagy.
4:641–649. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Ciardiello F and Tortora G: EGFR
antagonists in cancer treatment. N Engl J Med. 358:1160–1174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Yan J, Yang H, Wang G, Sun L, Zhou Y, Guo
Y, Xi Z and Jiang X: Autophagy augmented by troglitazone is
independent of EGFR transactivation and correlated with
AMP-activated protein kinase signaling. Autophagy. 6:67–73. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Cardone L, Bardelli A and Avvedimento VE:
Activation of β-catenin by oncogenic PIK3CA and EGFR promotes
resistance to glucose deprivation by inducing a strong antioxidant
response. PLoS One. 7:e375262012.
|
|
25.
|
Cao C, Lu S, Jiang Q, et al: EGFR
activation confers protections against UV-induced apoptosis in
cultured mouse skin dendritic cells. Cell Signal. 20:1830–1838.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Ghildiyal R, Dixit D and Sen E: EGFR
inhibitor BIBU induces apoptosis and defective autophagy in glioma
cells. Mol Carcinog. Jun 29–2012.(Epub ahead of print). View Article : Google Scholar
|
|
27.
|
Akhtar S, Yousif MH, Chandrasekhar B and
Benter IF: Activation of EGFR/ERBB2 via pathways involving ERK1/2,
P38 MAPK, AKT and FOXO enhances recovery of diabetic hearts from
ischemia-reperfusion injury. PLoS One. 7:e390662012. View Article : Google Scholar
|
|
28.
|
Chen KL, Chang WS, Cheung CH, et al:
Targeting cathepsin S induces tumor cell autophagy via the EGFR-ERK
signaling pathway. Cancer Lett. 317:89–98. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Yu Y, Fan SM, Ye YC, Tashiro S, Onodera S
and Ikejima T: The tyrphostin AG1478 augments oridonin-induced A431
cell apoptosis by blockage of JNK MAPK and enhancement of oxidative
stress. Free Radic Res. 46:1393–1405. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Dreier A, Barth S, Goswami A and Weis J:
Cetuximab induces mitochondrial translocalization of EGFRvIII, but
not EGFR: involvement of mitochondria in tumor drug resistance?
Tumour Biol. 33:85–94. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Cao X, Zhu H, Ali-Osman F and Lo HW: EGFR
and EGFRvIII undergo stress- and EGFR kinase inhibitor-induced
mitochondrial translocalization: a potential mechanism of
EGFR-driven antagonism of apoptosis. Mol Cancer. 10:262011.
View Article : Google Scholar
|
|
32.
|
Morris CR, Stanton MJ, Manthey KC, Oh KB
and Wagner KU: A knockout of the Tsg101 gene leads to decreased
expression of ErbB receptor tyrosine kinases and induction of
autophagy prior to cell death. PLoS One. 7:e343082012. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Li X and Fan Z: The epidermal growth
factor receptor antibody cetuximab induces autophagy in cancer
cells by downregulating HIF-α and Bcl-2 and activating the beclin
1/hVps34 complex. Cancer Res. 70:5942–5952. 2010.PubMed/NCBI
|
|
34.
|
Teixeira AL, Gomes M and Medeiros R: EGFR
signaling pathway and related-miRNAs in age-related diseases: the
example of miR-221 and miR-222. Front Genet. 3:2862012. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Hu YL, Jahangiri A, Delay M and Aghi MK:
Tumor cell autophagy as an adaptive response mediating resistance
to treatments such as antiangiogenic therapy. Cancer Res.
72:4294–4299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Zou Z, Yuan Z, Zhang Q, et al: Aurora
kinase A inhibition-induced autophagy triggers drug resistance in
breast cancer cells. Autophagy. 8:1798–1810. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Regales L, Balak MN, Gong Y, Politi K,
Sawai A, Le C, Koutcher JA, Solit DB, Rosen N, Zakowski MF and Pao
W: Development of new mouse lung tumor models expressing EGFR T790M
mutants associated with clinical resistance to kinase inhibitors.
PLoS One. 2:e8102007. View Article : Google Scholar
|
|
38.
|
Nguyen KS, Kobayashi S and Costa DB:
Acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in non-small-cell lung cancers dependent on the
epidermal growth factor receptor pathway. Clin Lung Cancer.
10:281–289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Moreira-Leite FF, Harrison LR, Mironov A,
Roberts RA and Dive C: Inducible EGFR T790M-mediated gefitinib
resistance in non-small cell lung cancer cells does not modulate
sensitivity to PI103 provoked autophagy. J Thorac Oncol. 5:765–777.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Eimer S, Belaud-Rotureau MA, Airiau K,
Jeanneteau M, Laharanne E, Veron N, Vital A, Loiseau H, Merlio JP
and Belloc F: Autophagy inhibition cooperates with erlotinib to
induce glioblastoma cell death. Cancer Biol Ther. 11:1017–1027.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Guo GF, Jiang WQ, Zhang B, Cai YC, Xu RH,
Chen XX, Wang F and Xia LP: Autophagy-related proteins Beclin-1 and
LC3 predict cetuximab efficacy in advanced colorectal cancer. World
J Gastroenterol. 17:4779–4786. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Nilsson JR: Does chloroquine, an
antimalarial drug, affect autophagy in Tetrahymena
pyriformis? J Protozool. 39:9–16. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Gunja N, Roberts D, McCoubrie D, Lamberth
P, Jan A, Simes DC, Hackett P and Buckley NA: Survival after
massive hydroxychloroquine overdose. Anaesth Intensive Care.
37:130–133. 2009.PubMed/NCBI
|
|
44.
|
Sasaki K, Tsuno NH, Sunami E, et al:
Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on
colon cancer cells. BMC Cancer. 10:3702010. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Sasaki K, Tsuno NH, Sunami E, et al:
Resistance of colon cancer to 5-fluorouracil may be overcome by
combination with chloroquine, an in vivo study. Anticancer Drugs.
23:675–682. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Goldberg SB, Supko JG, Neal JW, et al: A
phase I study of erlotinib and hydroxychloroquine in advanced
non-small-cell lung cancer. J Thorac Oncol. 7:1602–1608. 2012.
View Article : Google Scholar
|
|
47.
|
Hu YL, Jahangiri A, Delay M and Aghi MK:
Tumor cell autophagy as an adaptive response mediating resistance
to treatments such as antiangiogenic therapy. Cancer Res.
72:4294–4299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Li X, Lu Y, Pan T and Fan Z: Roles of
autophagy in cetuximab-mediated cancer therapy against EGFR.
Autophagy. 6:1066–1077. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Buchser WJ, Laskow TC, Pavlik PJ, Lin HM
and Lotze MT: Cell-mediated autophagy promotes cancer cell
survival. Cancer Res. 72:2970–2979. 2012. View Article : Google Scholar : PubMed/NCBI
|