Introduction
Small molecules non-endogenous to the biological
system, often referred to as xenobiotics, include a diverse variety
of molecules, simple organic toxicants with uncomplicated
structures and natural eukaryotic antibiotics and synthetic
molecules of more complicated structures and cytotoxic properties
(1), the latter of which have
chemotherapeutic properties. Although small molecule
chemoxenobiotics, of various traditional classes, form the basis of
present synergistic cancer treatment strategies, their clinical
efficacy remains questionable for the treatment of solid and
hematopoietic malignancies alike, upon surgical resection in the
former, and during bone marrow irradiation-transplantation and
after in the latter. For this reason, a better understanding of the
modes and character underlying molecular cellular interactions is
necessary, particularly for the purposes of improving the tumor
tissue selectiveness of enhanced permeation and retention
(EPR)-based chemotherapy (2), which
has a prolonged blood half-life but is non-selective for solid
tumor foci. Along these lines, there has been relatively recent
translational advancement towards the development of optimally
sized dendrimer nanoparticle-based small molecule chemotherapy at
~9 nanometers (nm) (HD) (3–7), which
selectively delivers small molecule chemoxenobiotics into solid
malignancies at effective concentrations without systemic toxicity
(4,8).
As such, with optimally sized dendrimer nanoparticle-based small
molecule chemotherapy, there is the potential for complete tumor
regression (3,9) in lieu of the possibility for the
development of drug resistance phenotypes (10,11) and
therapy-related malignancies (12) or
myelodysplastic syndromes (13).
During the discovery and developmental stages, the
testing of small molecule xenobiotics occurs at several levels: i)
at the naked target, on chromatin or on a protein receptor/enzyme
in isolation, which provides information on relative binding
affinities for intra-cellular proteins; ii) at the cellular level
in vitro, which provides information on the inhibitory
concentrations needed to achieve tumor cell death and the
overexpression status of induced pro- or anti-apoptotic protein
forms, but does not take into consideration cell membrane (CM)
phospholipid or CM protein receptor interactions, the most common
level of interaction for secondary intra-cellular effects of
non-permeable small molecule xenobiotics, either due to molecular
size restriction to permeation or charge restriction to permeation
(4,8);
iii) at the systemic level in vivo, which gives an idea of
the dosing range necessary to achieve a chemotherapeutic effect and
tumor regression, and in the case of more lipophilic
chemoxenobiotics, the dosing required to overcome serum protein
binding, this being the primary limitation to achieving effective
transvascular delivery and intra-tumoral concentrations in cases of
free small molecule xenobiotics, not linked to optimally sized
nanoparticles (3–7).
Most recently, the conserved biophysical
determinants for the interactions of small biomolecules, cations
and anions in the biological system in the physiologic state have
been elucidated, with respect to understanding permeation
thresholds across microvascular and epithelial barriers (8) as well as understanding the modes, the
levels and the character of interactions of small biomolecules with
and within cells. With this novel approach, the biological
interactions of any small molecule can be understood in terms of
its 2-dimensional structure. This requires taking into
consideration the following character of charge distribution over
molecular space, determinations of the predicted overall
octanol-to-water partition coefficient (Log OWPC; unitless), and
the predicted molecular size [van der Waals diameter (vdWD; nm)]
viz a viz the Log OWPC-to-vdWD (nm−1) parameter.
The biological interactions of the hydrophilic or hydrophobic parts
of the molecule can be understood in terms of the 2-dimensional
part-structures, and determinations of the interacting hydrophilic
moiety (or core)-to-vdWD ratio (nm−1) and the
incorporating lipophilicity of the hydrophobic core (or
moiety)-to-vdWD ratio (nm−1) parameters,
respectively.
For the proper determination of the apoptotic
potential of chemoxenobiotics in synergism, it is important to know
whether interactions at the cellular level are with and across CM
protein aqueous channels, with CM surface protein receptors and
endocytic, with CM surface protein receptors and non-endocytic or
directly with phospholipids. It is also important to know whether
interactions at the sub-cellular level are nuclear, mitochondrial
or microtubular. Therefore, in this research study, current small
molecule chemoxenobiotics and xenobiotics not traditionally
considered as chemoxenobiotics, are analyzed in terms of conserved
biophysical determinants to determine the modes, levels and
character of interactions of xenobiotics with cells and cell
organelles. The insight to be gained is to be applicable for the
selection of existing chemoxenobiotics most synergistic in
cytotoxic effects and the development of more effective existing
chemotherapy regimens utilizing small molecule chemoxenobiotics
(14,15). This knowledge is applicable for the
discovery of alternative xenobiotics with chemotherapeutic
potential to overcome chemotherapeutic resistance (16), as well as for the design and
development of next generation biocompatible optimally sized drug
carriers free from intravascular protein interactions, for
effective transvascular delivery into solid tumor tissue cells
(3–7).
Materials and methods
Data acquisition and determination of
principal components for analysis of small molecule
xenobiotics
Small molecule xenobiotics known to be
chemotherapeutic and those with chemotherapeutic potential were
identified for the database. Freely available and validated online
biochemical molecule databases including http://www.chemicalize.org and http://www.chemspider.com were utilized for
determinations of 2-dimensional (2-D) molecular structures and
ionization state at physiologic pH of ~7.4, analogous to previous
methodology for endogenous biomolecules (8). Molecular structure and configurations of
xenobiotics were assessed for the type of covalent bonds within the
structure, and to the backbone as is the presence or absence of
associated molecular charge. The presence of halogenation (0),
hydroxylation (0, −1), phosphorylation (−1, −2), carboxylation
(−1), carbonylation (C=O), sulfonation (0, +1) and amination (0,
+1), whether primary, secondary, tertiary or quaternary (+1) is
noted. The amount of polar surface area (psa) is also noted.
The predicted octanol-to-water partition coefficient
(poOWPC or OWPC; unitless), the predicted Log Pow, was applied for
molecules in neutral or unionized states, and the predicted Log
Dow, was applied for molecules in the ionized state. The predicted
vdWD (nm) was applied as the measure of estimated molecular size,
obtained from the predicted spherical van der Waals volume. The
predicted Log OWPC-to-vdWD (nm−1) was determined
for the whole molecule, while the predicted incorporating
lipophilicity octanol-to-water partition coefficient-to-van der
Waals diameter ratio (Log incorpOWPC-to-vdWD;
nm−1) was determined for the hydrophobic cores
and moieties (hydrophobic moiety/core Log OWPC-to-vdWD;
nm−1), and the predicted
interactability octanol-to-water partition coefficient-to-van der
Waals diameter ratio (Log interactOWPC-to-vdWD;
nm−1) was determined for the hydrophilic moieties
[or cores] (hydrophobic moiety Log OWPC-to-vdWD;
nm−1).
Classification of molecular philicity
and charge
The presence of charge and its distribution over
biomolecular space were assessed based on visual inspection of 2-D
molecular structures. The classification scheme, as devised with
slight modifications, was applied for the characterization of
molecular charge over molecular space, as follows:
I. No overall charge: neutral (0), which can be a
combination of hydroxylo (OH), carbonylo (C=O), etheroylo
(O-CH3), amidylo (N-C=O), where the presence of
peripheral neutral groups in a circumferential or
semi-circumferential arrangement around a lipophilic core is
designated as Peri or semi-Peri;
II. Sufficient molecular space separation of charge
(S) was defined as the presence of focal charges separated in
molecular space, in the form of sufficiently separated attractive +
or - charge that results in sufficiently separated contributions of
cationicity (S 1+ 1+) or of anionicity (S 1- 1-);
III. Insufficient molecular space separation of
charge (IS) was defined as the presence of focal charge in
molecular space, in the form of insufficiently separated attractive
+ or - charge that results in insufficiently separated
contributions of cationicity (IS 1+ 1+) or of anionicity (IS 1-
1-), where cationoneutral molecular charge is defined as IS 1+
1-.
Classification of small molecule
xenobiotic mode and character of CM protein channel or receptor
interaction
Small molecule xenobiotic mode and character of CM
cholesterol/phospholipid glycerol-to-fatty acid-ester, CM protein
channel or CM receptor interaction were classified, as follows:
I. Pure hydrophiles, defined as xenobiotics without
intervening lipophilicity with predicted hydrophilic (−) Log
OWPC-to-vdWD ratio (nm−1) at a physiologic pH of
7.4, with non-charged pure hydrophiles being CM aqueous channel
pore permeable at vdWDs <0.78 nm and intra-cellularly localizing
(0 1- 1+).
II. Hydro-lipophiles, defined as xenobiotics with
intervening lipophilicity (Log incorpOWPC-to-vdWD; nm−1)
with predicted hydrophilic (−) Log OWPC-to-vdWD ratio
(nm−1) at physiologic pH of 7.4, and being CM aqueous
channel pore impermeable, with CM non-channel receptor or CM
receptor interaction instead, as follows: a) cationic
polyhydroxylated/carbonylated/etheroylated [1+ Peri (0)]: channel
receptor-mediated CM endocytosis (i.e. doxorubicin); b)
cationic-cationic polyhydroxylated/carbonylated/etheroylated [S 1+
1+ Peri (0)]: channel receptor-mediated CM endocytosis (i.e.
vincristine); c) di-cationic-cationic [S IS 1+ 1+ IS 1+ 1+ (0)]:
effective cationicity 2+ 2+): non-channel receptor-mediated CM
endocytosis (i.e. AMD3100); d) di-carboxylated neutral [S 1- 1- 0
(0)]: non-channel folic acid receptor-mediated CM endocytosis (i.e.
methotrexate).
III. Lipohiles, defined as non-charged less
lipophilic toxicants with vdWDs <0.78 nm (CM channel pore
permeable sub-CM interactors), and as xenobiotics with vdWDs
>0.78 nm and overall lipophilicity [(+) Log OWPC-to-vdWD ratio
(nm−1)] with sufficient (S) intervening lipophilicity
(Log incorpOWPC-to-vdWD; nm−1) in the presence of
molecular hydrophilicity
(monohydroxylation/monocarbonylated/monoetheroylated/monocarboxylated;
polyhydroxylated/polycarbonylated/polyetheroylated; charge) either
present isotropically [CM receptor hydrophobic core interactors
with vdWDs >0.78 nm (receptor-mediated CM endocytosis)], or
present anisotropically (CM cholesterol/phospholipid
glycerol-to-fatty acid-ester or CM receptor hydrophobic core
interactors with vdWDs >0.78 nm): a) anisotrophic anionic [1-
(0)] with linear structure (i.e. chlorambucil), anisotrophic
cataniononeutral [IS 1+ 1- (0)] with linear structure (i.e.
melphalan), anisotropic polyneutral [0 (0)] with linear structure
(i.e. capecitabine), anisotropic polyneutral circular [circ (0)]
(i.e. cyclosporine A): CM cholesterol or phospholipid
glycerol-to-fatty acid-ester association or disruption; b)
isotropic di-neutral [0 (0) 0] with sterol structure: non-channel
steroid receptor-mediated pressuromodulation antagonism (i.e.
abiraterone); non-channel receptor-mediated pressuromodulation
(endogenous sex steroids, i.e. testosterone, estradiol); c)
isotropic cationic-neutral [1+ (0) 0] with sterol mimicking
structure: non-channel steroid receptor-mediated pressuromodulation
antagonism (i.e. hydroxytamoxifen); d) isotrophic cationic
semi-polyneutral [1+ semi-Peri (0)] with flexible linear structure:
potential for channel receptor-mediated CM endocytosis (i.e.
verapamil); e) esotrophic polyneutral [Peri (0)] with compact
non-linear structure: non-channel receptor-mediated CM endocytosis
(i.e. colchicine); isotrophic polyneutral [Peri (0)] with
non-compact non-linear structure: non-channel receptor-mediated CM
endocytosis (i.e. etoposide); f) anisotropic di-neutral [0 0 (0)]
with part-sterol structure: non-channel receptor P-glycoprotein
(P-gp)-mediated pressuromodulation (i.e. artemisinin); g)
anisotropic polyneutral [semi-Peri (0)] with sterol structure:
non-channel receptor-mediated pressuromodulation (endogenous
corticosteroids, i.e. cortisol, aldosterone); h) anisotropic
cationic neutral [1+ 0 (0)], isotropic cationic neutral [1+ (0) 0],
anisotropic non-cationic di-neutral [0 0 (0)] or isotropic
non-cationic di-neutral [0 (0) 0] with non-linear L-to-U-type
step-like structure: non-channel receptor-mediated
pressuromodulation antagonism (i.e. growth factor and cytokine
receptor antagonists).
Results and Discussion
Octanol-to-water partition
coefficient-to-van der Waals diameter ratio of whole and part
structures in the context of xenobiotic structures
The poOWPC or OWPC represents the presence of
molecular surface area hydrophilicity due to the presence of
functional groups of hydrophilic character, and in the case of
small molecule xenobiotics of hydrophilic character, the predicted
overall Log OWPC is the sole determinant of the overall tendency
for interaction with the aqueous phase and represented by the
predicted hydrophilic (−) Log OWPC-to-vdWD ratio
(nm−1), whereas, in the case of small molecule
xenobiotics of lipophilic character with the concomitant presence
of hydrophilic moieties sufficiently separated in molecular space
(S), the predicted incorporating Log OWPC of the hydrophobic
portion is the sole determinant of the specific tendency for
interaction with the hydrophobic phase and represented by the
predicted lipophilic (+) Log incorpOWPC-to-vdWD ratio
(nm−1) to interact or associate with the
hydrophobic constituents of the CM, either with the CM bilayer
cholesterols and phospholipid fatty acid-esters or with CM
protein/receptor hydrophobic cores.
Based on assessment of the 2-D molecular structures
of small molecule xenobiotics, those of pure hydrophilic character
have a cyclic or linear backbone containing hydrophilic groups, and
therefore, pure hydrophilic xenobiotics neither associate with CM
phospholipids nor CM proteins and receptors, while those with lower
hydrophilicity (−) Log OWPC-to-vdWD ratios (nm−1)
can permeate across CM protein channel aqueous pores analogous to
permeation across barrier pores, molecular size permitting
(8), which, in the case of the least
hydrophilic pure hydrophile xenobiotics is ≤0.78 nm (Tables I and II).
 | Table I.Cell membrane (CM) channel aqueous
pore permeation and DNA/RNA adduction. |
Table I.
Cell membrane (CM) channel aqueous
pore permeation and DNA/RNA adduction.
|
| Log OWPC
(unitless) | vdWD(nm) | Charge
distribution | Groups | (Log) OPWC-to-vdWD
ratio [per nm (nm−1)] | 1ary mode of
interaction | Interaction
level(s) | Mechanism(s) of
action |
|---|
|
Nitroso-N-methylurea (MNU) | −0.55 | 0.54 | 0 | CH3,
C=O, N=O, Ns | −1.02 | CM aqueous channel
pore permeation | DNA/RNA strand | Nuclear and
mitochondrial DNA/RNA alkyl adducts |
| Temozolamide
(TMZ) | −0.45 | 0.65 | 0 | CH3,
cyclic C=Os and Ns | −0.70 | CM aqueous channel
pore permeation | DNA/RNA strand | Nuclear and
mitochondrial DNA/RNA alkyl adducts |
| Nitroso-N-ethylurea
(ENU) | −0.22 | 0.57 | 0 | Ethyl, C=O, N=O,
Ns | −0.38 | CM aqueous channel
rore rermeation | DNA/RNA strand | Nuclear and
mitochondrial DNA/RNA alkyl adducts |
| Table II.Cell membrane (CM) channel aqueous
pore permeation and nucleoside substitution. |
Table II.
Cell membrane (CM) channel aqueous
pore permeation and nucleoside substitution.
|
| Log OWPC
(unitless) | vdWD (nm) | Charge
distribution | Groups | (Log) OPWC-to-vdWD
ratio [per nm (nm−1)] | 1ary mode of
interaction | Interaction
level(s) | Mechanism(s) of
action |
|---|
| Decitabine | −2.35 | 0.70 | 0 | Cyclic pyridine
w/Ns and C=O and NH2, cyclic ribose w/OH,
CH2-OH | −3.35 | CM aqueous channel
pore permeation | DNA/RNA Nucleobase
ribosylase DNA/RNA nucleoside phosphorylase (DNA/RNA strand) | Nuclear and
mitochondrial DNA/RNA synthesis inhibition; phosphorylation of
dehydroxylated nucleoside (in lieu of cytidine) |
| Gemcitabine | −1.65 | 0.72 | 0 | Cyclic pyridine
w/Ns and C=O and NH2, cyclic ribose w/F ×2, OH,
CH2-OH | −2.29 | CM aqueous channel
pore permeation | DNA/RNA nucleobase
ribosylase DNA/RNA nucleoside phosphorylase (DNA/RNA strand) | Nuclear and
mitochondrial DNA/RNA synthesis inhibition; phosphorylation of
fluorinated nucleoside (in lieu of cytidine) |
| 5-Fluorouracil
(5-FU) | −0.66 | 0.56 | 0 | Cyclic pyridine
w/C=Os and Ns, F | −1.16 | CM aqueous channel
pore permeation | RNA nucleobase
ribophosphorylase (RNA strand) | Nuclear and
mitochondrial RNA synthesis inhibition; ribophosphorylation of
fluorinated nucleobase (in lieu of RNA uracil) |
|
3-Methyladenine | −0.31 | 0.61 | 0 | Cyclic purine w/Ns,
N-CH3, NH2 | −0.51 | CM aqueous channel
pore permeation | DNA/RNA nucleobase
ribosylase DNA/RNA nucleoside phosphorylase (DNA/RNA strand) | Competitive
activation of poly(ADP-ribose) polymerase-1 (PARP-1) and futile
mRNA translation; mitochondrial oxidative stress and free MM AIF
w/generation of free caspace 3; 2arily increased free caspace
3-mediated cleavage of chromatin |
Xenobiotics of overall hydrophilic character with
incorporating lipophilicity (small molecule hydro-lipophiles) and
of overall lipophilic character in the absence or presence of
hydrophilic portions/hydrophilic functional groups (small molecule
lipophiles) that are larger than vdWD ~0.78 nm do not permeate
across CM aqueous channel pores due to a molecular size limitation
to permeation, and therefore, instead associate with CM
constituents, either with CM phospholipids or with CM phospholipid
bilayer bilayer-associated proteins. Based on the structural
arrangement of atoms, xenobiotics larger than vdWD ~0.78 nm can be
categorized, as follows:
i) Those with non-charged backbone atoms in central
linear arrangement in the form of cyclic rings alternating with
atoms in chains in the presence or absence of structural
anisotropic hydrophilicity (uni-polar hydrophilicity), which
results in unstable association with CM associated-protein core
hydrophobicity, whereby such xenobiotics associate within CM
phospholipid bilayers instead, and pertubation disrupt due to
non-biologic interaction spatial displacement of bilayer fatty
acid-esters, with the potential for 1ary indirect
pressuromodulation at lower concentrations (17) and include cyclosporine A with the
capacity for phospholipid interspace widening-disruption upon
insertion (Tables III–V).
| Table III.Cell membrane (CM) channel aqueous
pore permeation with the potential to bind to cytochrome P450s: DNA
adduction and/or crosslinking. |
Table III.
Cell membrane (CM) channel aqueous
pore permeation with the potential to bind to cytochrome P450s: DNA
adduction and/or crosslinking.
|
| Log OWPC
(unitless) | vdWD (nm) | Charge
distribution | Groups | (Log) OPWC-to-vdWD
ratio [per nm (nm−1)] | 1ary mode of
interaction | Interaction
level(s) | Mechanism(s) of
action |
|---|
| Procarbazine | −1.70 | 0.74 | 0 |
CH3-(NH2)2,
CH2-benzyl, amide isopropyl | −2.29 | CM aqueous channel
pore permeation >> CM receptor | Cytochrome P450s
glutathione transferase (GST) DNA strand | Activation by
cytochrome P450 (Cyp 1A1, Cyp 1A2, Cyp 2E1) and Cyp P450 induction;
glutathione transferase (GST) activation [GS-H > GS-procarbazine
and GSSG depletion]; GSSG depletion −> glutathione reductase
(GR) inhibition; nuclear DNA and mitochondrial DNA alkylation |
|
Procarbazine hydrophilic
moiety1 |
|
|
|
CH3-NH2-NH2 | −1.17 | CM receptors
(non-specific) CM receptor hydrophilicity |
|
|
|
~Procarbazine hydrophobic
moiety1 |
|
|
|
CH2-Benzyl | 4.43 | CM receptor
hydrophobicity |
|
|
|
Procarbazine hydrophilic
moiety2 |
|
|
| N-C=O | −2.61 | CM receptor
hydrophilicity |
|
|
|
Procarbazine hydrophobic
moiety2 |
|
|
| Isopropyl | 3.75 | CM receptor
hydrophobicity |
|
|
|
Cyclophosphamide | 0.10 | 0.73 | 0 | Cl-ethyl ×2, cyclic
O-P=O, Ns | 0.14 | CM aqueous pore
channel permeation >> CM association: <0.78 nm | Cytochrome P450s
glutathione transferase (GST) DNA strand | Activation by
cytochrome P450 (Cyp 2C9) binding and Cyp P450 induction;
glutathione transferase (GST) activation−> [GS-H > GS
cyclophosphamide/ifosfamide and GSSG depletion]; GSSG depletion
> glutathione reductase (GR) inhibition; direct non covalent or
covalent GR inhibition; nuclear DNA and mitochondrial DNA
alkylation and crosslinking; potential for 1ary indirect
pressuromodution (perturbomodulation) |
| Ifosfamide | ~0.10 | ~0.73 | 0 | Cl-ethyl ×2, cyclic
O-P=O, Ns | ~0.14 | CM aqueous channel
pore permeation >> CM association: <0.78 nm | Cytochrome P450s
glutathione transferase (GST) DNA strand | Activation by
cytochrome P450 (Cyp 2C9) binding and Cyp P450 induction;
glutathione transferase (GST) activation >[GS-H >
GS-cyclophosphamide/ifosfamide and GSSG depletion]; GSSG depletion
> glutathione reductase (GR) inhibition; direct non covalent or
covalent GR inhibition; nuclear DNA and mitochondrial DNA
alkylation and crosslinking' potential for 1ary indirect
pressuromodution (perturbomodulation) |
|
Cyclophosphamide/ifosfamide
hydrophobic moiety1 | 1.19 | 0.48 | 0 | Cl-ethyl | 2.49 | CM/Sub-CM
hydrophobicity |
|
|
|
Cyclophosphamide/ifosfamide
hydrophobic moiety2 | 1.19 | 0.48 | 0 | Cl-ethyl | 2.49 | CM/Sub-CM
hydrophobicity |
|
|
|
Cyclophosphamide/ifosfamide
hydrophilic core | −1.92 | 0.59 | 0 | Cyclic O-P=O,
Ns | −3.49 | CM/Sub-CM
phospholipids |
|
|
| Carmustine | 0.95 | 0.67 | 0 | Cl-ethyl ×2, N=O,
C=O, Ns | 1.41 | CM aqueous channel
pore permeation | Cytochrome P450s
glutathione transferase (GST) DNA strand | In-activation by
cytochrome P450 (Cyp 2B1) binding and Cyp P450 induction;
glutathione transferase (GST) activation > [GS-H >
GS-cyclophosphamide/ifosfamide and GSSG depletion]; GSSG depletion
> glutathione reductase (GR) inhibition; direct non covalent or
covalent GR inhibition; nuclear DNA and mitochondrial DNA
alkylation and crosslinking; greater potential for 1ary indirect
pressuromodution (perturbomodulation) |
|
Carmustine hydrophobic moiety1
Carmustine | 1.19 | 0.48 | 0 | Cl-ethyl | 2.49 | CM
hydrophobicity |
|
|
|
Carmustine hydrophobic
moiety2 | 1.19 | 0.48 | 0 | Cl-ethyl | 2.49 | CM
hydrophobicity |
|
|
|
Carmustine hydrophilic
core | −0.58 | −0.58 | 0 | N=O, C=O, Ns | 1.08 | CM
phospholipids |
|
|
| Table V.Cell membrane (CM)
cholesteroloassociation-to-CM phospholipidoassociation: cholesterol
removal-to-CM phospholipid pertubation and potential for 1ary
indirect pressuromodulation. |
Table V.
Cell membrane (CM)
cholesteroloassociation-to-CM phospholipidoassociation: cholesterol
removal-to-CM phospholipid pertubation and potential for 1ary
indirect pressuromodulation.
|
| Log OWPC
(unitless) | vdWD (nm) | Charge
distribution | Groups | (Log) OPWC-to-vdWD
ratio [per nm (nm−1)] | 1ary mode of
interaction | Interaction
level(s) | Mechanism(s) of
action |
|---|
| Nystatin | −2.00 | 1.18 | S 1+ 1-0 (0) | Parallel
polyunsaturated FA and polyhydroxyl chain w/carbonyl ester,
carboxyl, glucosamine | −1.70 | CM cholesterol
association | CM cholesterol | CM cholesterol
association and cholesterol removal; potential for delayed 1ary
indirect pressuromodution (perturbomodulation) |
| Amphotericin B | −1.30 | 1.17 | S 1+ 1-0 (0) | Parallel
polyunsaturated FA and polyhydroxyl chain w/carbonylester,
carboxyl, glucosamine | −1.11 | CM cholesterol
association | CM cholesterol | CM cholesterol
association and cholesterol removal; potential for delayed 1ary
indirect pressuromodution (perturbomodulation) |
| Filipin | −0.15 | 1.00 | 0 (0) | Parallel
polyunsaturated FA and polyhydroxyl chain w/carbonylester 5C
alkyl | −0.15 | CM cholesterol
association | CM cholesterol | CM cholesterol
association and cholesterol removal; potential for delayed 1ary
indirect pressuromodution (perturbomodulation) |
|
Nystatin/Ampho B/filipin
hydrophobic (inner crescent) moiety Eq | 6.73 | 0.81 | (0) | Polyunsaturated
long chain | 8.32 | CM cholesterol
hydrophobicity |
|
|
|
Nystatin/Ampho B/filipin
hydrophilic (outer crescent) moiety Eq | −3.60 | 0.83 | 0 | Polyhydroxyl-chain
w/carbonyl | 4.36 | CM phospholipid
head and extacellular hydrophilicity |
|
|
|
Nystatin/Ampho B hydrophilic
(outer crescent) moiety1 | 3.05 | 0.42 | 1+ | Hexose 1ary
N+ | −7.28 | Extacellular
hydrophilicity |
|
|
|
Nystatin/Ampho B hydrophilic
(outer crescent) moiety2 | −3.50 to −0.27 | 0.42 | 1- to
COO-Na+ | Carboxyl | −8.42 to −0.65 | Extacellular
hydrophilicity |
|
|
| Chlorambucil | 0.60 | 0.79 | 1-(0) | Cl-ethylsx2, single
benzyl, COO-, N | 0.76 | CM cholesterol
association | CM cholesterol | CM cholesterol
association and cholesterol removal; potential for delayed 1ary
indirect pressuromodution (perturbomodulation) |
|
Chlorambucil hydrophobic
core | 5.26 | 0.78 | (0) | Cl-ethylsx2, single
benzyl, N | 6.73 | CM cholesterol
hydrophobicity |
|
|
|
Chlorambucil hydrophilic
moiety | −3.50 to −0.27 | 0.42 | 1-to
COO-Na+ | Carboxyl | −8.42 to −0.65 | Extacellular
hydrophilicity |
|
|
|
Melphalan | 1.00 | 0.79 | IS 1+ 1-(0) | Cl-ethylsx2, N,
single benzyl, NH3+ COO− | 1.27 | CM
incorporo-association sub cellular mitochondrial membrane (MM)
incorporo association | CM phospholipids
Sub-CM MM phospholipids (via trans-CM displacement as does
thyroxine) | CM phospholipid
incorporoassociation −> CM trans-displacement; cytochrome P450
CYP 3A and CYP 27 inhibition w/oinduction; MM phospholipid
association > mitochondrial hyperdrive-mediated apoptosis in BCL
dependent cells; non potential for 1ary indirect pressuromodution
(perturbomodulation) |
|
Melphalan hydrophibic
core | 4.37 | 0.75 | (0) | Cl-ethylsx2, N,
single benzyl | 5.86 | CM phospholipid
tail hydrophobicity |
|
|
|
Melphalan hydrophilic
moiety | 1.19 | 0.48 | IS 1+ 1− | NH3+
COO− | −3.80 | CM phospholipid
head hydrophobicity |
|
|
| Ketoconazole (at pH
7.4) | 4.19 | 0.94 | 0 (0) 0 (0) | Di-chloro benzyl,
cyclopyridine, cycloether [O]Benzyl, cyclohexane w/Ns ×2,
acetate | 4.46 | CM
pertuboassociation | CM
phospholipids | CM phospholipid
bilayer pertubation and 2ary cholesterol removal; potential for
1ary indirect pressuromodution (perturbomodulation) |
|
Ketoconazole hydrophobic
moiety1 | 3.30 | 0.71 | (0) |
Pyridine-CH2-CH2-dichlorobenzyl | 4.64 | CM
hydrophobicity |
|
|
|
Ketoconazole
hydrophobic-isophilic moiety | 0.48 | 0.65 | 0 |
(CH3)2-cyclother-CH2-O-CH3 | 0.74 | CM
hydrophobicity-CM isophilicity |
|
|
|
Ketoconazole hydrophobic
moiety2 | 2.44 | 0.71 | (0) |
CH3-cyclohexane w/2
Ns-benzyl-CH3 | 3.42 | CM
hydrophobicity |
|
|
|
Ketoconazole hydrophilic
moiety | 0.38 | 0.44 | 0 | C(=O)C | −0.86 | CM
hydrophilicity |
|
|
| Capecitabine | 0.75 | 0.83 | 0 (0) | Ribose w/OHx2,
fluoruracil w/C=O, N, 5 C alkyl ester | 0.90 | CM
perturbo-association | CM
phospholipids | CM phospholipid
bilayer pertubation; potential for 1ary indirect pressuromodution
(perturbomodulation) |
|
Capecitabine hydrophobic
core | 1.94 | 0.61 | (0) | ~5 Carbon alkyl
ether | 3.19 | CM
hydrophobicity |
|
|
|
Capecitabine nucleoside
(FU) | 0.66 | 0.56 | 0 | Cyclic C=Os and Ns,
F | −1.18 | CM
hydrophilicity |
|
|
|
Capecitabine hydrophilic
moiety1 | −0.47 | 0.38 | 0 carbonylo (CM
esterase −> COO-) | C=O | −1.22 ×2 | CM
hydrophilicity |
|
|
|
Capecitabine hydrophilic
moiety2 | −0.33 | 0.32 | 0 Hydroxylo | Ribose OH | −1.05 ×2 | CM
hydrophilicity |
|
|
| Fluconazole | 0.56 | 0.77 | 0 (0) | Di-fluoro benzyl,
CH3-pyridines ×2, OH | 0.73 | CM channel
permeation; CM perturboassociation; Sub CM channel permeation; Sub
CM perturboassociation | CM pores CM
phospholipids Cytochrome P450s Sub-CM pores Sub-CM
phospholipids | Potential for
permeation across CM pores; potential for CM phospholipid
pertubation; cytochrome P450 inhibition (Cyp 2C9 and Cyp 2C19)
w/induction; endoGolgi SER Sub-CM phospholipid pertubation; less
potential for 1ary indirect pressuromodution
(perturbomodulation) |
| Fluconazole
hydrophobic core | 1.22 | 0.76 | (0) | Di-fluoro benzyl,
CH3-pyridines ×2 | 1.61 | CM hydrophobicity
Sub-CM hydrophobicity |
|
|
| Fluconazole
hydrophilic moiety | −0.33 | 0.32 | 0 Hydroxylo | OH | −1.05 | CM hydrophilicity
Sub-CM hydrophilicity |
|
|
ii) Those with backbone atoms arranged in non-linear
non-compact, or in a step-like configuration of inter-connected
cyclic rings, in the presence or absence of alternating with atoms
in linear arrangement (L-to-U-type), which results in stable
association with CM proteins/CM peptide receptors and
non-interaction with CM bilayers (Tables
VI–XI).
| Table VI.Divalent cationicity-mediated cell
membrane (CM) receptor vesiculo-vacuolization endocytosis,
sub-cellular vacuolization along with exosome formation with
potential for CM receptor-mediated 3ary indirect
pressuromodulation. |
Table VI.
Divalent cationicity-mediated cell
membrane (CM) receptor vesiculo-vacuolization endocytosis,
sub-cellular vacuolization along with exosome formation with
potential for CM receptor-mediated 3ary indirect
pressuromodulation.
|
| Log OWPC
(unitless) | vdWD (nm) | Charge
distribution | Groups | (Log) OPWC-to-vdWD
ratio [per nm (nm−1)] | 1ary mode of
interaction | Interaction
level(s) | Mechanism(s) of
action |
|---|
| AMD3100
(plerixafor) | −5.34 | 1.00 | S IS 1+ 1+ IS 1+ 1+
(0) | 2 cyclic carbon
rings, each w/Ns ×4 and 2ary N+ s ×2, intervening benzyl
×1 | −5.42 | Divalent
cationicity-mediated CM receptor endocytosis | CM CXCR4 endocytic
vesicles [in lieu of SDF-1 (CXCL 12)] |
Vesiculo-vacuolization endocytosis; CM
vacuolization > Sub-cellular vacuolization; 2ary CM exosome
formation; non-interaction w/lymphoinfiltrating cells in milieu;
potential for mitogenesis and loss of contact inhibition; (3ary
indirect shift pressuromodulation) |
| AMD3100 hydrophobic
core | 3.02 | 0.60 | (0) |
CH3-Benzyl-CH3 | 5.05 | CXCR4 chemokine
receptor core |
|
|
| AMD3100 hydrophilic
moiety1 | −6.55 | 0.76 | IS 1+ 1+: 2 | Cyclic carbon ring
with 2ary N+ ×2 | −8.63 | Cationic
interaction w/receptor (extracellular CM interface) |
|
|
| AMD3100 hydrophilic
moiety2 | −6.55 | 0.76 | IS 1+ 1+: 2 | Cyclic carbon ring
with 2ary N+ ×2 | −8.63 | Cationic
interaction w/receptor (extracellular CM interface) |
|
|
| Paraquat | 5.66 | 0.70 | IS 1+ 1+ (0) | Cyclopyridine
w/quat N+, single bond, cyclopyridine w/Quat
N+ | −7.83 | Divalent
cationicity-mediated CM receptor endocytosis | CM paraquat binding
protein (PBP) endocytic vesicles |
Vesiculo-vacuolization endocytosis; CM
vacuolization > Sub-cellular vacuolization; 2ary CM Exosome
formation; potential for mitogenesis and loss of contact
inhibition; (3ary indirect shift pressuromodulation) |
| Paraquat
hydrophobic core | 2.96 | 0.70 | (0) |
Cyclopyridinew/N-SingleBond-cyclopyridine
w/N | 4.25 | CM receptor
hydrophobicity |
|
|
| Paraquat
hydrophilic moiety1 | −3.97 | 0.56 | 1+ | ~ 4ary
N+ (in benzyl ring) | −7.08 |
| Cationic
interaction w/receptor (extracellular CM interface) |
|
| Paraquat
hydrophilic moiety2 | −3.97 | 0.56 | 1+ | ~ 4ary
N+ (in benzyl ring) | −7.08 | Cationic
interaction w/receptor (extracellular CM interface) |
|
|
| Bleomycin | 8.50 | 1.31 | IS 1+ 1+ semi-Peri
(0) | 1ary N+ and 3ary
S+, cyclo(sulfo)pyridine alkyl amide w/OHs, hexoses ×2,
cyclopyridines ×2, amides, OHs | −6.50 | Divalent
cationicity-mediated CM receptor endocytosis | CM bleomycin
binding protein (BBP) endocytic vesicles |
Vesiculo-vacuolization endocytosis; CM
vacuolization > Sub-cellular vacuolization; 2ary CM exosome
formation; potential for mitogenesis and loss of contact
inhibition; (3ary indirect shift pressuromodulation) |
|
|
|
| OR S 1+ 1+
semi-Peri (0) |
|
| OR Monovalent
cationicity-hydroxylation cum carbonylation-monovalent cationicity
mediated CM receptor endocytosis |
|
|
| Bleomycin
hydrophobic core | 3.14 | 0.94 | (0) |
Cyclo(sulfo)pyridines and alkyl of amide
alkyl chain | 3.35 | CM receptor
hydrophobicity |
|
|
| Bleomycin
hydrophilic moiety1 | −0.08 | 0.54 | 1+ | ~ 3ary
S+ | −0.15 | Cationic
interaction w/receptor (extracellular CM interface) |
|
|
| Bleomycin
hydrophilic moiety2 | 3.05 | 0.42 | 1+ | 1ary
N+ | −7.28 | Cationic
interaction w/receptor (extracellular CM interface) |
|
|
| Bleomycin
hydrophilic moiety3 | −0.47 | 0.38 | Semi-Peri
carbonylo | C=O (pentose); C=O
(core) | −1.22 ×8 (×1;
×7) | CM receptor
hydrophilicity |
|
|
| Bleomycin
hydrophilic moiety4 | −0.33 | 0.32 | Semi-Peri
hydroxylo | OH (pentoses); OH
(core) | −1.05 ×8 (×6;
×2) | CM receptor
hydrophilicity |
|
|
| Table XI.Cell membrane (CM) receptor-mediated
antagonism/partial antagonism of pressuromodulation
extracellulomodulation with concomitant receptor kinase inhibition:
antago-nism/partial antagonsim of direct CM receptor-mediated
pressuromodulation +/- external cationomodulation (≥ 3+ - >
1+). |
Table XI.
Cell membrane (CM) receptor-mediated
antagonism/partial antagonism of pressuromodulation
extracellulomodulation with concomitant receptor kinase inhibition:
antago-nism/partial antagonsim of direct CM receptor-mediated
pressuromodulation +/- external cationomodulation (≥ 3+ - >
1+).
|
| Log OWPC
(unitless) | vdWD (nm) | Charge
distribution | Groups | (Log) OPWC-to-vdWD
ratio [per nm (nm1)] | 1ary mode of
interaction | Interaction
level(s) | Mechanism(s) of
action |
|---|
| Gefitinib
(Iressa) | 3.75 | 0.89 | 1+ 0 (0) | Cycloether
protected 3ary N+, alkyl ether, benzyl pyridine,
halogenated benzyl | 4.21 | CM receptor
pressuromodulation antagonism | CM EGF
receptor | Stable binding to
EGFR complex (KD antagonist < KD EGF); non-interaction of
endogenous EGF; non-effective CM EGFR pressuromodulation; decreased
cell division; decreasedVEGF; noninteraction w/extracellular matrix
heparan |
| L-type gefitinib
hydrophobic core | 4.21 | 0.74 | (0) | Benzyl pyridine,
halogenated benzyl | 5.71 | CM receptor
Hydrophobicity |
|
|
| Gefitinib
hydrophilic moietyl | −3.05 | 0.42 | 1+ | Cycloether
protected 3ary N+ | −7.28 | CM receptor
hydrophilicity |
|
|
| Gefitinib
hydrophilic moiety2 | 0.12 to −0.52 | 0.46 to 0.41 | 0 Etherylo |
~O-CH3 | 0.26 to −1.27 | CM receptor
hydrophilicity |
|
|
| Ceritinib | 3.40 | 0.97 | 1+ (0) 0 | cyclo[NH2+]pentane,
CH3-benzyl ether propyl, N, CL-cyclopyridine, N, benzyl,
O=S[propyl]=O | 8.02 | CM receptor
pressuromodulation antagonism | CM ALK receptor (in
lieu of midkine or pleiotrophin) | Stable binding to
ALKR complex (KD antagonist < KD ALK); non-interaction of
endogenous ALK ligand; non-effective ALKR pressuromodulation;
decreased cell division; decreased VEGF; noninteraction
w/extracellular matrix heparan |
| L-type ceritinib
hydrophobic core | ~7.16 | ~0.89 | (0) | ~Cyclopentane,
CH3-benzyl-O-propyl, N, CL-cyclopyridine, N, benzyl | ~5.09 | CM receptor
hydrophobicity |
|
|
| Ceritinib
hydrophilic moietyl | −2.95 | 0.47 | 1+ | 2ary N+
(of cyclopentane) | −6.22 | CM receptor
hydrophilicity |
|
|
| ceritinib | −0.22 | 0.60 | 0 | −(O=S=O)C(C)C | −0.37 | CM receptor |
|
|
| hydrophilic moiety2
Erlotinib (Tarceva) | 3.20 | 0.87 | 0 0 (0) | Alkyl ether side
chains ×2, alkyne, benzyl, N, pyridine benzyl | 3.67 | hydrophilicity CM
receptor pressuromodulation antagonism | CM EGF
receptor | Stable binding to
EGFR complex (KD antagonist < KD EGF); non-interaction of
endogenous EGF; non-effective CM EGFR pressuromodulation decreased
cell division; decreased VEGF; noninteraction w/extracellular
matrix heparan |
| L-type erlotinib
gydrophobic core | 3.61 | 0.74 | (0) | Alkyne, benzyl, N,
pyridine benzyl | 4.88 | CM receptor
hydrophobicity |
|
|
| Erlotinib
gydrophobic moietyl | 0.13 | 0.56 | 0
Ethero-isoneutral | Alkyl ether | 0.23 | CM receptor
hydrophobicity |
|
|
| Erlotinib
hydrophobic moiety2 | 0.13 | 0.56 | 0
Ethero-isoneutral | Alkyl ether | 0.23 | CM receptor
hydrophobicity |
|
|
| Lapatinib | 2.12 | 0.96 | 1+ (0–1) 0
(0–2) | O=S[CH3,
(CH2) 2-NH2+]=0, cycloetherw/dbs, benzyl-cyclopyridine,
N, Cl-benzyl-ether-benzyl-F | 2.20 | CM receptor
pressuromodulation antagonism | CM EGF
receptor | Stable binding to
EGFR complex (KD antagonist <KD EGF); non-interaction of
endogenous EGF; non-effective CM EGFR pressuromodulation; decreased
cell division; decreased VEGF; non-interaction w/extracellular
matrix heparan |
| U-type lapatinib
hydrophobic core | 6.68 | 0.89 | (0–1); (0–2) | Cyclocarboether,
benzyl-cyclopyridine, C, Cl-benzyl-C-benzyl-F | 7.51 | CM receptor
hydrophobicity |
|
|
| Lapatinib
hydrophilic moietyl | −4.30 | 0.62 | 1+ | 0=S[CH3,
(CH2)2-NH2+]=0 | −6.96 | CM receptor
hydrophilicity |
|
|
| Lapatinib
hydrophilic moiety2 | −0.60 | 0.54 | 0 | Pyrindine-NH2 | −1.11 | CM receptor
hydrophilicity |
|
|
| Lapatinib
hydrophobic moietyl | 0.12 | 0.46 | (0–1)
cycloEthero-isoneutral |
~CycloCH3-O-CH3 | 0.26 | CM receptor
hydrophilicity |
|
|
| MK-2206 | 1.80 | 0.87 | 1+ (0) 0 | 1ary N+,
cylcoquatrane, benzyls ×2, cyclopyridines ×3, OH | 2.07 | CM receptor partial
Pressuromodulation antagonism | CM GM-CSF
receptor | Stable binding to
GM-CSF complex (KD antagonist < KD GM-CSF); lesser affinity
concomitant binding of GM-CSF to GM-CSFR; partially-effective
inhibition of endogenous pressuromodulation; decreased potential
for cell division |
| Modified L-type
MK-2206 hydrophobic core | 6.71 | 0.82 | (0) | Cylcoquatrane,
benzyls ×2, cyclopyridines ×2 | 8.14 | CM receptor
hydrophobicity |
|
|
| MK-2206 hydrophilic
moietyl | −3.05 | 0.42 | 1+ | 1ary
N+ | −7.28 | CM receptor
hydrophilicity |
|
|
| MK-2206 hydrophilic
moiety2 | −0.47 | 0.38 | 0 Carbonylo | C=O | −1.22 | CM receptor
hydrophilicity |
|
|
| MK-2206 hydrophilic
moiety3 | −0.41 | 0.47 | 0 Pyridino | Cyclo
N-NH-CH2 -N-CH2 | −0.87 | CM receptor
hydrophilicity |
|
|
| Staurosporine | 1.20 | 0.91 | 1+ (0) 0 | (2ary
N+)- cyclopentylether-(OCH3),
cycloquatraneNthers ×2, cyclobenzyls ×3, carbonylopyridine | 1.32 | CM receptor
pressuromodulation antagonism | CM
asialoglycoprotein receptor (AGR) (in lieu of endogenous cationic
peptide) | Stable binding to
AGR complex (KD antagonist > KD cationic AGR ligand);
non-endocytosis of AGR complex; non-3ary indirect
pressuromodulation −> non-mitogenesis non-(cell) division |
| ~Staurosporine
hydrophobic core | 5.72 | 0.81 | (0) | CycloquatraneNthers
×2, cyclobenzyls ×3 | 7.08 | CM receptor
hydrophobicity |
|
|
| Staurosporine
hydrophilic moiety1 | −2.95 | 0.47 | 1+ | 2ary
N+ | −6.22 | CM receptor
hydrophilicity |
|
|
| Staurosporine
hydrophilic moiety2 | −0.47 | 0.38 | 0 Carbonylo | C=O | −1.22 | CM receptor
hydrophilicity |
|
|
| Staurosporine
hydrophilic moiety3 | 0.12 to −0.52 | 0.46 to 0.41 | 0 Etherylo |
~O-CH3 | 0.26 to −1.27 | CM receptor
hydrophilicity |
|
|
| Afatinib | 1.11 | 0.91 | 1+ 0 (0) | 3ary N+
alkene amide (acrylamide), cycloether ether, benzyl cyclopyridine,
N, F-benzyl-Cl | 1.22 | CM receptor
pressuromodulation antagonism | CM EGF receptor
(covelent) | Stable binding to
EGFR complex (KD antagonist < KD EGF); non-interaction of
endogenous EGF; non-effective CM EGFR pressuromodulation; decreased
cell division; decreased VEGF; noninteraction w/extracellular
matrix heparan |
| L-type afatinib
hydrophobic core | 4.21 | 0.74 | (0) | Benzyl pyridine,
halogenated benzyl | 5.71 | CM receptor
hydrophobicity |
|
|
| Afatinib
hydrophilic moiety1 | −3.20 | 0.65 | 1+ | 3ary N+
alkene amide (acrylamide) | −4.93 | CM receptor
hydrophilicity |
|
|
| Afatinib
hydrophilic moiety2 | −0.62 | 0.54 | 0 | Cycloether
ether | −1.15 | CM receptor
hydrophilicity |
|
|
| Imatinib
(Gleevac) | 1.50 | 0.94 | 1+ (0–1) 0
(0–2) | 3ary N+
in cyclic hexane w/2 Ns, benzyl amide, CH3-benzyl, N,
trans pyridinepyridine | 1.59 | CM receptor partial
pressuromodulation antagonism | CM PDGF
receptor | Stable binding to
PDGFR complex (KD antagonist < KD PDGF); lesser affinity
concomitant binding of PDGF to PDGFR; partially-effective
inhibition of endogenous pressuromodulation; decreased potential
for cell division |
| L-type imatinib
hydrophobic core | 5.21 | 0.87 | (0–1); (0–2) |
CH3-benzyl-CH2, N of
amide, CH3-benzyl, N, trans pyridine-to-pyridine | 6.00 | CM receptor
hydrophobicity |
|
|
| Imatinib
hydrophilic moietyl | −2.80 | 0.52 | 1+ | ~3ary N+
of cyclic hexane w/2 Ns | −5.42 | CM receptor
hydrophilicity |
|
|
| Imatinib
hydrophilic moiety2 | −0.47 | 0.38 | 0 Carbonylo | C=O | −1.22 | CM receptor
hydrophilicity |
|
|
| Crizotinib | 1.00 | 0.88 | 1+ 0 (0) | Cyclo[NH2+]hexane,
penta-and hexa-pyridine[NH2], ether, (Cl)2-benzyl-F | 1.13 | CM receptor
pressuromodulation antagonism | CM ALK receptor (in
lieu of midkine or pleiotrophin) | Stable binding to
ALKR complex (KD antagonist < KD ALK); non-interaction of
endogenous ALK ligand; non-effective ALKR pressuromodulation;
decreased cell division; decreased VEGF; noninteraction
w/extracellular matrix heparan |
| L-type crizotinib
hydrophobic core | 5.96 | 0.88 | 0 (0) | Cyclohexane, penta-
and hexa-pyridine[NH2], ether, (Cl)2-benzyl-F | 6.80 | CM receptor
Hydrophobicity |
|
|
| Crizotinib
hydrophilic moiety | −2.95 | 0.47 | 1+ | 2ary
N+ | −6.22 | CM receptor
hydrophilicity |
|
|
|
Hydroxy-camptothecin (10-HCT) | 0.92 | 0.82 | 0 (0) 0 | OH-benzyl,
pyridines ×3 (2 Ns), C=O, OH-cylic ester | 1.12 | CM receptor partial
pressuromodulation antagonism | CM TRAIL R2 | Stable binding to
TRAIL receptor complex (KD antagonist < KD TRAIL); lesser
affinity concomitant binding of TRAIL to TRAIL R2;
partially-effective inhibition of pressuromodulation; decreased
potential for cell division |
| ~L-type 10-HCT
hydrophobic core | 4.19 | 0.80 | (0) | Benzyl of phenol,
pyridines ×3, cyclic ester O | 5.26 | CM receptor
hydrophobicity |
|
|
| 10-HCT hydrophilic
moietyl | −0.33 | 0.32 | 0 Hydroxylo | OH | −1.05 ×2 | CM receptor
hydrophilicity |
|
|
| 10-HCT hydrophilic
moiety2 | −0.47 | 0.38 | 0 Carbonylo | C=O | −1.22 ×2 | CM receptor
hydrophilicity |
|
|
| AMD070 | −0.14 | 0.85 | 1+ 0 (0) | 1ary N+,
benzyl ×1, N, pyridine ×2, cyclohexane | −0.16 | CM receptor
pressuromodulation antagonism | CM CXCR4 [in lieu
of SDF-1 (CXCL 12)] | Stable binding to
CXCR4 complex (KD antagonist < KDSDF-1); non-interaction of
endogenousSDF-1; non-effective CM CXCR4 pressuromodulation;
decreased cell division; decreased VEGF; non-interaction
w/extracellular matrix heparan |
| AMD070 hydrophobic
core | 4.33 | 0.85 | 0 (0) | Benzyl ×1, N,
pyridine ×2, cyclohexane | 5.19 | CM receptor
hydrophobicity |
|
|
| AMD070 hydrophilic
moiety | −3.05 | 0.42 | 1+ | 1ary
N+ | −7.28 | CM receptor
hydrophilicity |
|
|
| Topotecan | −2.10 | 0.88 | 1+ (0) 0 | 3ary N+
ethyl-phenol, pyridines ×3 (2 Ns), C=0, OH-cylic ester | −2.39 | CM receptor partial
Pressuromodulation antagonism | CM TRAIL R2 | Stable binding to
TRAIL receptor complex (KD antagonist < KD TRAIL); lesser
affinity concomitant binding of TRAIL to TRAIL R2;
partially-effective inhibition of pressuromodulation; decreased
potential for cell division |
| ~L-type topotecan
hydrophobic core | 4.71 | 0.82 | (0) |
~CH2-benzyl of phenol,
pyridines ×3, cyclic ester O | 5.77 | CM receptor
hydrophobicity |
|
|
| 10-HCT hydrophilic
moietyl | −3.05 | 0.42 | 1+ | 3ary
N+ | −7.28 | CM receptor
hydrophilicity |
|
|
| 10-HCT hydrophilic
moiety2 | −0.33 | 0.32 | 0 Hydroxyl o | OH | −1.05 ×2 | CM receptor
hydrophilicity |
|
|
| 10-HCT hydrophilic
moiety3 | −0.47 | 0.38 | 0 Carbonyl o | c=o | −1.22 ×2 | CM receptor
hydrophilicity |
|
|
Small molecule xenobiotics that
permeate across CM channel aqueous pores to arrest nuclear and
mitochondrial function
This category includes the least hydrophilic pure
hydrophile small molecule xenobiotics that permeate across CM
protein channel aqueous pore nuclear membrane (NM) fenestrations
and outer-to-inner mitochondrial membrane (MM) aqueous pores to
arrest nuclear DNA transcription and RNA translation function (i.e.
Ki67) as well as mitochondrial DNA transcription and RNA
translation function, followed by arrest of DNA replication
function (Tables I and II, Figs. 1
and 2).
This category of CM, NM and MM pore permeable small
molecule xenobiotics include:
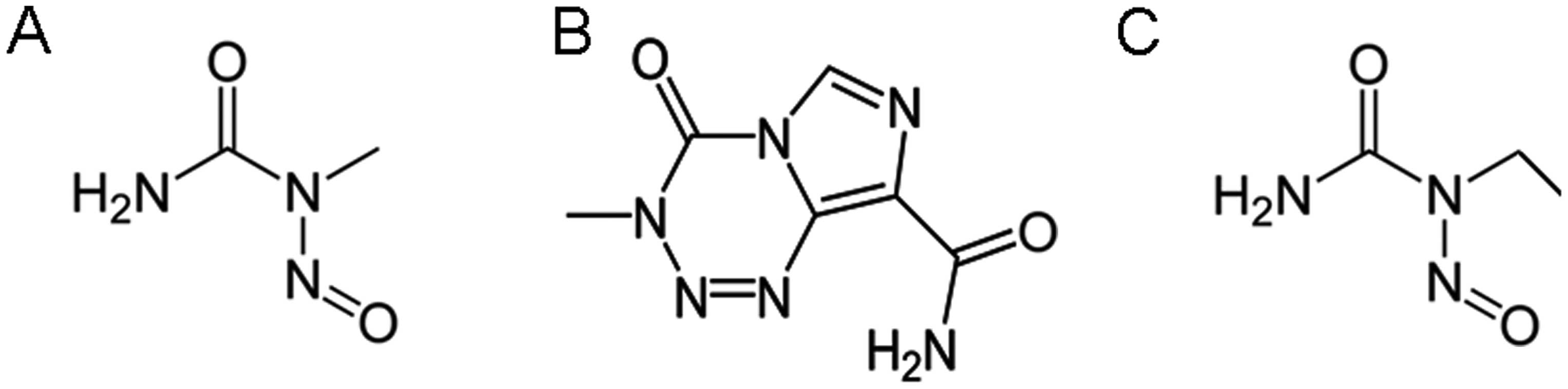
i) The DNA and RNA alkylating adductors,
nitroso-N-methylurea (EMU) (Log OWPC, −0.55; vdWD, 0.54 nm; Log
OWPC-to-vdWD ratio, −1.20 nm−1), temozolomide
(TMZ) (Log OWPC, −0.45; vdWD, 0.65 nm; Log OWPC-to-vdWD ratio,
−0.70 nm−1) (18)
and nitroso-N-ethylurea (ENU) (Log OWPC, −0.22; vdWD, 0.57 nm; Log
OWPC-to-vdWD ratio, −0.38 nm−1) (19,20). EMU,
TMZ and ENU arrest replicative nuclear function via association
with nitrogenous bases, of DNA, and RNA, strand nucleotides with
sufficient affinity to alkylate nucleotide nucleosides involved in
base pairing hydrogen bonding, which renders alkylated base
segments of DNA, and RNA, non-functional; these DNA and RNA
alkylating small molecule xenobiotics, but are not P450 cytochrome
inducers due to insufficient lipophilicities for size of their
hydrophobic moiety (Table I and
Fig. 1).
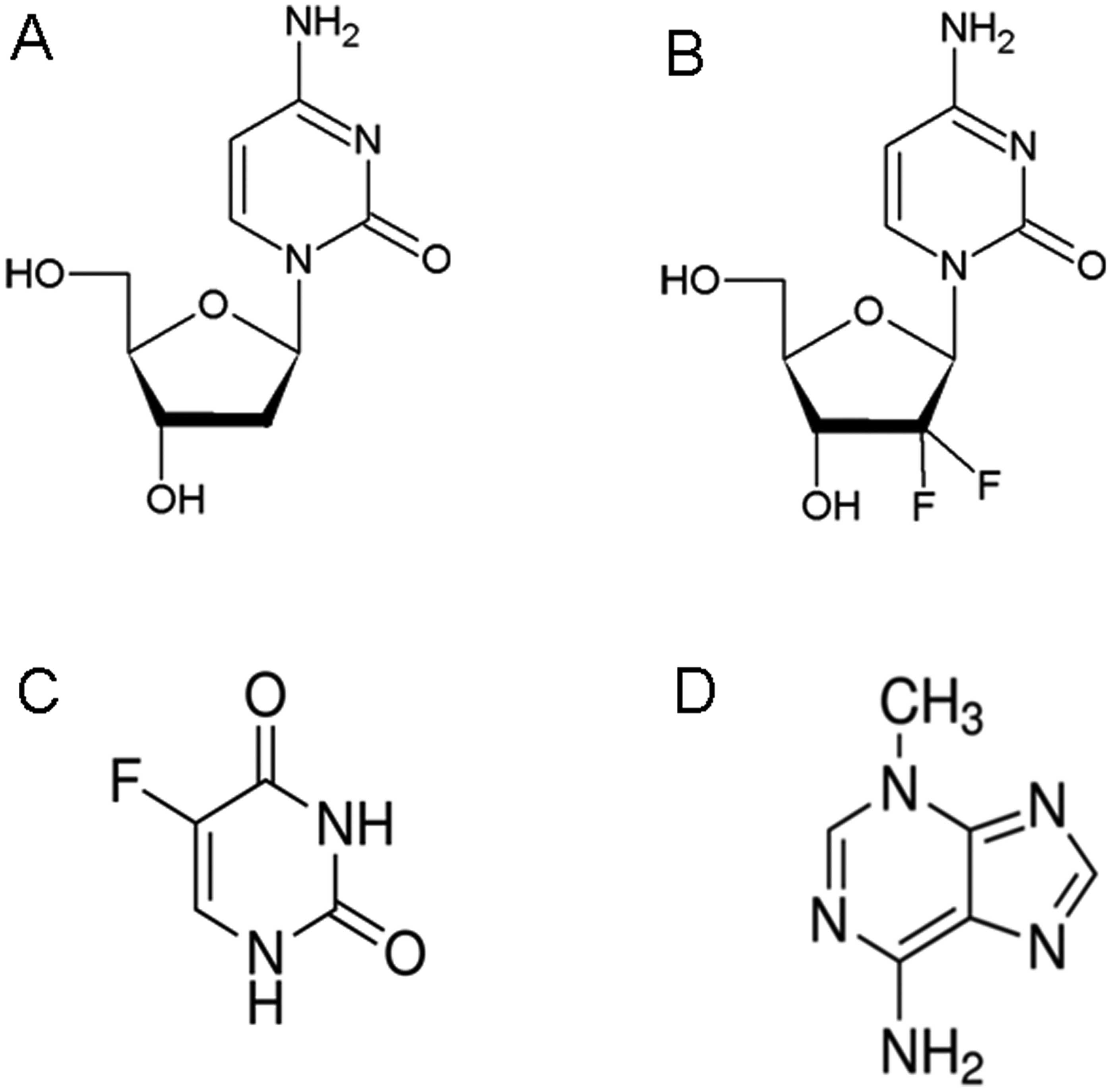
ii) The nucleoside substitutors, decitabine (Log
OWPC, −2.35; vdWD, 0.70 nm; Log OWPC-to-vdWD ratio, −3.35
nm−1), gemcitabine (Log OWPC, −1.65; vdWD, 0.72
nm; Log OWPC-to-vdWD ratio, −2.29 nm−1),
5-fluorouracil (5-FU) (Log OWPC, −0.66; vdWD, 0.56 nm; Log
OWPC-to-vdWD ratio, −1.18 nm−1) (21) and 3-methyladenine (3-MA) (Log OWPC,
−0.31; vdWD, 0.61 nm; Log OWPC-to-vdWD ratio, −0.51
nm−1). Decitabine, gemcitabine and 5-FU arrest
replicative function via competitive inhibition with endogenous
nitrogenous bases for phosphorylation and/or ribosylation enzyme
active sites, by substitution into DNA and RNA strands, thereby
rendering substituted nucleotide segments of DNA and RNA
non-functional. 3-MA, although a nucleotide substitute for
adenosine upon ribosylation phosphorylation, does not interfere
with DNA helix base pairing upon substitution due to the exterior
presence of the methyl group (CH3) (as opposed to
interior), whereby, the functionality of 3-MA substituted
nucleotide segments of DNA is maintained, that which results in
increased burst nuclear DNA transcription and competitive
activation of mRNA polyadenylation poly(ADP-ribose) polymerase-1
(PARP-1) (22) and in MM oxidative
stress with subsequent release of MM apoptosis inducing factor
(AIF), AIF binding of X-linked inhibitor of apoptosis factor (XIAF)
and generation of free caspases (i.e. caspase-3) with resultant
cleavage of transcriptionally active nuclear chromatin (Table II and Fig.
2).
Of such small molecule hydrophilic xenobiotics,
those that permeate through CM channel aqueous pores via
unrestricted diffusion include those of diameters ranging between
0.54 and 0.65 nm, in the context of overall hydrophilicities for
molecular size ranging between −0.38 and −1.20
nm−1. Those that permeate through CM channel
aqueous pores with a tendency towards restricted diffusion include
those of larger diameters, 0.70 and 0.72 nm, in the context of
greater overall hydrophilicities for molecular size of −2.29 and
−3.35 nm−1. Such small molecule xenobiotics are
permeable across CM protein channel aqueous pores due to the
absence of charge, either anionic or cationic, analogous to
endogenous molecules such as the nitrogenous bases and nucleosides,
which have similar functional groups and fall within a similar
range of vdWDs and hydrophilicities for size in the absence of
charge, and primarily accumulate in the nucleus, which has NM
fenestrations with a functional upper limit of pores of ~9 nm
(23), for which the permeability
surface area product is greater than that of the MM pores. Although
such small molecule xenobiotics primarily accumulate within the
nucleoplasm, they can also accumulate within the mitochondrial
cytosol, via permeation across outer-to-inner MM protein channel
pores [voltage dependent anion channel (VDAC)] (24,25),
across which small molecule endogenous hydrophiles of similar vdWDs
and hydrophilicities for size, are permeable (26); meanwhile, larger and charged
endogenous small molecule hydrophiles more hydrophilic for size,
such as ATP (3- and Mg2+ → 1-), are not, while smaller
and charged endogenous small molecule hydrophiles less hydrophilic
for size such as citrate (3- and Ca2+ → 1-), succinate
(2- → SuccinateH 1- at pH ~7) and HPO4 (2- →
H2PO4 1- at pH ~7) are insignificantly more
permeable, as these have permeabilities that fall within the order
of magnitude of that of ATP (24), in
contrast to chloride (Cl−), an anion that is not
significantly permeable across CM pores with an
anionization-to-atomic diameter ratio (AI-to-AD; nm−1)
of 4.90 nm−1, which is 3 orders of magnitude more
permeable than ATP with a permeability coefficient of
1.1×10−12 cm3/sec (24), findings that which implicate
‘kiss-and-run’ exocytosis as the basis for ATP release into the
cytosol, for secondary permeation into the nucleus.
Small molecule xenobiotics of this category, being
of pure hydrophilic character with lesser overall hydrophilicity,
are exquisitely permeable across CM aqueous channel pores and into
the intra-nuclear compartment, but also into the
intra-mitochondrial compartment (27). Thus, such xenobiotic chemotherapies
have shown the potential to be uniformly cytotoxic to tumor cells,
via effects in both nuclear and mitochondrial compartments in the
setting of an actively maintained concentration gradient in
vitro, particularly synergistically (28,29).
Therefore, in order to demonstrate similar effectiveness in the
clinical setting, such small molecule xenobiotics, as well as
others discussed herein, must be made to first and foremost
selectively accumulate to effective concentrations within tumor
tissue, which can only be accomplished upon labile linking to
optimally-sized, and designed, nanoparticles within the 8- to
9-nm-size range delivered transvascularly (3–7), to ensure
uniform cytotoxicity to all tumor tissue cells and minimal risk for
subsequent neoplastic transformation of tumor-associated cells
including tumor stem cells.
Small molecule xenobiotics that
permeate across CM channel aqueous pores to bind to cytochrome
P450s: DNA adduction and/or crosslinking
This category includes small molecule xenobiotics,
hydro-lipophiles and lipophiles with hydrophilicity, with vdWDs in
the 0.74 to 0.67 nm range, which can permeate across CM, NM and MM
aqueous pores with the potential to bind to cytochrome P450s due to
the hydrophobicity for size of their hydrophobic moieties, with the
potential to arrest nuclear and mitochondrial replication via
DNA/RNA alkylation or DNA strand-to-DNA strand crosslinking
(Table III and Fig. 3).
This category of CM, NM and MM pore permeable small
molecule xenobiotics include:
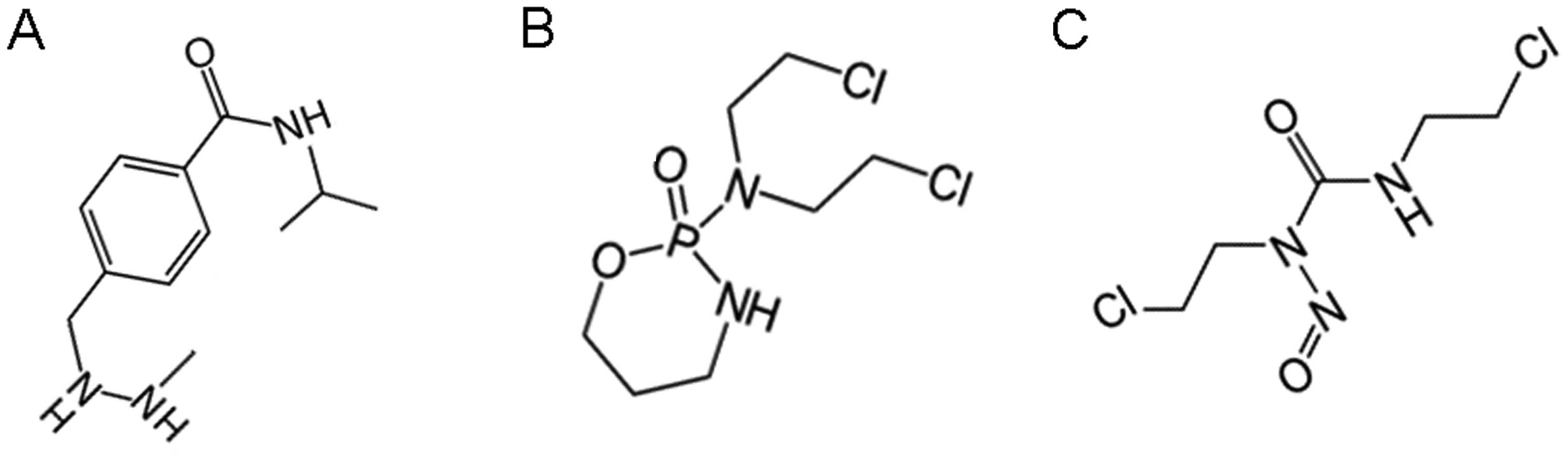
i) The intra-CM-associated cytochrome P450
hydroxylation-activated (30,31) and intra-MM-associated cytochrome P450
hydroxylation-inactivated (31,32),
sub-classified as
a) The intra-CM-associated cytochrome P450
hydroxylation-activated DNA/RNA guanosine guanine alkylator
procarbazine (33) (Log OWPC, −1.70;
vdWD, 0.74 nm; Log OWPC-to-vdWD ratio, −2.29
nm−1); procarbazine hydrophilic moiety1 (Log
OWPC, −0.59; vdWD, 0.51 nm; Log OWPC-to-vdWD ratio, −1.17
nm−1); procarbazine hydrophobic moiety1 (Log
OWPC, 2.51; vdWD, 0.57 nm; Log OWPC-to-vdWD ratio, 4.43
nm−1); procarbazine hydrophilic moiety2 (Log
OWPC, −1.11; vdWD, 0.43 nm; Log OWPC-to-vdWD ratio, −2.61
nm−1); procarbazine hydrophobic moiety2 (Log
OWPC, 1.82; vdWD, 0.48 nm; Log OWPC-to-vdWD ratio, 3.75
nm−1): procarbazine alkylates DNA/RNA guanosine
guanines via its CH3-NH2-NH2-
terminal hydrophilic moiety1, but without the potential to
crosslink (Table III and Fig. 3).
(b) The intra-CM-associated cytochrome P450
hydroxylation-activated DNA strand-to-DNA strand crosslinkers,
cyclophosphamide (34) (Log OWPC,
0.10; vdWD, 0.73 nm; Log OWPC-to-vdWD ratio, 0.14
nm−1); cyclophosphamide hydrophilic core (Log
OWPC, −1.92; vdWD, 0.59 nm; Log OWPC-to-vdWD ratio, −3.25
nm−1); cyclophosphamide hydrophobic moieties 1
and 2 (Log OWPC, 1.19; vdWD, 0.48 nm; Log OWPC-to-vdWD ratio, 2.49
nm−1) and ifosfamide (Log OWPC, 0.10; vdWD, 0.73
nm; Log OWPC-to-vdWD ratio, 0.14 nm−1);
ifosfamide hydrophilic core (Log OWPC, −1.92; vdWD, 0.59 nm; Log
OWPC-to-vdWD ratio, −3.25 nm−1); ifosfamide
hydrophobic moieties 1 and 2 (Log OWPC, 1.19; vdWD, 0.48 nm; Log
OWPC-to-vdWD ratio, 2.49 nm−1). Cyclophosphamide
and ifosfamide crosslink DNA strand-to-DNA strand guanine N7s due
to the length of their 2 reactive hydrophobic moieties,
CH2-CH2-CL ×2, rendering G-to-G cross linked
segments inseparable, non-functional and prone to strand breaks,
and can directly inhibit glutathione reductase (37–41)
(Table III and Fig. 3).
ii) The intra-CM-associated cytochrome P450
hydroxylation-inactivated (31,32,35), DNA
strand-to-DNA strand crosslinker carmustine (BCNU) (Log OWPC, 0.95;
vdWD, 0.67 nm; Log OWPC-to-vdWD ratio, 1.41
nm−1). Carmustine crosslinks DNA strand-to-DNA
strand guanine N7s (36) due to the
length of its 2 reactive hydrophobic moieties,
CH2-CH2-Cl ×2, rendering G-to-G cross linked
segments inseparable, non-functional and prone to strand breaks,
and can directly inhibit glutathione reductase (37–41)
(Table III and Fig. 3).
Of such small molecule xenobiotics with
intra-structural hydrophobicity, those that are most likely to
achieve intra-cellular levels are those that are both of smaller
size and relatively less lipophilic, as these can diffuse through
CM channel aqueous pores without restriction and partition to a
sufficient extent into the aqueous phase, which is the case for
procarbazine (Log OWPC-to-vdWD ratio, −2.29 nm−1;
vdWD, 0.74 nm) while those most likely to interact with the CM
phospholipid bilayer are those that are of larger size as these are
restricted to diffusion through CM channel aqueous pores, and
importantly, those with greater overall molecular lipophilicity cum
larger size, as these have the tendency to associate into the
phospholipid bilayer among fatty acid-esters. This is the case of
those within the overall lipophilicity range of
cyclophosphamide/ifosfamide (Log OWPC-to-vdWD ratio, 0.14
nm−1; vdWD, 0.73 nm) (34,42,43) which
are of larger size, and carmustine (Log OWPC-to-vdWD ratio, 1.41
nm−1; vdWD, 0.67 nm) (44), which is more lipophilic (44), for which there is a greater tendency
to associate with and perturb CM phospholipids and the potential
for a 1ary indirect pressuromodulation-mediated secondary increase
in very high molecular weight (MW) protein transcription (17), while such small molecule xenobiotics
of lipophilic character may only achieve intra-cellular levels at
significant extracellular concentrations (45), which makes the more lipophilic
xenobiotics of this category marginal chemoxenobiotics for
effective transvascular delivery into solid tumor tissue (3–7) .
Small molecule xenobiotics that insert
in-between CM phospholipids with the potential for 1ary indirect
pressuromodulation
This category includes small molecule xenobiotics
with hollow isophilic interiors and molecular lipophilicity in the
form of the circumferential presence of lipophilic moieties on the
exterior, insert in-between CM phospholipids at the phospholipid
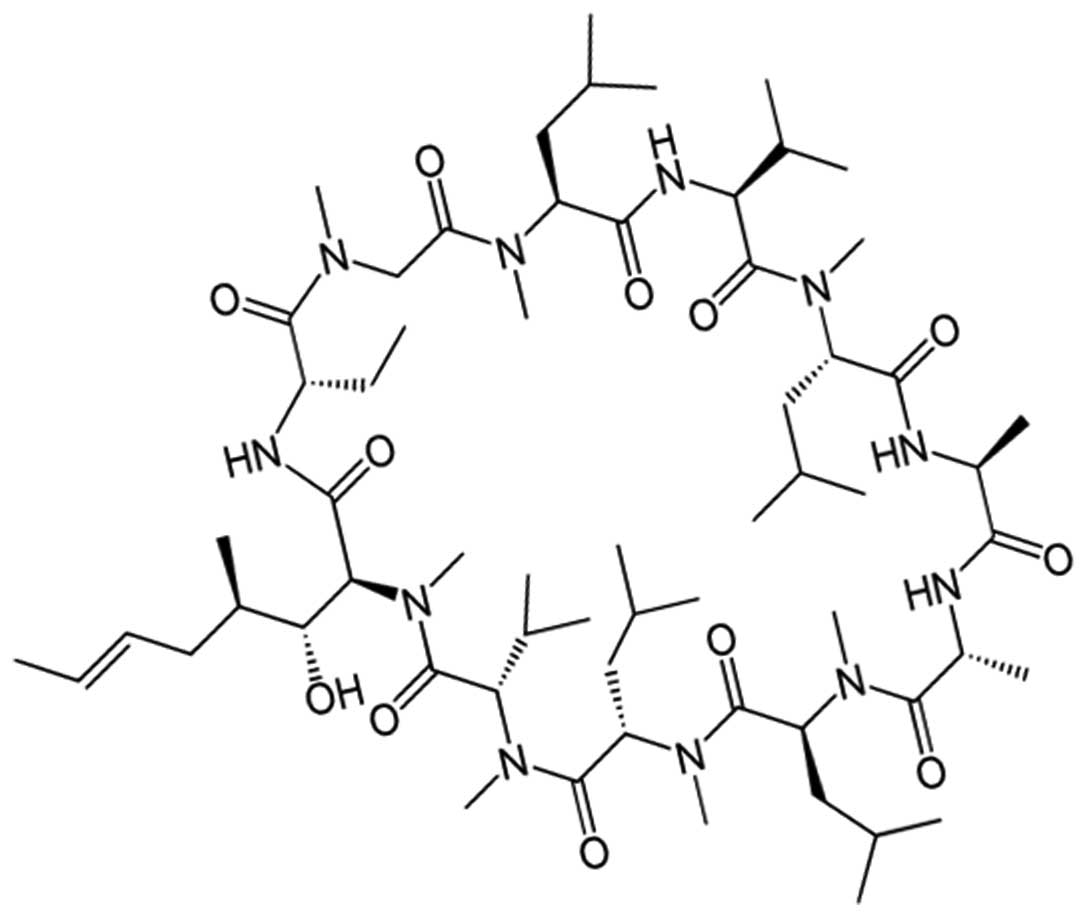
glycerol-fatty acid ester junctions (46), and include, cyclosporine A (Log OWPC,
3.64; vdWD, 1.31 nm; Log OWPC-to-vdWD ratio, 2.78
nm−1), which has a 5 carbon (5C) alkene tail that
predisposes to CM phospholipid glycerol-fatty acid ester
association rather than with CM receptor protein hydrophobicity
(Table IV and Fig. 4).
| Table IV.Cell membrane (CM) insertoassociation
and phospholipid interspace widening with potential for 1ary
indirect pressuromodulation. |
Table IV.
Cell membrane (CM) insertoassociation
and phospholipid interspace widening with potential for 1ary
indirect pressuromodulation.
|
| Log OWPC
(unitless) | vdWD (nm) | Charge
distribution | Groups | (Log) OPWC-to-vdWD
ratio [per nm (nm−1)] | 1ary mode of
interaction | Interaction
level(s) | Mechanism(s) of
action |
|---|
| Cyclosporine A | 3.64 | 1.31 | Circ (0) | Outer ring
isopropyls ×6, CH3s and alkyl and 6C w/alkyene; Inner
ring intervening CH3s, amides and OH | 2.78 | CM phospholipid
inter-insertion | Glycerol-FA-ester
junction (stable) | Potential for
mitochondria-mediated apoptosis in synergism with CM receptor
mediated pressuromodulation 2ary to futile synthesis of
intermediate MW proteins; potential for 1ary indirect
pressuromodulation at low concentration; [> increased
transcription of very high MW nuclear division proteins] |
| ~Cyclosporine A
hydrophobic tail moiety | ~2.5 | ~0.66 | (0) | 6C chain
alkyene | ~3.79 | CM phospholipid
ester tail hydrophobicity |
|
|
| Cyclosporine A
hydrophobic (outer ring) moieties | n/a | n/a | 0 Outer Circ | Outer ring
isopropyls ×6, CH3s and N-C=Os | n/a | CM
interphospholipid glycerol junction isophilicity n/a |
|
|
| Cyclosporine A
hydrophobic (inner ring) moieties | n/a | n/a | 0 Inner circ | Inner ring
intervening CH3s and OH |
|
| CM
inter-phospholipid glycerol junction isophilicity |
|
Cyclosporine A has been shown to decrease the rate
of cell division only in the presence of powerful CM receptor
pressuromodulators (47,48), which is due to generation of nuclear
transcription-driven burst mitochondrial reactive O2
species (47,49) secondary to the increased transcription
of the intermediate MW proteins (i.e. p53) and BCL depletion, or in
the presence of mitochondrial-associated microtubule network
disruptors (49), which is due to
concomitant mitochondrial anchorage-mediated MM
disruption/dissolution toxicity and generation of free caspases
(i.e. free caspase-3). At low intra-tumoral concentrations
cyclosporine A has more potential to cause 1ary indirect
pressuromodulation (perturbomodulation) (17), that would actually cause an increase
in the transcription of very high MW proteins such as secretory
proteins (i.e. fibronectin, 240 kDa) and nuclear division proteins
(Ki67, 359 kDa; separase, 230 kDa) (17). Furthermore, as cyclosporine A is
highly serum protein-bound secondary to its overall lipophilicity
for size (Log OWPC-to-vdWD ratio, 2.78 nm−1), it
is difficult to obtain µM local cyclosporine A intra-tumoral
concentrations in standard free drug intravenous chemotherapeutic
regimens.
Small molecule xenobiotics that
associate with CM cholesterol or phospholipid fatty acid-esters
with the potential for 1ary indirect pressuromodulation
This category includes small molecule xenobiotics,
hydro-lipophiles with anisotropic molecular hydrophilicity cum
lipophilicity to lipophiles with anisotropic molecular
hydrophilicity or absence of, with vdWDs in the 1.18–0.77 nm range
(Table V and Fig. 5).
Small molecule xenobiotic hydro-lipophiles with
anisotropic molecular hydrophilicity cum lipophilicity include: i)
nystatin (Log OWPC, −2.00; vdWD, 1.18 nm; Log OWPC-to-vdWD ratio,
−1.70 nm−1); ii) amphotericin B (Log OWPC, −1.30;
vdWD, 1.17 nm; Log OWPC-to-vdWD ratio, −1.11
nm−1); iii) filipin (Log OWPC, −0.15; vdWD, 1.00
nm; Log OWPC-to-vdWD ratio, −0.15 nm−1), with
their hydrophobic inner crescent having a lipophilicity for size of
8.32 nm−1 (Log OWPC, 6.73; vdWD, 0.81 nm).
Nystatin, amphotericin B and filipin associate with
CM bilayer cholesterol with their hydrophobic inner crescent over
the exposed outer surface area of CM cholesterol, which is within
the range of lipophilicity for the size of the CM bilayer surface
cholesterol portion of between 9.25–7.7 nm−1 (Log
OWPC, 8.5–7.11; vdWD, 0.92 nm), but do not physically incorporate
into CM-associated protein receptor isophilic-to-hydrophobic
interiors (cores) due to the presence of anisotropic molecular
hydrophilicity cum lipophilicity. Thus, such small molecule
xenobiotics remove CM cholesterol via cholesteroloassociation with
the more exteriorly protruding cholesterol portion to decrease CM
cholesterol concentration, thereby, de-stabilizing the CM
sufficiently enough to concomitantly cause CM
destabilization-mediated extrusion of CM cholesterol-associated
proteins/receptors, and as a result, also have the potential to
cause a subsequent 1ary indirect pressuromodulation
(perturbomodulation)-mediated secondary increase in very high MW
protein transcription (17,50,51)
(Table V and Fig. 5).
Small molecule xenobiotic lipophiles with
anisotropic molecular hydrophilicity include:
i) Chlorambucil [(Log OWPC, 0.60; vdWD, 0.79 nm; Log
OWPC-to-vdWD ratio, 0.76 nm−1); chlorambucil
hydrophobic core (Log OWPC, 5.26; vdWD, 0.79 nm; Log OWPC-to-vdWD
ratio, 6.75 nm−1)], in the case of small molecule
xenobiotic lipophile with anisotropic molecular hydrophilicity,
chlorambucil [(Log OWPC, 0.60; vdWD, 0.79 nm; Log OWPC-to-vdWD
ratio, 0.76 nm−1); chlorambucil hydrophobic core
(Log OWPC, 5.26; vdWD, 0.79 nm; Log OWPC-to-vdWD ratio, 6.75
nm−1)], associates into the CM closely with the
CM phospholipid layer-spanning portion of cholesterol that has a
lipophilicity of ~7.26 nm−1 which is close to its
hydrophobic core lipophilicity of 6.75 nm−1,
while transiently stabilizing itself in the layer by concomitantly
interacting anisotropically with the layer phospholipid head groups
viz a viz its mono-anionic hydrophilic-interacting COO−
moiety, to remove cholesterol via cholesteroloassociation secondary
to unstable structural association with phospholipid layer fatty
acid-ester tails in context of an inability to glyceroloesterify.
Therefore, chlorambucil is not an effective intra-cellularly
localizing mitochondrial or nuclear DNA/RNA
alkylating/bi-functional DNA crosslinking chemoxenobiotic, but
instead removes CM cholesterol via cholesteroloassociation with the
CM phospholipid layer-spanning interior cholesterol portion to
decrease CM cholesterol concentration (52,53) and as
a result, has the potential to cause a subsequent 1ary indirect
pressuromodulation (perturbomodulation)-mediated secondary increase
in very high MW protein transcription (17) (Table V
and Fig. 5).
ii) Melphalan [(Log OWPC, 1.00; vdWD, 0.79 nm; Log
OWPC-to-vdWD ratio, 1.27 nm−1); melphalan
hydrophobic core (Log OWPC, 4.42; vdWD, 0.75 nm; Log OWPC-to-vdWD
ratio, 5.86 nm−1)]. In the case of small molecule
xenobiotic lipophile with anisotropic molecular hydrophilicity,
melphalan [(Log OWPC, 1.00; vdWD, 0.79 nm; Log OWPC-to-vdWD ratio,
1.27 nm−1); melphalan hydrophobic core (Log OWPC,
4.42; vdWD, 0.75 nm; Log OWPC-to-vdWD ratio, 5.86
nm−1)], associates directly into the CM among the
CM phospholipid layer fatty acid-ester tails, rather than with the
CM phospholipid layer-spanning interior cholesterol portion, along
with more stable association within the layer by virtue of the
concomitant anisotropic interaction of its cataniononeutral
hydrophilic-interacting NH3+ COO− moiety with
layer phospholipid head groups, that importantly results in its
ability to secondarily subsequently trans-displace across CM
phospholipid layers and bilayer over time into the intra-cellular
compartment. For this reason, melphalan achieves perceptible
intra-cellular concentrations, to which specific cytochrome P450s
exist (CYP 3A and CYP 27), but without induction. As a result of
only a transient cytochrome P450 association in the context of
higher affinity association with outer mitochondrial membrane (OMM)
bilayer layer fatty acid-ester tails due to a hydrophobic core
lipophilicity of 5.86 nm−1, melphalan is not an
effective intra-cellularly localizing nuclear DNA/RNA
alkylating/bi-functional DNA crosslinking chemoxenobiotic.
Therefore, the primary mode of melphalan cellular toxicity is
mitochondrial (54–56) and mitochondrially-mediated, via
initiation of the mitochondrial cellular apoptosis cascade,
beginning at the level of the OMM bilayer (57) upon the displacement of AIF (58,59) from
the OMM, as follows: i) disassociation of OMM AIF into the cytosol
and binding to XIAF (60) with
affinity, thereby disassociating XIAF-bound cytosolic-to-nuclear
caspases (caspase-3) (61,62) from bound to free caspase forms, which
are pro-membranocytotoxic and pro-nucleochromatotoxic and
pro-apoptotic in their free forms; in tandem with ii) association
of cytosolic-to-nuclear AIF homolog, BCL (63) with the OMM in place of AIF, thereby,
shifting the nuclear BCL bound-p53 equilibrium to free p53
(64–66), being the primary constitutive nuclear
transcription factor for PUMA/BIM-like proteins (67,68) and
BAX/BID-like proteins (66), whereby,
avid binding of BCL by the PUMA and BIM-like proteins, being
singular α-helix peptides, leads to further depletion of free BCL
(PUMA-BCL and BIM-BCL). The combination of i) and ii) results in
mitochondrially-mediated cellular apoptosis (Table V and Fig.
5).
iii) Ketaconazole [(Log OWPC, 4.19; vdWD, 0.94 nm;
Log OWPC-to-vdWD ratio, 4.46 nm−1); ketaconazole
hydrophobic moiety1 (Log OWPC, 3.30; vdWD, 0.71 nm; Log
OWPC-to-vdWD ratio, 4.64 nm−1)]. In the case of
small molecule xenobiotic lipophile without anisotropic molecular
hydrophilicity, ketaconazole [(Log OWPC, 4.19; vdWD, 0.94 nm; Log
OWPC-to-vdWD ratio, 4.46 nm−1); ketaconazole
hydrophobic moiety1 (Log OWPC, 3.30; vdWD, 0.71 nm; Log
OWPC-to-vdWD ratio, 4.64 nm−1)], associates
directly into the CM among the CM phospholipid layer fatty
acid-ester tails, but the least stably of those in this category,
due to an absence of structural hydrophilic anisotropy. Thus,
ketoconazole perturbs the CM bilayer sufficiently enough to
secondarily dissociate CM cholesterol from the bilayer, which is
the primary mode of ketoconazole cellular toxicity and is
attributable to its CM cholesterol removal secondary to
perturbation of the CM bilayer with an associated initial decrease
in CM and whole cell/intra-cellular pressuromodulation (17,69,70)
followed by a secondary increase in very high MW protein
transcription due to 1ary indirect pressuromodulation
(perturbomodulation) (17), which may
result in burst nuclear transcription-associated with mitochondrial
oxidative phosphorylation-MM-mediated nuclear and cellular
cytotoxicity/apoptosis (17,71) particularly, in the setting of
pre-existent concomitant CM receptor pressuromodulation antagnonism
such as that of nocodazole (72), a
CM receptor pressuromodulation antagonist (Table V and Fig.
5).
iv) Capecitabine [(Log OWPC, 0.75; vdWD, 0.83 nm;
Log OWPC-to-vdWD ratio, 0.90 nm−1); capecitabine
hydrophobic core (Log OWPC, 1.94; vdWD, 0.61 nm; Log OWPC-to-vdWD
ratio, 3.19 nm−1)].
v) Fluconazole [(Log OWPC, 0.56; vdWD, 0.77 nm; Log
OWPC-to-vdWD ratio, 0.73 nm−1); fluconazole hydrophobic
core (Log OWPC, 1.22; vdWD, 0.76 nm; Log OWPC-to-vdWD ratio, 1.41
nm−1)].
In the case of small molecule xenobiotic lipophiles
with anisotropic molecular hydrophilicity with less incorporating
lipophilicity, capecitabine [(Log OWPC, 0.75; vdWD, 0.83 nm; Log
OWPC-to-vdWD ratio, 0.90 nm−1); capecitabine
hydrophobic core (Log OWPC, 1.94; vdWD, 0.61 nm; Log OWPC-to-vdWD
ratio, 3.19 nm−1)] and fluconazole [(Log OWPC,
0.56; vdWD, 0.77 nm; Log OWPC-to-vdWD ratio, 0.73
nm−1); fluconazole hydrophobic core (Log OWPC,
1.22; vdWD, 0.76 nm; Log OWPC-to-vdWD ratio, 1.41
nm−1)], both associate directly into the CM among
the CM phospholipid layer fatty acid-ester tails, but due to
insufficient hydrophobic portion incorporating lipophilicities in
the range of between 3.19 and 1.41 nm−1, only
temporarily associate into the CM bilayer, to transiently perturb
and disorder CM phospholipids, and of the two, fluconazole (vdWD,
0.77 nm) has the potential for CM channel aqueous pore permeation
(73), with the potential to also
perturb sub-cellular membrane phospholipids including those of the
endoGolgi smooth endoplasmic reticulum (SER) with high CM-derived
sub-cellular membrane cholesterol phospholipid turnover rates and
lower incorporating lipophilicities. In the case of both,
capecitabine and fluconazole, the primary mode of cellular toxicity
is attributable to perturbation of the CM bilayer with an
associated initial decrease in CM and whole cell/intra-cellular
pressuromodulation (17,74), in which case, of the two, capecitabine
has the greater potential to cause a subsequent 1ary indirect
pressuromodulation (perturbomodulation)-mediated secondary increase
in very high MW protein transcription (17), that will result in burst nuclear
transcription-associated with mitochondrial oxidative
phosphorylation-MM-mediated nuclear and cellular
cytotoxicity/apoptosis (17,74). In contrast, fluconazole, which also
has the potential to cause a subsequent 1ary indirect
pressuromodulation (perturbomodulation)-mediated secondary increase
in very high MW protein transcription, associated with burst
nuclear transcription-associated with mitochondrial oxidative
phosphorylation-MM-mediated nuclear and cellular
cytotoxicity/apoptosis, does so to a lesser extent in comparison to
voriconazole (75), which is
structurally similar but more lipophilic than fluconazole and
itraconazole (75), which is
structurally similar to ketoconazole, as fluconazole also
concomitantly decreases sub-cellular membrane compliance (Table V and Fig.
5).
Small molecule xenobiotics that cause
divalent cationicity-mediated CM receptor vesiculo-vacuolization
endocytosis with secondary exosome formation with potential for CM
receptor-mediated 3ary indirect pressuromodulation
This category includes small molecule xenobiotic
hydro-lipophiles with dual cationicity insufficiently separated
(IS) in molecular space and backbone lipophilicity, with vdWDs in
the 0.70–1.31 nm range. By being cationicity-excluded from
permeation across CM protein channel aqueous pores, xenobiotics of
this category bind to CM protein receptors, ligand-receptor
complexes that endocytose upon xenobiotic binding (Table VI and Fig.
6).
The CM pore impermeable dually-cationic small
molecule xenobiotics include:
i) AMD3100 (plerixafor) [(Log OWPC, −5.40; vdWD,
1.00 nm; Log OWPC-to-vdWD ratio, −5.42 nm−1);
AMD3100 hydrophobic core (Log OWPC, 3.56; vdWD, 0.81 nm; Log
OWPC-to-vdWD ratio, 4.39 nm−1); AMD3100
hydrophilic moiety ×2 (Log OWPC, −6.55; vdWD, 0.76 nm; Log
OWPC-to-vdWD ratio, −8.63 nm−1 ×2, +2 cationicity
per moiety)];
ii) Paraquat [(Log OWPC, −5.56; vdWD, 0.70 nm; Log
OWPC-to-vdWD ratio, −7.98 nm−1); paraquat
hydrophobic core (Log OWPC, 2.96; vdWD, 0.70 nm; Log OWPC-to-vdWD
ratio, 4.25 nm−1)];
iii) Bleomycin [(Log OWPC, −8.50; vdWD, 1.31 nm;
Log OWPC-to-vdWD ratio, −6.50 nm−1); bleomycin
hydrophobic core (Log OWPC, 3.14; vdWD, 0.94 nm; Log OWPC-to-vdWD
ratio, 3.35 nm−1)].
AMD3100, paraquat and bleomycin associate with CM
CXCR4 (76–78), CM paraquat binding protein (PBP)
receptor and CM bleomycin binding protein (BBP) (79,80),
respectively, and cause CM receptor-mediated CM
vesiculo-vacuolization endocytosis and sub-cellular
vacuolization-mediated intra-cellular cytotoxicity (81,82), and
at least in the case of CM CXCR4 endocytosing xenobiotic, AMD3100,
in non-cross talk with lympho-infiltrating cells (T cells) in the
milieu, which are necessary for the mounting of an appropriate
cytotoxic T cell response (83–85).
Furthermore, and importantly, the process of CM receptor-mediated
CM vesiculo-vacuolization endocytosis and sub-cellular
vacuolization with secondary CM exosome release, is a process that
rapidly evolves towards a significant decrease in whole cell
compliance in tandem with a significant increase in the
transcription of very high MW secretory proteins including the
collagens (120–190 kDa) and fibronectins (230 kDa) as well as the
nuclear cell division-associated proteins including Ki67 (359 kDa)
and separase (230 kDa) (CM receptor-mediated 3ary indirect shift
pressuromodulation: receptor endocytic 2+) (17), that are associated with a
therapy-related risk for fibrosis (86–88) and
mitogenesis division (81,89), respectively, along with the increased
risk of metastases due to a concomitant decrease in the
transcription of intermediate MW cell surface adhesion proteins
(Table VI and Fig. 6).
Due to the risk of endocytosis-mediated 3ary
indirect pressuromodulation (17), CM
pore impermeable dually-cationic small molecule xenobiotics such as
AMD3100, paraquat and bleomycin are tumoropotentiators as
monochemotherapies, in a concentration dependent manner (48) (atrial naturetic peptide is an
endogenous CM receptor 3ary indirect pressuromodulator (17), which at a higher concentration,
likely, induces mitochondrial division), the risk of which can be
oviated with synergistic additional direct CM receptor-mediated
pressuromodulation as burst nuclear transcription-associated with
mitochondrial oxidative phosphorylation-MM-mediated nuclear and
cellular cytotoxicity/apoptosis results (89)[dexamethasone is an endogenous CM
receptor direct pressuromodulator (17)], as it can also be oviated by
synergistic microtubule network inhibition, at the level of the MM
VDAC γ-tubulin (Tables VII and
IX, Figs.
7 and 9) (90).
| Table VII.Carbonylation/hydroxylation cum
cationicity-facilitated cell membrane (CM) channel endocytosis and
mitochondrial VDAC association: non-association of microtubule
tubulin to mitochondrial membrane (MM) and mitochondrial
anchorage-mediated MM disruption/mitochondria-mediated
apoptosis. |
Table VII.
Carbonylation/hydroxylation cum
cationicity-facilitated cell membrane (CM) channel endocytosis and
mitochondrial VDAC association: non-association of microtubule
tubulin to mitochondrial membrane (MM) and mitochondrial
anchorage-mediated MM disruption/mitochondria-mediated
apoptosis.
|
| Log OWPC
(unitless) | vdWD (nm) | Charge
distribution | Groups | (Log) OPWC-to-vdWD
ratio [per nm (nm−1)] | 1ary mode of
interaction | Interaction
level(s) | Mechanism(s) of
action |
|---|
| Vincristine | −3.00 | 1.11 | S 1+ 1+ Peri
(0) | 3ary N+ ×2 in
polycyclic w/poly C=O and OH, benzyl, cyclic cyclopentanes,
cycloctane | −2.69 | Cationicity cum
hydroxylation (and carbonylation CM channel endocytosis)
facilitated | CM Ca2+
channel endocytic vesicles mitochondrial VDAC channel (γ-tubulin α
interface) | CM Ca2+
channel α helixes destabilization endocytosis (−> vincristine
release −> MM VDAC); non-recrutiment non-association of
microtubular network to MM VDAC; mitochondrial anchorage and
immobility; MM AIF release binding XIAF (−> XIAF free
caspase-3); MM and mitochondrial dissolution; potential for
cellular apoptosis |
|
Vincristine complete dual
hydrophobic core | 5.14 | 0.96 | (0) | 3ary N ×2 in
polycyclic, benzyl, cyclic cyclopentanes, cycloctane | 5.34 | CM Ca2+
channel hydrophobicity MM VDAC internal little α ball
hydrophobicity |
|
|
|
Vincristine complete dual core
hydrophilic moieties1 | 2.80 | 0.52 | S 1+ 1+ | ~ 3ary N+ in -
cyclopyridine ×2 | −5.42 | Ca2+
channel hydrophilicity |
|
|
|
Vincristine complete dual core
hydrophilic moieties2 | 0.47 | 0.38 | Peri carbonylo | C=O (core part 1);
C=O (core part II) | −1.22 (×3; ×1) |
PeripheralCa2+ channel α
helices |
|
|
|
Vincristine complete dual core
hydrophilic moieties3 | −0.33 | 0.32 | Peri hydroxylo | OH (core part I);
OH (core part II) | −1.05 (×1; ×1) | Peripheral
Ca2+ channel α helices |
|
|
|
Vincristine complete dual core
hydrophilic moieties4 | 0.12 to 0.52 | 0.46 to 0.41 | Peri etherylo | ~O-CH3 (core part
I); ~O-CH3 (core part II) | 0.26 to −1.27 (×3;
×1) | Peripheral
Ca2+ channel α helices |
|
|
| Doxorubicin | −0.79 | 0.95 | 1+ Peri (0) | 1aryN+
cycloether w/OH, benzyls ×2 w/ether and OHs, cyclic hexanes ×2 w/o
and w/C=Os, alkyl C=O and OH | −0.83 | Cationicity cum
hydroxylation (and carbonylation)-facilitated CM channel
endocytosis | CM
Na+/K+ ATPase channel endocytic vesicles
mitochondrial VDAC channel (γ-tubulin α interface) | CM
Na+/K+ channel α helixes destabilization
endocytosis (−> doxorubicin release > MM VDAC);
non-recrutiment non-association of microtubular network to MM VDAC;
mitochondrial anchorage and immobility; MM AIF release ninding XIAF
(> XIAF free caspase-3); MM and mitochondrial dissolution;
potential for cellular apoptosis |
| Doxorubicin
hydrophobic core | −0.79 | 0.95 | (0) | Benzyls ×2, cyclic
hexanes ×2, ether | 6.36 | CM
Na+/K+ ATPase channel hydrophobicity MM VDAC
internal little α ball hydrophobicity |
|
|
| Doxorubicin
hydrophilic moiety1 | 3.05 | 0.42 | 1+ | 1ary N+ | −7.28 |
Na+/K+ channel
hydrophilicity |
|
|
| Doxorubicin
hydrophilic moiety2 | −0.47 | 0.38 | Peri carbonylo | C=O | −1.22×3 | Peripheral
Na+/K+ channel α helices |
|
|
| Doxorubicin
hydrophilic moiety3 | −0.33 | 0.32 | Peri hydroxylo | OH | −1.05×5 | Peripheral
Na+/K+ channel α helices |
|
|
| Table IX.Hydroxylation/carbonylation/dual
carboxylation-facilitated cell membrane (CM) receptor endocytosis:
tubulin polymerization re-polymerization inhibition and
mitochondria-mediated apoptosis to rapamycin-associated protein
binding tubulin non-binding |
Table IX.
Hydroxylation/carbonylation/dual
carboxylation-facilitated cell membrane (CM) receptor endocytosis:
tubulin polymerization re-polymerization inhibition and
mitochondria-mediated apoptosis to rapamycin-associated protein
binding tubulin non-binding
|
| Log OWPC
(unitless) | vdWD (nm) | Charge
distribution | Groups | (Log) OPWC-to-vdWD
ratio [per nm (nm1)] | 1ary mode of
interaction | Interaction
level(s) | Mechanism(s) of
action |
|---|
| Etoposide
(VP16) | 1.16 | 0.97 | Peri (0) | Polycyclic ethers
w/OHs ×2, benzyl, phenol w/CH3 ethers ×2 | 1.20 | Hydroxylation cum
carbonylation-facilitated CM receptor endocytosis | CM teniposide
(VM26) ‘receptor’ endocytic vesicles β-tubulin free ends
(soluble) | CM teniposide
(VM26) ‘receptor’ endocytosis [−> etoposide (VP16) release];
VM16 free β-tubulin binding; decreased ß-tubulin-to-ß-tubulin
affinity (−> inability for new microtubule gormation);
mitochondrial snchorage and immobility MM disruption/dissolution
(−> MM AIF-XIAF and free caspace-3 −> mitochondria-mediated
nuclear and cellular apoptosis) |
| ~Etoposide
hydrophobic core | ~3.88 | ~0.97 | (0) | Benzyl
w/cycloethero, benzyl of phenol | ~5.09 | CM receptor
hydrophobicity |
|
|
| Etoposide isophilic
moiety | ~0.02 | ~0.50 | Peri
cycloethero |
~-O-CH3-O− | 0.04 | CM receptor
isophilicity |
|
|
| Etoposide
hydrophilic moietyl | −0.33 | 0.32 | Peri hydroxylo | OH ×3 | −1.05 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Etoposide
hydrophilic moiety2 | −0.47 | 0.38 | Peri carbonylo | C=O | −1.22 | intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Etoposide
hydrophilic moiety3 | 0.12 to −0.52 | 0.46 to 0.41 | Peri etherylo |
~O-CH3 | 0.26 to −1.27
×2 | Intra CM receptor
hydrophilicity (inter-α helix) |
|
|
| Teniposide
(VM26) | 2.78 | 1.00 | Peri (0) (0) | Polycyclic ethers
w/OHs ×2, benzyl, phenol w/CH3 ethers ×2, cyclic ring
w/S | 2.79 | hydroxylation cum
carbonylation-facilitated CM receptor endocytosis | CM teniposide
(VM26) ‘receptor’ endocytic vesicles β-tubulin free ends
(soluble) | CM Teniposide
(VM26) ‘receptor’ endocytosis [−> etoposide (VP16) release];
VM16 free ß-tubulin binding; decreased ß-tubulin-to-ß-tubulin
affinity (−> inability for new microtubule formation);
mitochondrial anchorage and immobility MM disruption/dissolution
(−> MM AIF-XIAF and free caspace-3 −> mitochondria-mediated
nuclear and cellular apoptosis) |
| ~Teniposide
hydrophobic core | ~3.88 | ~0.97 | (0) | Benzyl
w/cycloethero, benzyl of phenol | ~5.09 | CM receptor
hydrophobicity |
|
|
| Teniposide
hydrophobic moiety | 2.40 | 0.55 | (0) |
CH3-cyclic ring w/S | 4.37 | CM receptor
hydrophobicity |
|
|
| Teniposide
isophilic moiety | ~0.02 | ~0.50 | Peri
cycloethero | ~
−O-CH3-O− | 0.04 | CM receptor
isophilicity |
|
|
| Teniposide
hydrophilic moietyl | −0.33 | 0.32 | Peri hydroxylo | OH | −1.05 ×3 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Teniposide
hydrophilic moiety2 | −0.47 | 0.38 | Peri carbonylo | C=O | −1.22 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Teniposide
hydrophilic moiety3 | 0.12 to −0.52 | 0.46 to 0.41 | Peri etherylo |
~O-CH3 | 0.26 to −1.27
×2 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Colchicine | 1.42 | 0.87 | Peri (0) | Benzyl heptane
w/C=O and ethers, heptane w/amidoacetate, benzyl w/poly
CH3 ethers | 1.62 | Hydroxylation cum
carbonylation-facilitated CM receptor endocytosis | CM colchicine
‘receptor’ endocytic vesicles ß-tubulin free ends (soluble) | CM colchicine
‘receptor’ endocytosis −> colchicine release); colchicine free
ß-tubulin binding; decreased ß-tubulin-to-ß-tubulin affinity (−>
inability for new microtubule formation); mitochondrial anchorage
and immobility MM disruption/dissolution (−> MM AIF-XIAF and
free caspase-3 −> mitochondria-mediated nuclear and cellular
apoptosis) |
| Colchicine
hydrophobic core | 4.98 | 0.72 | (0) | Benzyl heptane,
heptane, benzyl | 6.88 | CM receptor
hydrophobicity |
|
|
| Colchicine
hydrophilic moiety1 | −1.11 | 0.43 | Peri amidylo | N-C=O | −2.61 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Colchicine
hydrophilic moiety2 | −0.47 | 0.38 | Peri carbonylo | C=O | −1.22 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Colchicine
hydrophilic moiety3 | 0.12 to −0.52 | 0.46 to 0.41 | Peri etherylo |
~O-CH3 | 0.26 to −1.27
×4 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Paclitaxel | 3.54 | 1.12 | Peri (0) (0) (0)
(0) | Polycyclics w/poly
C=O esters (×4), benzyls ×3, OH | 3.16 | Hydroxylation cum
carbonylation-facilitated CM receptor endocytosis | CM paclitaxel
‘receptor’ endocytic vesicles ß-tubulin free ends (soluble) | CM paclitaxel
‘receptor’ endocytosis (−> paclitaxel release); paclitaxel free
ß-tubulin binding; decreased ß-tubulin-to-ß-tubulin affinity (−>
inability for new microtubule formation); mitochondrial anchorage
and immobility MM disruption/dissolution (−> MM AIF-XIAF and
free caspase-3 −> mitochondria-mediated nuclear and cellular
apoptosis) |
| Paclitaxel
hydrophobic core | 5.65 | 0.87 | (0) | Central
polycyclics | 6.46 | CM receptor
hydrophobicity |
|
|
| Paclitaxel
hydrophobic moieties | 2.85 | 0.59 | (0) |
~benzyl-CH2 | 4.83 ×3 | CM receptor
hydrophobicity |
|
|
| Paclitaxel
hydrophilic moietyl | −0.47 | 0.38 | Peri carbonylo | C=O | −1.22 ×6 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Paclitaxel
hydrophilic moiety2 | −0.33 | 0.32 | Peri hydroxylo | OH | −1.05 ×3 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Ixabepilone | 3.39 | 0.97 | Peri (0) (0) | Cycloamide (15 Cs)
epoxide w/CH3 sulfocyclopyridine alkene, CH3
and asymmetric C=O, OH side groups | 3.51 | Hydroxylation cum
carbonylation-facilitated CM receptor endocytosis | CM Ixabepilone
‘receptor’ endocytic vesicles ß-tubulin free ends (soluble) | CM ixabepilone
‘receptor’ endocytosis (−> ixabepilone release); ixabepilone
fret ß-tubulin binding; decreased ß-tubulin-to-ß-tubulin affinity
(−> inability for new microtubule formation); mitochondrial
anchorage and immobility MM disruptio dissolution (−> MM
AIF-XIAF and free caspase-3 −> mitochondrial-mediated nuclear
and cellular apoptosis) |
| Ixabepilone
hydrophobic core | 6.21 | 0.90 | (0) | Cycloamide (15 Cs)
epoxide w/CH3 sulfocyclopyridine alkene | 3.19 | CM receptor
hydrophobicity |
|
|
| Ixabepilone
hydrophobic moiety | 0.79 | 0.54 | (0) | Pentane w/sulfur
and nitrogen | 1.46 | CM receptor
hydrophobicity |
|
|
| Ixabepilone
hydrophilic moiety1 | −0.47 | 0.38 | Peri carbonylo | C=O | −1.22 ×2 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Ixabepilone
hydrophilic moiety2 | −0.33 | 0.32 | Peri hydroxylo | OH | −1.05 ×2 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| (+/-)
Spiro-oxanthromicin A | 2.40 | 1.01 | S 1- 1- Peri (0)
(0) (0) | (+)
CH3-benzyl-OH (−COO), (+) cyclohexane=O, (+)
CH3-benzyl-OH, Interconnecting
cyclohexane-OCH3, (−) CH3-benzyl-OH (−COO),
(−) cyclohexane=O, | 2.38 | Dual carboxylation
cum hydroxylation (and carbonylation)-facilitated CM receptor
endocytosis | CM (+/-)
Spiro-oxanthromicin endocytic vesicles ß-tubulin free ends
(soluble) | CM (+/-)
Spiro-oxanthromicin A ‘receptor’ endocytosis (−> (+/-)
Spiro-oxanthromicin A release); (+/-) Spiro-oxanthromicin A free
ß-tubulin binding; decreased ß-tubulin-to-ß-tubulin affinity (−>
inability for new microtubule formation); mitochondrial anchorage
and |
|
|
|
|
| (−)
CH3-benzyl-OH |
|
|
| Immobility MM
disruption/dissolution (−> MM AIF-XIAF and free caspace-3 −>
mitochondria-mediated nuclear and cellular apoptosis) |
| (+)
Spiro-oxanthromicin A orthagonal hydrophobic core | 5.47 | 0.74 | (0) | (+)
CH3-benzyls ×2, (+) cyclohexane | 7.39 | CM receptor
hydrophobicity |
|
|
| (+/-)
Spiro-oxanthromicin A interconnecting hydrophobic moiety | 2.67 | 0.57 | (0) | Cyclohexane | 4.66 | CM receptor
hydrophobicity |
|
|
| (+/-)
Spiro-oxanthromicin A interconnecting hydrophilic moiety | 0.12 to −0.52 | 0.46 to 0.41 | Peri
Etherylo-R |
~O-CH3-R | 0.26 to −1.27 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| (−)
Spiro-oxanthromicin A orthagonal hydrophobic core | 5.47 | 0.74 | (0) | (−)
CH3-benzyls ×2, (−) cyclohexane | 7.39 | CM receptor
hydrophobicity |
|
|
| (+/-)
Spiro-oxanthromicin A hydrophilic moietiesl | −0.47 | 0.38 | Peri carbonylo | C=O | −1.22 (xl; xl) | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| (+/-)
Spiro-oxanthromicin A hydrophilic moietiesl | −0.33 | 0.32 | Peri hydroxylo | (+) OH; (−) OH | −1.05 (×2; ×2) | Intra CM receptor
hydrophilicity |
|
|
| (+/-)
Spiro-oxanthromicin A hydrophilic moieties2 | −3.50 | 0.42 | Peri S 1- 1− | (+) COO-; (−)
COO− | −8.42 (×1; ×1) | (inter-α helixes)
Na+-CM receptor hydrophilicity (×1); Na+-CM receptor hydrophilicity
(×1) (inter-α helices) |
|
|
| Tacrolimus
(FK506) | 5.59 | 1.13 | Peri (0) (0) | Cycloester
cycloamide cycloether (20 C) (−alkyl, -alkene, OH, C=O),
CH3-O-cyclohexane-OH (−alkene) | 4.94 | Hydroxylation cum
carbonylation-facilitated CM receptor endocytosis | CM Tacrolimus
‘receptor’ endocytic vesicles FKBP52 binding (soluble ß-tubulin
non-binding) | CM tacrolimus
‘receptor’ endocytosis (−> tacrolimus release); tacrolimus
binding to FKBP12/inter-domain (I–II/III) of FKBP52; disassociation
of FKBP52 domain III from a-/ß-tubulin (−> decreased propensity
for tubulin polymerization −> decreased intracellular
pressuromodulation); disassociation of FKBP52 domain IV from BCL
(−> free BCL and MM stabilization −> mitochondrial
anti-apoptosis) |
| ~Tacrolimus
hydrophobic core | ~9.07 | −1.13 | (0) | Cycloester
cycloamide (21 C) w/alkyl, alkene side groups | −9.14 | CM receptor
hydrophobicity |
|
|
| Tacrolimus
hydrophobic moiety | 0.85 to 1.66 | 0.63 to 0.69 | (0) |
CH3-O-cyclohexane -OH
CH3-O-Cyclohexane -OH-(alkene) | 1.35 to 2.39 | CM receptor
hydrophobicity |
|
|
| Tacrolimus
hydrophilic moiety2 | −0.47 | 0.38 | Peri carbonylo | C=O | −1.22 ×4 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Tacrolimus
hydrophilic moiety1 | −0.33 | 0.32 | Peri hydroxylo | OH | −1.05 ×3 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
| Tacrolimus
hydrophilic moiety3 | 0.12 to −0.52 | 0.46 to 0.41 | Peri etherylo |
−O-CH3 | 0.26 to −1.27
×3 | Intra CM receptor
hydrophilicity (inter-α helixes) |
|
|
Small molecule xenobiotics that cause
carbonylation/hydroxylation cum cationicity-facilitated CM channel
endocytosis to localize to the MM VDAC at the γ-tubulin association
interface
This category includes the small molecule
xenobiotic hydro-lipophiles with CM channel endocytosing capability
due to the presence of sufficient inner incorporating lipophilicity
in the context of interacting outer polyhydroxylated carbonylated
hydrophilicity in the form of circumferential polyhydroxylated
carbonylated hydrophilicity, with vdWDs in the 0.95–1.11 nm range
(17). By being hydrophilic and due
to the CM protein channel aqueous pore widths, xenobiotics of this
category associate with CM protein channel α-helix cum α-helix
isophilic aqueous pores to induce CM channel endocytosis of the
Ca2+ channel (i.e. vincristine) and the
Na+/K+ ATPase (doxorubicin and daunorubicin),
and upon CM channel endocytosis disassociation, associate
promiscuously with the inner-to-outer MM VDAC, a non-endocytosable
circular β-helix with voltage-gateable internal short α-helix-type
channel (91) (Table VII and Fig. 7).
The CM channel endocytosing small molecule
xenobiotic hydro-lipophiles include: i) vincristine [(Log OWPC,
−3.00; vdWD, 1.11 nm; Log OWPC-to-vdWD ratio, −2.69
nm−1); vincristine hydrophobic core (Log OWPC,
5.14; vdWD, 0.96 nm; Log OWPC-to-vdWD ratio, 5.34
nm−1)]; ii) doxorubicin (adriamycin) [(Log OWPC,
−0.79; vdWD, 0.95 nm; Log OWPC-to-vdWD ratio, −0.83
nm−1); doxorubicin hydrophobic core (Log OWPC,
4.88; vdWD, 0.77 nm; Log OWPC-to-vdWD ratio, 6.96
nm−1)].
Vincristine and doxorubicin associate into the
inter-α-helix isophilic pores of the Ca2+ channel
(92) and
Na+/K+ ATPase (17,50,51),
respectively, to de-stabilize the CM interaction of the
multi-inter-α-helix constructs of such trans-membrane proteins,
that results in ligand-associated CM protein channel endocytosis
(93,94), followed by intra-cellular ligand CM
channel disassociation, and then, subsequent promiscuous MM VDAC
channel isophilic pore association (95): thus, the primary mechanism of cellular
toxicity for vincristine and doxorubicin is at the level of the
mitochondria and MM, and due to the concomitant presence of
sufficiently separated (SS) 1+ cationicity in the former
(vincristine) and exteriorly protruding 1+ cationicity in the
latter (doxorubicin), that results in non-association of
microtubule γ-tubulin at MM VDAC and non-recruitment of renewed
microtubule networks to the MM, mitochondrial anchorage
immobility-associated MM disruption/dissolution with liberation of
MM AIF and initiation of the mitochondrially-mediated nuclear
apoptosis cascade, with the significant potential for inducing
cellular apoptosis (96–101), which is due to no significant
(in-significant) potential for CM receptor endocytosis-mediated
(Pseudo) 3ary indirect pressuromodulation (17) (Table
VII and Fig. 7).
The presence of structural cationicity is a pivotal
structure distinction between CM channel endocytosing small
molecule xenobiotic hydro-lipophiles such as vincristine and
doxorubicin that induce non-association of microtubule γ-tubulin at
MM VDAC and result in CM mitochondrial anchorage
non-mobility-mediated MM disruption/mitochondrial-mediated
apoptosis, in contrast to those that do not but with instead have
mitogenic potential, which is the case for ouabain (102), a non-cationic circumferentially
carbonylated/hydroxylated hydro-lipophile endocytoser of the
Na+/K+ ATPase, with potential to cause
epithelilal-to-mesenchymal transformation (EMT) as well as
tumoropropagation via CM receptor endocytosis-mediated (Pseudo)
3ary indirect pressuromodulation (17).
The xenobiotics of this category induce endocytosis
of their respective channels, the Ca2+ channel (123 kDa
N/P/Q α subunit MW) (92)
(vincristine > verapamil) and the Na+/K+
ATPase channel (112 kDa α subunit MW) (103) (ouabain, momensin, doxorubicin and
daunorubicin), which are present on the CM in juxtaposition to one
another and to P-gp (non-glycosylated MW 140 kDa) (104), whereby, ligand-mediated
Ca2+ channel-endocytosis of associated CM results in
concomitant endocytosis of P-gp (105), that decreases immediate P-gp CM
expression. Thus, P-gp overexpression and associated multi-drug
resistance emerges, secondarily, in surviving tumor cells with
lower compliance set points than before therapy due to the
increased transcription of higher MW proteins such as P-gp
(11), in which case,
intra-cellularly accumulating drug fraction is extruded by its
ability to interact with intra-CM P-gp α-helixes viz a viz
hydro-lipophile incorporating lipophilicity, and induce, α-helix
apposition pump functionality (104).
Furthermore, true CM P-gp small molecule ligands
that associate with the P-gp extracellular loop-α-helix are few and
far between, and include artemsinin (106), which functions as a P-gp inhibitor
but also as a pressuromodulator, thus, P-gp inducer, as do ones
with hollow interiors including QZ59-RRR and QZ59-SSS (104); whereas, small molecule ligands with
less polyneutral exterior hydrophilicity than vincristine, include
quinidine (105), a competitive
antagonist of vincristine at the Ca2+ channel, with the
ability to obstruct the channel, but without the ability to
endocytose it, whereby, it functions as a Ca2+ channel
pressuromodulator and inducer, and thus, indirectly as an inducer
of Na+/K+ ATPase and P-gp, all present in
close CM pressuromodulating proximity.
Small molecule xenobiotics that cause
dual carboxylation-facilitated CM folic acid receptor-mediated
endocytosis followed by MM and the rough endoplasmic reticulum
membrane folic acid receptor-mediated vesiculization with the
potential for CM receptor-mediated (Pseudo) 3ary indirect
pressuromodulation
This category includes the small molecule
xenobiotic hydro-lipophiles with CM folic acid receptor (FAR)
endocytosing capability due to the presence of incorporating
lipophilicity in the context of anisotropic interacting outer dual
carboxylation hydrophilicity, with vdWDs at 0.89 nm. The
xenobiotics of this category associate into the FAR hydrophobic
core while concomitantly associating with the hydrophilic exterior
FAR cationic (+) R groups via the carboxyl (COO−) groups
(107), which results in a
contraction-pull down of the loose non-aligned multi-α-helix FAR
towards the CM. This results in the endocytosis of the ligand-FAR
complex at a much faster rate than that induced by endogenous
ligand folic acid (folate), which has a lower kDa than that of the
competitive pro-endocytic xenobiotics of this category (108) (Table
VIII and Fig. 8).
| Table VIII.Dual carboxylation-facilitated cell
membrane (CM) receptor endocytosis: mitochondrial membrane (MM) and
nuclear rough endoplasmic reticulum (RER) membrane (RERM)
vesiculization. |
Table VIII.
Dual carboxylation-facilitated cell
membrane (CM) receptor endocytosis: mitochondrial membrane (MM) and
nuclear rough endoplasmic reticulum (RER) membrane (RERM)
vesiculization.
|
| Log OWPC
(unitless) | vdWD (nm) | Charge
distribution | Groups | (Log) OPWC-to-vdWD
ratio [per nm (nm−1)] | 1ary mode of
interaction | Interaction
level(s) | Mechanism(s) of
action |
|---|
| Methotrexate | −6.60 | 0.89 | S 1- 1- 0 (0) | NH2 ×2,
cyclopyridines ×2, CH3-N, benzyl, N C=O, COO- ×2 | −7.40 | Dual carboxylation
cum hydroxylation (and carbonylation) facilitated CM receptor
endocytosis | CM folic acid
receptor (FAR) endocytic vesicles MM FAR endocytic vesicles nuclear
RER FAR endocytic vesicles | CM FAR pull-down
endocytosis vesiculization (−> MTX release); MTX FAR-mediated MM
vesiculization and MTX FAR-mediated nuclear RERM vesiculization; MM
AIF release binding XIAF; XIAF free caspase 3; MM and mitochondrial
dissolution; potential for cellular apoptosis |
|
Methotrexate hydrophobic
core | 1.23 | 0.77 | (0) | Cyclopyridines ×2,
CH3-N, benzyl | 1.60 | FAR
hydrophobicity |
|
|
|
Methotrexate hydrophilic
moiety1 | −1.11 | 0.43 | 0 Amidylo | N-C=O | −2.61 | FAR
hydrophilicity |
|
|
|
Methotrexate hydrophilic
moiety2 | 3.50 | 0.42 | 1− | COO− | −8.42 | FAR receptor
cationic R group |
|
|
|
Methotrexate hydrophilic
moiety3 | −3.50 | 0.42 | 1− | COO− | −8.42 | FAR receptor
cationic R group |
|
|
| Raltitrexed | −0.79 | 0.95 | S 1- 1- 0 (0) | CH3 ×1,
cyclopyridine −N C=O, CH3−N, cyclosulfodine, benzyl, N
C=O, COO− ×2 | −4.93 | Dual carboxylation
cum hydroxylation (and carbonylation) facilitated CM receptor
endocytosis | CM folic acid
receptor (FAR) endocytic vesicles MM FAR endocytic vesicles nuclear
RER FAR endocytic vesicles | CM FAR pull-down
endocytosis vesiculization (−> raltitrexad release); raltitrexad
FAR-mediated MM vesiculization and raltitrexad FAR-mediated nuclear
RERM vesiculization; MM AIF release binding XIAF; XIAF free caspase
3; MM and mitochondrial dissolution; potential for cellular
apoptosis |
|
Raltitrexed hydrophobic
core | 3.67 | 0.78 | (0) | CH3 ×1,
cyclopyridine, CH3-N, cyclosulfodine, benzyl | −4.93 | FAR
hydrophobicity |
|
|
|
Raltitrexed hydrophilic
moiety1 | −1.11 | 0.43 | 0 Amidylo | N-C=O | −2.61 (×1; ×1) | FAR receptor
hydrophilicity |
|
|
|
Raltitrexed hydrophilic
moiety2 | −3.50 | 0.42 | 1− |
COO− | −8.42 | FAR receptor
cationic R group |
|
|
|
Raltitrexed hydrophilic
moiety3 | −3.50 | 0.42 | 1− |
COO− | −8.42 | FAR receptor
cationic R group |
|
|
The CM FAR endocytosing small molecule xenobiotic
hydro-lipophiles include: i) methotrexate [(Log OWPC, −6.60; vdWD,
0.89 nm; Log OWPC-to-vdWD ratio, −7.40 nm−1);
methotrexate hydrophobic core (Log OWPC, 1.23; vdWD, 0.77 nm; Log
OWPC-to-vdWD ratio, 1.60 nm−1)]; ii) raltitrexed
(Tomudex) [(Log OWPC, −4.40; vdWD, 0.89 nm; Log OWPC-to-vdWD ratio,
−4.93 nm−1); raltitrexed hydrophobic core (Log
OWPC, 3.67; vdWD, 0.78 nm; Log OWPC-to-vdWD ratio, 4.70
nm−1)].
Both methotrexate and raltitrexed endocytose the
ligand-FAR complex, initially at the level of the CM, the CM FAR,
and upon CM FAR endocytosis followed by intra-cellular
disassociation, re-associate/bind to the sub-cellular membrane
FARs, the MM FAR and the rough endoplasmic reticulum (RER) FAR
inducing their endocytosis. This cumulates in increased
sub-cellular vesiculization-mediated generation of pro-oxidant
reactive oxygen species (109,110),
and in case of the mitochondria, vesiculization-mediated loss of MM
electromotive potential (109,111–113).
Therefore, the mechanism for anti-folate chemoxenobiotic-mediated
cellular cytotoxicity is FAR endocytosis-driven sub-cellular
pro-oxidant oxidative stress (114–117),
particularly mitochondrial, whereby there is only limited potential
for CM receptor endocytosis-mediated (Pseudo) 3ary indirect
pressuromodulation (110,118) (Table
VIII and Fig. 8).
Small molecule xenobiotics that cause
hydroxylation/carbonylation/dual carboxylation-facilitated CM
receptor endocytosis and tubulin polymerization re-polymerization
inhibition and mitochondrial-mediated apoptosis
This category includes the small molecule
xenobiotic lipophiles with CM receptor endocytosing capability,
with vdWDs in the 0.87–1.01 nm range. Due the presence of inner
incorporating lipophilicity in the context of outer circumferential
hydrophilicity in the form of
hydroxylation/carbonylation/etheroylation/dual carboxylation, the
xenobiotics of this category associate into multiple α-helix-type
protein receptor hydrophobic cores to destabilize the protein
receptor construct sufficiently enough to cause ligand-associated
CM receptor endocytosis followed by intra-cellular disassociation.
Re-association with affinity to soluble β-tubulin inhibits
microtubule de-/re-polymerization function and results in
mitochondrial anchorage resultant MM disruption/mitochondrial
dissolution (119,120). For this reason, small molecule
xenobiotics in this category have no significant (in-significant)
risk for secondary CM receptor endocytosis-mediated (Pseudo) 3ary
indirect pressuromodulation resultant increase in nuclear
transcription of very high MW proteins (17) (Table IX
and Fig. 9).
The small molecule xenobiotic lipophiles with CM
receptor endocytosing capability include: i) etoposide (VP16) [(Log
OWPC, 1.16; vdWD, 0.97 nm; Log OWPC-to-vdWD ratio, 1.20
nm−1); etoposide hydrophobic core (Log OWPC,
3.88; vdWD, 0.76 nm; Log OWPC-to-vdWD ratio, 5.09
nm−1)]; ii) teniposide (VM26) [(Log OWPC, 2.78;
vdWD, 1.00 nm; Log OWPC-to-vdWD ratio, 2.79
nm−1); teniposide hydrophobic core (Log OWPC,
3.88; vdWD, 0.76 nm; Log OWPC-to-vdWD ratio, 5.09
nm−1)]; iii) colchicine [(Log OWPC, 1.42; vdWD,
0.87 nm; Log OWPC-to-vdWD ratio, 1.62 nm−1);
colchicine hydrophobic core (Log OWPC, 4.98; vdWD, 0.72 nm; Log
OWPC-to-vdWD ratio, 6.88 nm−1)]; iv) paclitaxel
[(Log OWPC, 3.54; vdWD, 1.12 nm; Log OWPC-to-vdWD ratio, 3.16
nm−1); paclitaxel hydrophobic core (Log OWPC,
5.65; vdWD, 0.87 nm; Log OWPC-to-vdWD ratio, 6.46
nm−1)]; v) ixabepilone [(Log OWPC, 3.39; vdWD,
0.97 nm; Log OWPC-to-vdWD ratio, 3.51 nm−1);
ixabepilone hydrophobic core (Log OWPC, 6.21; vdWD, 0.90 nm; Log
OWPC-to-vdWD ratio, 6.91 nm−1)]; vi) (+/-)
spiro-oxanthromicin A [(Log OWPC, 2.40; vdWD, 1.01 nm; Log
OWPC-to-vdWD ratio, 2.38 nm−1); (+/-) orthogonal
spiro-oxanthromicin A hydrophobic cores (Log OWPC, 5.47; vdWD, 0.74
nm Log OWPC-to-vdWD ratio, 7.39 nm−1/core)].
For i through vi, CM receptor
de-stabilization-mediated endocytosis and intra-cellular
re-association with soluble β-tubulin results in inhibition of
microtubule polymerization de-/re-polymerization causing
sub-cellular organelle anchorage non-motility, and generates
intra-cellular reactive pro-oxidant species concomitant with
mitochondrial anchorage immobility-associated MM
disruption/dissolution with liberation of MM AIF and initiation of
the mitochondrial-mediated nuclear apoptosis cascade (120–131),
which have the significant potential for inducing cellular
apoptosis, particularly those in incorporating lipophilicities in
the intermediate range (Table IX and
Fig. 9).
vii) Tacrolimus (FK506) [)Log OWPC, 5.59; vdWD,
1.13 nm; Log OWPC-to-vdWD ratio, 4.94 nm−1);
tacrolimus hydrophobic core (Log OWPC, 9.07; vdWD, 0.99 nm; Log
OWPC-to-vdWD ratio, 9.14 nm−1)] and everolimus
(132), with greater incorporating
lipophilicity (9.14 nm−1) associate with the
FKBP12/inter-domain (I–II/III) of FKBP52 (133).
In contrast to i) through vi) of this category, the
CM chemokine receptor endocytosing immunosuppressives, tacrolimus,
in the case of xenobiotics such as tacrolimus, xenobiotic-induced
disassociation of the FKBP52 domain III from α/β-tubulin results in
a slight propensity for inhibition of tubulin de-polymerization
(propensity for tubulin polymerization) (133), while the concomitant disassociation
of the PUMA-like FKBP52 domain IV from BCL results in generation of
cytosolic free BCL, which overcomes mitochondrial anchorage
immobility-associated MM disruption due to the MM stabilizing
effect of BCL (134–137) (Table
IX and Fig. 9).
The CM receptor endocytosing small molecule
xenobiotic lipophiles of this category have significant potential
to be tumorocytotoxic, particularly in synergism with other
tumorocytotoxic chemoxenobiotics [nuclear (i.e. 5-FU) (138) and mitochondrial (i.e. doxorubicin)
(9,138)]. The primary limitation to efficacy
when administered as a part of current free drug chemotherapy
regimens is secondary to a high level of serum protein-binding due
to overall lipophilicity for size (Log OWPC-to-vdWD ratio range,
1.2–3.51 nm−1), which makes it is difficult to
obtain micromolar local intra-tumoral concentrations at which they
are most effective. Therefore, the overall efficacy of such
tumorocytotoxic small molecule xenobiotic lipophiles can be
significantly improved via the EPR effect, as has shown to be the
case for abraxane, a semi-polydisperse paclitaxel (non-covalent)
albumin-microaggregate nanoparticulate (HD ~120
nm) (139,140), which proteolytically degrades into
smaller globular paclitaxel (non-covalent) albumin
(HD ~7–10 nm) to enter the tumor interstitium,
that results in greater local intra-tumoral concentration of
improved efficacy of such small molecule chemoxenobiotics (3–7).
Small molecule xenobiotics that cause
CM receptor-mediated pressuromodulation antagonism/partial
antagonism of direct CM receptor-mediated pressuromodulation
This category includes the small molecule
xenobiotic lipophiles with CM receptor pressuromodulation
antagonism/partial antagonism capability, with vdWDs in the
0.86–0.89 nm range. Due the presence of inner incorporating
lipophilicity commensurate with that of the sex steroid receptor
hydrophobic cores, the xenobiotics of this category bind to sex
steroid receptor complexes with less affinity than endogenous sex
steroids, and thus, function as short-duration pressuromodulators,
in contrast to the endogenous sex steroids, which are prolonged
duration CM receptor pressuromodulators (17) (Table X
and Fig. 10).
| Table X.Cell membrane (CM) receptor-mediated
pressuromodulation antagonism/partial antagonism:
antagonism/partial antagonism of direct CM receptor-mediated
pressuro-modulation. |
Table X.
Cell membrane (CM) receptor-mediated
pressuromodulation antagonism/partial antagonism:
antagonism/partial antagonism of direct CM receptor-mediated
pressuro-modulation.
|
| Log OWPC
(unitless) | vdWD (nm) | Charge
distribution | Groups | (Log) OPWC-to-vdWD
ratio [per nm (nm1)] | 1ary mode of
interaction | Interaction
level(s) | Mechanism(s) of
action |
|---|
|
Hydroxy-tamoxifen | 3.00 | 0.89 | 1+ (0) 0 | 3ary N+
to ether, OH of phenol, benzyl-cis-benzyl,
CH2-CH3 | 3.37 | CM estrogen
receptor (ER) complex partial | CM ER complex | Less-stable binding
to ‘ER complex’ (KD antagonist > KD estradiol and shorter
antagonist tl/2 at receptor) decreased cell division |
|
|
|
|
|
|
|
Pressuromodulation |
|
|
| Hydroxy-tamoxifen
hydrophobic core | 5.36 | 0.75 | (0) |
Benzyl-cis-benzyl,
CH2-CH3, CH3 | 7.14 | CM receptor
hydrophobicity |
|
|
| Hydroxy-tamoxifen
hydrophilic moietyl | −1.05 | 0.68 | 1+ | 3ary N+
to ether with benzyl | −1.54 | CM receptor
hydrophilicity (unstable) |
|
|
| Hydroxy-tamoxifen
hydrophilic moiety2 | −0.33 | 0.32 | Hydroxylo | OH | −1.01 | CM receptor
hydrophilicity |
|
|
| Abiraterone | 3.81 | 0.86 | 0 (0) 0 | CH2-N-CH
pyridine, sterol backbone, CHs ×2, OH | 4.41 | CM testosterone
receptor complex Partial pressuromodulation | CM DHTR
complex | Less-stable binding
to DHT complex (KD antagonist > KD DHT and shorter antagonist
tl/2 at receptor) decreased cell division |
| Abiraterone
hydrophobic core | 5.42 | 0.80 | (0) | ~Benzyl, sterol
backbone, CHs ×2, OH | 6.74 | CM receptor
hydrophobicity |
|
|
| Abiraterone
hydrophobic moietyl | 0.76 | 0.52 | 0 |
~CH2-N-CH pyridine | 1.45 | CM receptor
hydrophobicity (unstable) |
|
|
| Abiraterone
hydrophilic moiety2 | −0.33 | 0.32 | Hydroxylo | OH | −1.05 | CM receptor
hydrophilicity |
|
|
The small molecule xenobiotic lipophiles with CM
receptor pressuromodulation antagonism/partial antagonism
capability include:
i) Hydroxytamoxifen (afimoxitene) [(Log OWPC, 3.00;
vdWD, 0.89 nm; Log OWPC-to-vdWD ratio, 3.37
nm−1); hydroxytamoxifen hydrophobic core (Log
OWPC, 5.36; vdWD, 0.75 nm; Log OWPC-to-vdWD ratio, 7.14
nm−1)], with hydrophobic core lipophilicity
similar to that of the estradiol and estriol sterol backbone (Log
OWPC, 5.28; vdWD, 0.78 nm) at 6.81 nm−1 (17), which has a bulky unstable exterior
extracellularly-interacting hydrophilicity and insufficient for
prolonged duration binding to the estrogen receptor (ER) complex
(141).
ii) Abiraterone [(Log OWPC, 3.81; vdWD, 0.86 nm;
Log OWPC-to-vdWD ratio, 4.41 nm−1); abiraterone
hydrophobic core (Log OWPC, 5.42; vdWD, 0.80 nm; Log OWPC-to-vdWD
ratio, 6.74 nm−1)], with hydrophobic core
lipophilicity similar to that of the dihydrotestosterone (DHT)
sterol backbone (Log OWPC, 5.82; vdWD, 0.81 nm) at 7.18
nm−1 (17), which
has lesser exterior extracellularly interacting hydrophilicity and
insufficient for prolonged duration binding at the DHT receptor
complex (142).
The sex steroid competitive antagonist
chemoxenobiotics are pressuromodulation/partial pressuromodulation
antagonists of the respective α-helix-based sex steroid receptor
types due to the non-binding of the endogenous small molecule sex
steroid pressuromodulators, but their competitive binding to the
respective sex steroid hormone receptor complexes still results in
impartial pressuromodulation of the α-helix-based small molecule
sex steroid receptor complex. Thus, the overall effect of CM
receptor pressuromodulation antagonism at the sex steroid hormone
receptor is sufficient enough to decrease the rate of mitogenesis
and cell division by decreasing the rate of protein transcription
of very high MW nuclear division proteins, Ki67 (359 kDa) and
separase (230 kDa). As such, the sex steroid hormone CM receptor
pressuromodulation/partial pressuromodulation antagonists have the
potential for temporizing solid tumor growth/progression (143,144),
as do upstream releasing hormone inhibitors of luteinising hormone
(LH) action on the gonadal sex steroid axis (145), however, in the physiologic state are
not tumorocidal (146–148), which is due to the presence of a
plethora of CM receptor pressuromodulators in the solid tumor
milieu, paracrine and autocrine, and therefore, an abundance of
tumor CM receptor-mediated pressuromodulation escape mechanisms,
there is a high risk of local and/or metastatic re-occurence upon
cessation of sex steroid CM receptor pressuromodulation/partial
pressuromodulation antagonist-based chemotherapy (Table X and Fig.
10).
Small molecule xenobiotics that cause
CM receptor-mediated pressuromodulation antagonism/partial
antagonism of CM receptor-mediated pressuromodulation cum
extracellulomodulation with concomitant receptor kinase
inhibition
This category includes the small molecule
xenobiotic lipophiles and hydro-lipophiles with CM receptor
pressuromodulation antagonism/partial antagonism cum
pressuromodulation extracellulomodulation and concomitant receptor
kinase inhibition capability, with vdWDs in the 0.82–0.97 nm range.
Due to L-to-U-type step-like backbone structural configurations, in
context of the presence of inner incorporating lipophilicity
commensurate with that of the growth factor and cytokine CM
receptor hydrophobic cores, the xenobiotics of this category bind
to growth factor and cytokine CM receptor subunits with affinity.
In the case of the endogenous growth factors and cytokines [i.e.
EGF, ALK, SDF-1 and asialoglycoprotein receptor (AGR) ligand] that
bind to α-helix-rich receptors (17),
the presence of the CM pressuromodulator antagonist/partial
antagonist results in non-binding of the endogenous growth factor
or cytokine, which results in partial ligand antagonist-mediated CM
receptor pressuromodulation; while, in the case of the endogenous
growth factors and cytokines (i.e. PDGF, GM-CSF and TRAIL) that
bind to β-helix-based subunit receptors (17), the concomitant presence of the CM
pressuromodulator antagonist/partial antagonist results in less
effective, lower affinity binding of the endogenous growth factor
or cytokine, which results in partial endogenous growth factor or
cytokine-mediated CM receptor pressuromodulation (Table XI and Fig.
11).
 | Figure 11.Cell membrane (CM) receptor-mediated
antagonism/partial antagonism of pressuromodulation
extracellulomodulation with concomitant receptor kinase inhibition:
antagonism/partial antagonsim of direct CM receptor-mediated
pressuromodulation ± external cationomodulation (≥3+ ≥1+). (A)
Gefitinib (Iressa), (B) ceritinib, (C) erlotinib (Tarceva), (D)
lapatinib, (E) MK-2206, (F) staurosporine, (G) afatinib, (H)
imatinib (Gleevac; CGP 57148), (I) crizotinib, (J)
hydroxycamptothecin, (K) AMD070, (L) topotecan. |
The small molecule xenobiotic lipophiles or
hydro-lipophiles that bind to α-helix-rich receptors in lieu of
endogenous growth factors or cytokines as direct CM receptor
partial pressuromodulation antagonists (17) include:
i) The EGF receptor antagonists (149–156),
gefitinib (Iressa) [(Log OWPC, 3.75; vdWD, 0.89 nm; Log
OWPC-to-vdWD ratio, 4.21 nm−1); gefitinib
hydrophobic core (Log OWPC, 4.21; vdWD, 0.74 nm; Log OWPC-to-vdWD
ratio, 5.71 nm−1)], erlotinib (Tarceva) [(Log
OWPC, 3.20; vdWD, 0.87 nm; Log OWPC-to-vdWD ratio, 3.67
nm−1); erlotinib hydrophobic core (Log OWPC,
3.61; vdWD, 0.74 nm; Log OWPC-to-vdWD ratio, 4.88
nm−1)], lapatinib [(Log OWPC, 2.12; vdWD, 0.96
nm; Log OWPC-to-vdWD ratio, 2.20 nm−1); lapatinib
hydrophobic core (Log OWPC, 6.68; vdWD, 0.89 nm; Log OWPC-to-vdWD
ratio, 7.51 nm−1)] and afatinib [(Log OWPC, 1.11;
vdWD, 0.91 nm; Log OWPC-to-vdWD ratio, 1.22
nm−1); afatinib hydrophobic core (Log OWPC, 4.21;
vdWD, 0.74 nm; Log OWPC-to-vdWD ratio, 5.71
nm−1)].
ii) The ALK receptor antagonists (157–160),
ceritinib [(Log OWPC, 3.40; vdWD, 0.97 nm; Log OWPC-to-vdWD ratio,
3.67 nm−1); ceritinib hydrophobic core (Log OWPC,
7.16; vdWD, 0.89 nm; Log OWPC-to-vdWD ratio, 8.02
nm−1)] and crizotinib [(Log OWPC, 1.00; vdWD,
0.88 nm; Log OWPC-to-vdWD ratio, 1.13 nm−1);
crizotinib hydrophobic core (Log OWPC, 5.96; vdWD, 0.88 nm; Log
OWPC-to-vdWD ratio, 6.88 nm−1)].
iii) The CXCR receptor antagonist (76,77,161,162),
AMD070 [Log OWPC, −0.14; vdWD: 0.85 nm; Log OWPC-to-vdWD ratio,
−0.16 nm−1; AMD070 hydrophobic core (Log OWPC,
4.33; vdWD, 0.84 nm; Log OWPC-to-vdWD ratio, 5.13
nm−1)].
iv) AGR antagonist (163–169),
staurosporine [(Log OWPC, 1.20; vdWD, 0.91 nm; Log OWPC-to-vdWD
ratio, 1.32 nm−1); staurosporine hydrophobic core
(Log OWPC, 5.72; vdWD, 0.81 nm; Log OWPC-to-vdWD ratio, 7.08
nm−1)].
The EGF receptor antagonists, ALK receptor
antagonists and CXCR receptor antagonists cause for a decrease the
rate of protein transcription of synthesizable proteins by CM
receptor partial pressuromodulation antagonism of tumor CM
receptors. The decrease in the transcription of the very high MW
nuclear cell division proteins, Ki67 (359 kDa) and separase (230
kDa) (170) results in decreased
mitogenesis and cell division, while the decrease in the
transcription of the lower MW protein forms including VEGF (20 kDa)
necessary for autocrine angiogenesis (154,171),
and importantly, BCL (26 kDa) necessary for maintenance of MM
integrity in tumor cells (BCL-dependent) (59), results in increased
mitochondria-mediated apoptosis, particularly in p53-deficient
tumor cells (172–174) (Table
XI and Fig. 11).
The small molecule xenobiotic lipophiles that bind
to multi-subunit polymeric β-helix β-helix-based receptors to
decrease the binding affinity of endogenous growth factors or
cytokines and function as direct CM receptor
pressuromodulation/partial pressuromodulation antagonists (17) include:
i) The PDGF receptor antagonist (175–177),
imatinib [Gleevac CGP 57148) [(Log OWPC, 1.50; vdWD, 0.94 nm; Log
OWPC-to-vdWD ratio, 1.59 nm−1); imatinib
hydrophobic core (Log OWPC, 5.21; vdWD, 0.87 nm; Log OWPC-to-vdWD
ratio, 6.00 nm−1)].
ii) The GM-CSF receptor antagonist (178,179),
MK-2206 [(Log OWPC, 1.80; vdWD, 0.87 nm; Log OWPC-to-vdWD ratio,
2.07 nm−1); MK-2206 hydrophobic core (Log OWPC,
6.71; vdWD, 0.82 nm; Log OWPC-to-vdWD ratio, 8.14
nm−1)]:
The PDGF receptor antagonist(s) and GM-CSF receptor
antagonist(s), imatinib and MK-2206, still permit concomitant
receptor subunit binding of the respective endogenous growth
factors, PDGF and GM-CSF, which are prolonged duration endogenous
direct CM receptor pressuromodulators of multi-subunit polymeric
β-helix-based receptors, but decrease the potency of the
pressuromodulation effect by causing a decrease in the affinity of
the receptors for endogenous PDGF and GM-CSF, respectively.
Therefore, a decrease the rate of protein transcription of
synthesizable proteins, results in decreased mitogenesis and cell
division due to a decrease in the transcription of the very high MW
nuclear cell division proteins, Ki67 (359 kDa) and separase (230
kDa) (180), and increases the
likelihood of mitochondria-mediated apoptosis in more BCL-dependent
tumor cells (181) due to a decrease
in the transcription of BCL (26 kDa) (181) (Table
XI and Fig. 11).
The small molecule xenobiotic lipophiles or
hydro-lipophiles that bind to multi-subunit polymeric β-helix-based
receptors to decrease the binding affinity of endogenous growth
factors or cytokines and function as 2ary indirect quad receptor
internal pseudo-cationomodulator (SS 1+) pressuromodulation/partial
pressuromodulation antagonists (17)
include.
The TRAIL receptor antagonists (182–186),
a) hydroxycamptothecin [(Log OWPC, 0.92; vdWD, 0.82 nm; Log
OWPC-to-vdWD ratio, 1.12 nm−1);
hydroxycamptothecin hydrophobic core (Log OWPC, 4.19; vdWD, 0.80
nm; Log OWPC-to-vdWD ratio, 5.24 nm−1)]; b)
topotecan [(Log OWPC, −2.10; vdWD, 0.88 nm; Log OWPC-to-vdWD ratio,
−2.39 nm−1); topotecan hydrophobic core (Log
OWPC, 4.71; vdWD, 0.82 nm; Log OWPC-to-vdWD ratio, 5.77
nm−1)].
The TRAIL receptor antagonists, hydroxycamptothecin
and topotecan, still permit the concomitant binding of the
endogenous TRAIL, but decreases the potency of the
pressuromodulation effect by causing for a decrease in the affinity
of TRAIL R2 for endogenous TRAIL, which is an extremely portent
prolonged duration 2ary indirect pressuromodulator multi-subunit
poly-meric β-helix-based receptors (187–191).
As such, hydroxycamptothecin and topotecan-mediated CM receptor
pressuromodulation antagonism of TRAIL results in TRAIL
pressuromodulation-mediated TRAIL R2 chromatin transcription and in
auto-induction of TRAIL R2 at the CM (192) as well as in that of the
lower-to-higher MW proteins (i.e. VEGF, 20 kDa; p21; p53;
topoisomerase I, 90 kDa; HIF1-α, 93 kDa; P-gp, 140 kDa;
topoisomerase II-α, 174 kDa) (186,193),
but not of the highest MW nuclear division-associated proteins
(Ki67, 359 kDa; separase, 230 kDa), in which case, the increased
transcription of intermediate MW protein, p53 (53 kDa) (184,186),
results in the depletion of free BCL (p53-BCL) and in
mitochondrial-mediated oxidative stress sufficient to induce
apoptosis (182,183) that maybe associated with a secondary
decrease in mitogenesis and cell division (182) in synergism with additional CM
receptor pressuromodulation antagonism (185) (Table
XI and Fig. 11).
The CM receptor growth factor or cytokine
pressuromodulation/partial pressuromodulation antagonists,
including the direct CM pressuromodulation/partial
pressuromodulation antagonists of the hepatocyte growth factor
(HGF)/scatter factor (SF) receptor family (194–197),
have the potential for halting solid tumor growth/progression
during the treatment phase (150).
However, due to significant concentrations of growth factor and
cytokine CM receptor pressuromodulators in the solid tumor milieu,
paracrine and autocrine, in context of the significant redundancy
of growth factor and cytokine receptors on tumor CMs for CM
receptor-mediated pressuromodulation, are not tumorocidal in the
physiologic state, as evidenced by the high incidence of local
solid tumor re-occurence upon cessation of treatment phase, due to
the remaining presence of resistant tumor cells overexpressing
mutated receptor subtypes (198) and
the EMT transformation of epithelium of tumorgenic potential
(199), for example, in the case of
weak CM pressuromodulator antagonists such as gefitinib (152,198)
with greater binding affinities for their receptors.
CM receptor growth factor or cytokine
pressuromodulation/partial pressuromodulation antagonism-based
synergistic approaches with the potential for tumorocidal
tumorocytotoxicity include those employing ones such as
staurosporine (59,164,167,168),
imatinib (200), MK-2206 (201) and topotecan (186), particularly, upon effective
transvascular delivery into solid tumors across VEGF-derived and
maintained diaphragm fenestrated tumor blood capillaries (3–8).
In conclusion, based on these observations herein,
on the modes, levels and character of interactions of xenobiotics
and chemoxenobiotics with cells, analyzed in terms of the predicted
conserved biophysical properties, insight has been gained into the
specific mechanisms by which chemoxenobiotics enter cells and the
organelles with which they interact to induce cytotoxcity. This
knowledge is applicable towards improving the effectiveness of
combination small chemotherapy regimens in present clinical use,
for the treatment of solid and hematopoietic malignancies,
including the order in which chemoxenobiotics are administered in
combination treatment regimens, in temporal proximity. It is
anticipated that by the application of this study's findings on the
modes and character of cellular interactions, existing combination
chemotherapy regimens can be designed to be more efficacious, and
furthermore, that by the incorporation of this knowledge into the
algorithms for the design of personalized cancer treatments, the
predictive accuracy of such algorithms can be further
optimized.
The observations of this study also underscore the
importance of focusing attention on specific CM receptors that
mediate the cellular interactions of the various classes of
molecular size-restricted chemoxenobiotics, particularly that of
pro-endocytic xenobiotics, as the proteomic profiling of the
density of these protein receptors across tumor types and grades
will result in most sensitive personalized cancer treatments
employing existing chemotherapeutic regimens. This being stated,
for the curative treatment of solid malignancies, small molecule
chemoxenobiotics must be made to selectively accumulate within the
micromolar concentrations in the tumor milieu, where they must
remain for prolonged duration in order for uniform tumorocidal
cytotoxcity to tumor and tumor-associated cells, which will require
the further translational development of novel chemotherapeutic
regimens employing optimally-sized and -designed biocompatible
imageable dendrimer-based nanoparticles bearing labilely attached
small molecule chemoxenobiotics.
Glossary
Abbreviations
Abbreviations:
|
CM
|
cell membrane
|
|
poOWPC or OWPC
|
predicted overall Log
octanol-to-water partition coefficient (unitless)
|
|
vdWD
|
van der Waals diameter; predicted Log
OWPC-to-vdWD ratio (nm−1)
|
|
Log incorpOWPC-to-vdWD ratio
|
predicted Log incorporating
lipophilicity-to-vdWD ratio (nm−1)
|
|
Log interactOWPC-to-vdWD ratio
|
predicted Log interactability
hydrophilicity-to-vdWD ratio (nm−1)
|
|
S
|
Sufficient separation of charge
|
|
IS
|
Insufficient separation of charge
|
|
Peri
|
peripheral/circumferential
polyhydroxylation, carbonylation and/or etheroylation
|
References
|
1
|
Klassen CD and Watkins IB III: Casarett
and Doull's Essentials of Toxicology. New York, NY: McGraw-Hill.
2010. View Article : Google Scholar
|
|
2
|
Matsumura Y and Maeda H: A new concept for
macromolecular therapeutics in cancer chemotherapy: Mechanism of
tumoritropic accumulation of proteins and the antitumor agent
Smancs. Cancer Res. 46:6387–6392. 1986.PubMed/NCBI
|
|
3
|
Sarin H: Recent progress towards
development of effective systemic chemotherapy for the treatment of
malignant brain tumors. J Transl Med. 7:772009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sarin H: Overcoming the challenges in the
effective delivery of chemotherapies to CNS solid tumors. Ther
Deliv. 1:289–305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sarin H: On the future development of
optimally-sized lipid-insoluble systemic therapies for CNS solid
tumors and other neuropathologies. Recent Patents CNS Drug Discov.
5:239–252. 2010. View Article : Google Scholar
|
|
6
|
Sarin H: Effective transvascular delivery
of chemotherapy into cancer cells with imageable nanoparticles in
the 7 to 10 nanometer size range. Current Advances in the Medical
Application of Nanotechnology. Bentham Science Publishers Ltd.
10–24. 2012.
|
|
7
|
Sarin H: Permeation tt n Silico Pharmacol.
3:52015. View Article : Google Scholar
|
|
8
|
Sarin H: Translational theranostic
methodology for diagnostic imaging and the concomitant treatment of
malignant solid tumors. Neurovascular Imaging. 1:32015. View Article : Google Scholar
|
|
9
|
Lee CC, Gillies ER, Fox ME, Guillaudeu SJ,
Fréchet JM, Dy EE and Szoka FC: A single dose of
doxorubicin-functionalized bow-tie dendrimer cures mice bearing
C-26 colon carcinomas. Proc Natl Acad Sci USA. 103:16649–16654.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Trédan O, Galmarini CM, Patel K and
Tannock IF: Drug resistance and the solid tumor microenvironment. J
Natl Cancer Inst. 99:1441–1454. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rosier RN, Teot LA, Hicks DG, Schwartz C,
O'Keefe RJ and Puzas JE: Multiple drug resistance in osteosarcoma.
Iowa Orthop J. 15:66–73. 1995.PubMed/NCBI
|
|
12
|
Kleinschmidt-Demasters BK, Kang JS and
Lillehei KO: The burden of radiation-induced central nervous system
tumors, A single institution experience. J Neuropathol Exp Neurol.
65:204–216. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Verhoef GE: DeW olf-Peeters C, Ferrant A,
Deprez S, Meeus P, Stul M, Zacheé P, Cassiman JJ, Van den Berghe H
and Boogaerts MA: Myelodysplastic syndromes with bone marrow
fibrosis: A myelodysplastic disorder with proliferative features.
Ann Hematol. 63:235–241. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Doudican NA, Kumar A, Singh NK, Nair PR,
Lala DA, Basu K, Talawdekar AA, Sultana Z, Tiwari KK, Tyagi A, et
al: Personalization of cancer treatment using predictive
simulation. J Transl Med. 13:432015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pingle SC, Sultana Z, Pastorino S, Jiang
P, Mukthavaram R, Chao Y, Bharati IS, Nomura N, Makale M, Abbasi T,
et al: In silico modeling predicts drug sensitivity of
patient-derived cancer cells. J Transl Med. 12:1282014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peacock JD, Cherba D, Kampfschulte K,
Smith MK, Monks NR, Webb CP and Steensma M: Molecular-guided
therapy predictions reveal drug resistance phenotypes and treatment
alternatives in malignant peripheral nerve sheath tumors. J Transl
Med. 11:2132013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sarin H: Pressuromodulation a. a Transl
Med. 13:3722015. View Article : Google Scholar
|
|
18
|
Newlands ES, Stevens MF, Wedge SR,
Wheelhouse RT and Brock C: Temozolomide: A review of its discovery,
chemical properties, pre-clinical development and clinical trials.
Cancer Treat Rev. 23:35–61. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Johnson RE, Campbell RJ and Laws ER Jr:
The cytotoxic effect of ethylnitrosourea on the developing rat
cerebellum. Histopathology. Acta Neuropathol. 55:257–261. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kolarić K, Roth A and Fuss V: Combination
chemotherapy with 1-methyl-1-nitrosourea and cyclophosphamide in
metastatic melanoma. Tumori. 64:89–94. 1978.PubMed/NCBI
|
|
21
|
An Q, Robins P, Lindahl T and Barnes DE:
5-Fluorouracil incorporated into DNA is excised by the Smug1 DNA
glycosylase to reduce drug cytotoxicity. Cancer Res. 67:940–945.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tentori L, Forini O, Fossile E, Muzi A,
Vergati M, Portarena I, Amici C, Gold B and Graziani G:
N3-methyladenine induces early poly(ADP-ribosylation), reduction of
nuclear factor-kappa B DNA binding ability, and nuclear
up-regulation of telomerase activity. Mol Pharmacol. 67:572–581.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Paine PL, Moore LC and Horowitz SB:
Nuclear envelope permeability. Nature. 254:109–114. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rostovtseva T and Colombini M: VDAC
channels mediate and gate the flow of ATP: Implications for the
regulation of mitochondrial function. Biophys J. 72:1954–1962.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Colombini M, Yeung CL, Tung J and König T:
The mitochondrial outer membrane channel, VDAC, is regulated by a
synthetic polyanion. Biochim Biophys Acta. 905:279–286. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nobel PS: Mitochondrial permeability for
alcohols aldoses, and amino acids. J Membr Biol. 12:287–299. 1973.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kondo Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Griffin RJ, Arris CE, Bleasdale C, Boyle
FT, Calvert AH, Curtin NJ, Dalby C, Kanugula S, Lembicz NK, Newell
DR, et al: Resistance-modifying agents. 8. Inhibition of
O(6)-alkylguanine-DNA alkyltransferase by O(6)-alkenyl-,
O(6)-cycloalkenyl-, and O(6)-(2-oxoalkyl)guanines and potentiation
of temozolomide cytotoxicity in vitro by
O(6)-(1-cyclopentenylmethyl)guanine. J Med Chem. 43:4071–4083.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Long L and Dolan ME: Role of cytochrome
P450 isoenzymes in metabolism of O(6)-benzylguanine: Implications
for dacarbazine activation. Clin Cancer Res. 7:4239–4244.
2001.PubMed/NCBI
|
|
30
|
Ortiz de and Montellano PR: Cytochrome
P450-activated prodrugs. Future Med Chem. 5:213–228. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Meyer RP, Podvinec M and Meyer UA:
Cytochrome P450 CYP1A1 accumulates in the cytosol of kidney and
brain and is activated by heme. Mol Pharmacol. 62:1061–1067. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sangar MC, Anandatheerthavarada HK, Tang
W, Prabu SK, Martin MV, Dostalek M, Guengerich FP and Avadhani NG:
Human liver mitochondrial cytochrome P450 2D6 - individual
variations and implications in drug metabolism. FEBS J.
276:3440–3453. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pletsa V, Valavanis C, van Delft JH,
Steenwinkel MJ and Kyrtopoulos SA: DNA damage and mutagenesis
induced by procarbazine in lambda lacZ transgenic mice, Evidence
that bone marrow mutations do not arise primarily through miscoding
by O6-methylguanine. Carcinogenesis. 18:2191–2196. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Crook TR, Souhami RL and McLean AE:
Cytotoxicity, DNA cross-linking, and single strand breaks induced
by activated cyclophosphamide and acrolein in human leukemia cells.
Cancer Res. 46:5029–5034. 1986.PubMed/NCBI
|
|
35
|
Weber GF and Waxman DJ: Denitrosation of
the anti-cancer drug 1,3-bis(2-chloroethyl)-1-nitrosourea catalyzed
by microsomal glutathione S-transferase and cytochrome P450
monooxygenases. Arch Biochem Biophys. 307:369–378. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Linfoot PA, Gray JW, Dean PN, Marton LJ
and Deen DF: Effect of cell cycle position on the survival of 9L
cells treated with nitrosoureas that alkylate, cross-link, and
carbamoylate. Cancer Res. 46:2402–2406. 1986.PubMed/NCBI
|
|
37
|
Doroshenko N and Doroshenko P: The
glutathione reductase inhibitor carmustine induces an influx of
Ca2+ in PC12 cells. Eur J Pharmacol. 497:17–24. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kehrer JP: The effect of BCNU (carmustine)
on tissue glutathione reductase activity. Toxicol Lett. 17:63–68.
1983. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
FitzGerald GB, Bauman C, Hussoin MS and
Wick MM: 2,4-Dihydroxybenzylamine: A specific inhibitor of
glutathione reductase. Biochem Pharmacol. 41:185–190. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Babson JR and Reed DJ: Inactivation of
glutathione reductase by 2-chloroethyl nitrosourea-derived
isocyanates. Biochem Biophys Res Commun. 83:754–762. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bizzozero OA and Ziegler JL: DeJ esus G
and Bolognani F: Acute depletion of reduced glutathione causes
extensive carbonylation of rat brain proteins. J Neurosci Res.
83:656–667. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Street JC, Mahmood U, Matei C and Koutcher
JA: In vivo and in vitro studies of cyclophosphamide chemotherapy
in a mouse mammary carcinoma by 31P NMR spectroscopy. NMR Biomed.
8:149–158. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Street JC and Koutcher JA: Effect of
radiotherapy and chemotherapy on composition of tumor membrane
phospholipids. Lipids. 32:45–49. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jilani K and Lang F: Carmustine-induced
phosphatidylserine translocation in the erythrocyte membrane.
Toxins (Basel). 5:703–716. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Westphal M, Hilt DC, Bortey E, Delavault
P, Olivares R, Warnke PC, Whittle IR, Jääskeläinen J and Ram Z: A
phase 3 trial of local chemotherapy with biodegradable carmustine
(BCNU) wafers (Gliadel wafers) in patients with primary malignant
glioma. Neuro Oncol. 5:79–88. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lambros MP and Rahman YE: Effects of
cyclosporin A on model lipid membranes. Chem Phys Lipids.
131:63–69. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Birraux J, Kirby JA, Thomason JM and
Taylor JJ: The effect of cyclosporin on cell division and apoptosis
in human oral keratinocytes. J Periodontal Res. 41:297–302. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bokemeyer D, Kramer HJ and Meyer-Lehnert
H: Atrial natriuretic peptide blunts the cellular effects of
cyclosporine in smooth muscle. Hypertension. 21:166–172. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Raymond MA, Mollica L, Vigneault N,
Désormeaux A, Chan JS, Filep JG, Hébert MJ, et al: Blockade of the
apoptotic machinery by cyclosporin A redirects cell death toward
necrosis in arterial endothelial cells: regulation by reactive
oxygen species and cathepsin D. FASEB J. 17:515–517.
2003.PubMed/NCBI
|
|
50
|
Laursen M, Yatime L, Nissen P and Fedosova
NU: Crystal structure of the high-affinity
Na+K+-ATPase-ouabain complex with
Mg2+ bound in the cation binding site. Proc Natl Acad
Sci USA. 110:10958–10963. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu J, Kesiry R, Periyasamy SM, Malhotra
D, Xie Z and Shapiro JI: Ouabain induces endocytosis of
plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent
mechanism. Kidney Int. 66:227–241. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Suwalsky M, Hernandez P, Villena F and
Sotomayor CP: The anticancer drug chlorambucil interacts with the
human erythrocyte membrane and model phospholipid bilayers. Z
Naturforsch C. 54:1089–1095. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Krigel R, Liebes LF, Pelle E and Silber R:
Chlorambucil therapy in hairy cell leukemia, Effects on lipid
composition and lymphocyte subpopulations. Blood. 60:272–275.
1982.PubMed/NCBI
|
|
54
|
Matsura T, Kai M, Jiang J, Babu H, Kini V,
Kusumoto C, Yamada K and Kagan VE: Endogenously generated hydrogen
peroxide is required for execution of melphalan-induced apoptosis
as well as oxidation and externalization of phosphatidylserine.
Chem Res Toxicol. 17:685–696. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tilby MJ, Lawley PD and Farmer PB:
Alkylation of DNA by melphalan in relation to immunoassay of
melphalan-DNA adducts, Characterization of mono-alkylated and
cross-linked products from reaction of melphalan with dGMP and GMP.
Chem Biol Interact. 73:183–194. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Van den Driessche B and Lemière F: VanD
ongen W and Esmans EL: Alkylation of DNA by melphalan:
Investigation of capillary liquid chromatography-electrospray
ionization tandem mass spectrometry in the study of the adducts at
the nucleoside level. J Chromatogr B Analyt Technol Biomed Life
Sci. 785:21–37. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Rodriguez RJ and Acosta D Jr: Inhibition
of mitochondrial function in isolated rate liver mitochondria by
azole antifungals. J Biochem Toxicol. 11:127–131. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Maté MJ, Ortiz-Lombardía M, Boitel B,
Haouz A, Tello D, Susin SA, Penninger J, Kroemer G and Alzari PM:
The crystal structure of the mouse apoptosis-inducing factor AIF.
Nat Struct Biol. 9:442–446. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
59
|
Daugas E, Susin SA, Zamzami N, Ferri KF,
Irinopoulou T, Larochette N, Prévost MC, Leber B, Andrews D,
Penninger J, et al: Mitochondrio-nuclear translocation of AIF in
apoptosis and necrosis. FASEB J. 14:729–739. 2000.PubMed/NCBI
|
|
60
|
Lewis EM, Wilkinson AS, Davis NY, Horita
DA and Wilkinson JC: Nondegradative ubiquitination of apoptosis
inducing factor (AIF) by X-linked inhibitor of apoptosis at a
residue critical for AIF-mediated chromatin degradation.
Biochemistry. 50:11084–11096. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wei Y, Fox T, Chambers SP, Sintchak J,
Coll JT, Golec JM, Swenson L, Wilson KP and Charifson PS: The
structures of caspases-1, −3, −7 and −8 reveal the basis for
substrate and inhibitor selectivity. Chem Biol. 7:423–432. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Riedl SJ and Shi Y: Molecular mechanisms
of caspase regulation during apoptosis. Nat Rev Mol Cell Biol.
5:897–907. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lindsay J, Esposti MD and Gilmore AP:
Bcl-2 proteins and mitochondria - specificity in membrane targeting
for death. Biochim Biophys Acta. 1813:532–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Miyashita T and Reed JC: Tumor suppressor
p53 is a direct transcriptional activator of the human bax gene.
Cell. 80:293–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cho Y, Gorina S, Jeffrey PD and Pavletich
NP: Crystal structure of a p53 tumor suppressor-DNA complex,
Understanding tumorigenic mutations. Science. 265:346–355. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Billen LP, Shamas-Din A and Andrews DW:
Bid A Bax-like BH3 protein. Oncogene. 27((Suppl 1)): S93–S104.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chipuk JE and Green DR: PUMA cooperates
with direct activator proteins to promote mitochondrial outer
membrane permeabilization and apoptosis. Cell Cycle. 8:2692–2696.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Nakano K and Vousden KH: PUMA a novel
proapoptotic gene, is induced by p53. Mol Cell. 7:683–694. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Loose DS, Kan PB, Hirst MA, Marcus RA and
Feldman D: Ketoconazole blocks adrenal steroidogenesis by
inhibiting cytochrome P450-dependent enzymes. J Clin Invest.
71:1495–1499. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Greenblatt DJ, Zhao Y, Venkatakrishnan K,
Duan SX, Harmatz JS, Parent SJ, Court MH and von Moltke LL:
Mechanism of cytochrome P450-3A inhibition by ketoconazole. J Pharm
Pharmacol. 63:214–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ho YS, Tsai PW, Yu CF, Liu HL, Chen RJ and
Lin JK: Ketoconazole-induced apoptosis through P53-dependent
pathway in human colorectal and hepatocellular carcinoma cell
lines. Toxicol Appl Pharmacol. 153:39–47. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wang YJ, Jeng JH, Chen RJ, Tseng H, Chen
LC, Liang YC, Lin CH, Chen CH, Chu JS, Ho WL, et al: Ketoconazole
potentiates the antitumor effects of nocodazole: In vivo therapy
for human tumor xenografts in nude mice. Mol Carcinog. 34:199–210.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Pascual A, García I, Conejo C and Perea
EJ: Uptake and intracellular activity of fluconazole in human
polymorphonuclear leukocytes. Antimicrob Agents Chemother.
37:187–190. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ciccolini J, Fina F, Bezulier K,
Giacometti S, Roussel M, Evrard A, Cuq P, Romain S, Martin PM and
Aubert C: Transmission of apoptosis in human colorectal tumor cells
exposed to capecitabine, Xeloda, is mediated via Fas. Mol Cancer
Ther. 1:923–927. 2002.PubMed/NCBI
|
|
75
|
Baltch AL, Smith RP, Ritz WJ, Bopp LH and
Michelsen PB: Intracellular activity of voriconazole, fluconazole,
and itraconazole against Candida albicans in human monocytes
with and without activation by GM-CSF and TNF-alpha. J Appl Res.
5:42005.
|
|
76
|
Murphy JW, Cho Y, Sachpatzidis A, Fan C,
Hodsdon ME and Lolis E: Structural and functional basis of CXCL12
(stromal cell-derived factor-1 α) binding to heparin. J Biol Chem.
282:10018–10027. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Debnath B, Xu S, Grande F, Garofalo A and
Neamati N: Small molecule inhibitors of CXCR4. Theranostics.
3:47–75. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Hatse S, Princen K and Bridger G: DeC
lercq E and Schols D: Chemokine receptor inhibition by AMD3100 is
strictly confined to CXCR4. FEBS Lett. 527:255–262. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Pron G, Belehradek J Jr and Mir LM:
Identification of a plasma membrane protein that specifically binds
bleomycin. Biochem Biophys Res Commun. 194:333–337. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Pron G, Belehradek J Jr, Orlowski S and
Mir LM: Involvement of membrane bleomycin-binding sites in
bleomycin cytotoxicity. Biochem Pharmacol. 48:301–310. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Krishan A and Whitlock S:
Bleomycin-induced fine structural alterations in cultured mouse
fibroblasts and human lymphocytes of neoplastic origin. Cancer Res.
33:777–785. 1973.PubMed/NCBI
|
|
82
|
Pron G, Mahrour N, Orlowski S, Tounekti O,
Poddevin B, Belehradek J Jr and Mir LM: Internalisation of the
bleomycin molecules responsible for bleomycin toxicity: A
receptor-mediated endocytosis mechanism. Biochem Pharmacol.
57:45–56. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Ascierto ML, Kmieciak M, Idowu MO, Manjili
R, Zhao Y, Grimes M, Dumur C, Wang E, Ramakrishnan V, Wang XY, et
al: A signature of immune function genes associated with
recurrence-free survival in breast cancer patients. Breast Cancer
Res Treat. 131:871–880. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Chen L, Huang Z, Yao G, Lyu X, Li J, Hu X,
Cai Y, Li W, Li X and Ye C: The expression of CXCL13 and its
relation to unfavorable clinical characteristics in young breast
cancer. J Transl Med. 13:1682015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Stamatopoulos B, Meuleman N, De Bruyn C,
Pieters K, Mineur P, Le Roy C, Saint-Georges S, Varin-Blank N,
Cymbalista F, Bron D, et al: AMD3100 disrupts the cross-talk
between chronic lymphocytic leukemia cells and a mesenchymal
stromal or nurse-like cell-based microenvironment: Pre-clinical
evidence for its association with chronic lymphocytic leukemia
treatments. Haematologica. 97:608–615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Chen LJ, Ye H, Zhang Q, Li FZ, Song LJ,
Yang J, Mu Q, Rao SS, Cai PC, Xiang F, et al: Bleomycin induced
epithelial-mesenchymal transition (EMT) in pleural mesothelial
cells. Toxicol Appl Pharmacol. 283:75–82. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Wu Z, Yang L, Cai L, Zhang M, Cheng X,
Yang X and Xu J: Detection of epithelial to mesenchymal transition
in airways of a bleomycin induced pulmonary fibrosis model derived
from an alpha-smooth muscle actin-Cre transgenic mouse. Respir Res.
8:12007. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yamada A, Aki T, Unuma K, Funakoshi T and
Uemura K: Paraquat induces epithelial-mesenchymal transition-like
cellular response resulting in fibrogenesis and the prevention of
apoptosis in human pulmonary epithelial cells. PLoS One.
10:e01201922015. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Dik WA, McAnulty RJ, Versnel MA, Naber BA,
Zimmermann LJ, Laurent GJ and Mutsaers SE: Short course
dexamethasone treatment following injury inhibits bleomycin induced
fibrosis in rats. Thorax. 58:765–771. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Domanska UM, Timmer-Bosscha H, Nagengast
WB, Munnink Oude TH, Kruizinga RC, Ananias HJ, Kliphuis NM, Huls G,
De Vries EG, de Jong IJ, et al: CXCR4 inhibition with AMD3100
sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia.
14:709–718. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ujwal R, Cascio D, Colletier JP, Faham S,
Zhang J, Toro L, Ping P and Abramson J: The crystal structure of
mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into
metabolite gating. Proc Natl Acad Sci USA. 105:17742–17747. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Catterall WA: Functional subunit structure
of voltage-gated calcium channels. Science. 253:1499–1500. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Barańska W, Kujawa M and Kujawska E:
Influence of vincristine on the Golgi apparatus in
preimplantation development of the mouse embryo. Gegenbaurs Morphol
Jahrb. 134:175–184. 1988.PubMed/NCBI
|
|
94
|
Kujawa M, Ochocka M and Moskalewski S:
Influence of vincristine on the Golgi complex of leukaemic
lymphoblasts. Folia Haematologica. 107:193–203. 1980.PubMed/NCBI
|
|
95
|
Carré M, André N, Carles G, Borghi H,
Brichese L, Briand C and Braguer D: Tubulin is an inherent
component of mitochondrial membranes that interacts with the
voltage-dependent anion channel. J Biol Chem. 277:33664–33669.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Groninger E, Meeuwsen-De Boer GJ, De Graaf
SS, Kamps WA and De Bont ES: Vincristine induced apoptosis in acute
lymphoblastic leukaemia cells: A mitochondrial controlled pathway
regulated by reactive oxygen species? Int J Oncol. 21:1339–1345.
2002.PubMed/NCBI
|
|
97
|
Eom YW, Kim MA, Park SS, Goo MJ, Kwon HJ,
Sohn S, Kim WH, Yoon G and Choi KS: Two distinct modes of cell
death induced by doxorubicin, Apoptosis and cell death through
mitotic catastrophe accompanied by senescence-like phenotype.
Oncogene. 24:4765–4777. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Gamen S, Anel A, Lasierra P, Alava MA,
Martinez-Lorenzo MJ, Piñeiro A and Naval J: Doxorubicin-induced
apoptosis in human T-cell leukemia is mediated by caspase-3
activation in a Fas-independent way. FEBS Lett. 417:360–364. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Kuznetsov AV, Margreiter R, Amberger A,
Saks V and Grimm M: Changes in mitochondrial redox state, membrane
potential and calcium precede mitochondrial dysfunction in
doxorubicin-induced cell death. Biochim Biophys Acta.
1813:1144–1152. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Mizutani H, Tada-Oikawa S, Hiraku Y,
Kojima M and Kawanishi S: Mechanism of apoptosis induced by
doxorubicin through the generation of hydrogen peroxide. Life Sci.
76:1439–1453. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Wang S, Konorev EA, Kotamraju S, Joseph J,
Kalivendi S and Kalyanaraman B: Doxorubicin induces apoptosis in
normal and tumor cells via distinctly different mechanisms.
Histopathology. J Biol Chem. 279:25535–25543. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Golomb E, Hill MR, Brown RG and Keiser HR:
Ouabain enhances the mitogenic effect of serum in vascular smooth
muscle cells. Am J Hypertens. 7:69–74. 1994.PubMed/NCBI
|
|
103
|
Kanai R, Ogawa H, Vilsen B, Cornelius F
and Toyoshima C: Crystal structure of a Na+-bound
Na+, K+-ATPase preceding the E1P state.
Nature. 502:201–206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Aller SG, Yu J, Ward A, Weng Y,
Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL,
et al: Structure of P-glycoprotein reveals a molecular basis for
poly-specific drug binding. Science. 323:1718–1722. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Sehested M, Jensen PB, Skovsgaard T,
Bindslev N, Demant EJ, Friche E and Vindeløv L: Inhibition of
vincristine binding to plasma membrane vesicles from
daunorubicin-resistant Ehrlich ascites cells by multidrug
resistance modulators. Br J Cancer. 60:809–814. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Srivalli KMR and Lakshmi P: Overview of
P-glycoprotein inhibitors: A rational outlook. Braz J Pharm Sci.
48:353–367. 2012. View Article : Google Scholar
|
|
107
|
Chen C, Ke J, Zhou XE, Yi W, Brunzelle JS,
Li J, Yong EL, Xu HE and Melcher K: Structural basis for molecular
recognition of folic acid by folate receptors. Nature. 500:486–489.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Nandini-Kishore SG and Frazier WA:
[3H]Methotrexate as a ligand for the folate receptor of
Dictyostelium discoideum. Proc Natl Acad Sci USA.
78:7299–7303. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Herman S, Zurgil N and Deutsch M: Low dose
methotrexate induces apoptosis with reactive oxygen species
involvement in T lymphocytic cell lines to a greater extent than in
monocytic lines. Inflamm Res. 54:273–280. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ramadan AA, Yousif WB and Ali AM: The
effect of methotrexate (MTX) on the small intestine of the mouse.
IV. The Golgi apparatus, phosphatases and esterases. Funct
Dev Morphol. 2:111–119. 1992.PubMed/NCBI
|
|
111
|
Ramadan AA, Yousif WB and Ali AM: The
effect of methotrexate (MTX) on the small intestine of the mouse.
Histopathology. Funct Dev Morphol. 2:3–9. 1992.PubMed/NCBI
|
|
112
|
Pritchard DM, Bower L, Potten CS, Jackman
AL and Hickman JA: The importance of p53-independent apoptosis in
the intestinal toxicity induced by raltitrexed (ZD1694, Tomudex):
genetic differences between BALB/c and DBA/2 mice. Clin Cancer Res.
6:4389–4395. 2000.PubMed/NCBI
|
|
113
|
Xue S, Chen YX, Qin SK, Yang AZ, Wang L,
Xu HJ and Geng HY: Raltitrexed induces mitochondrial mediated
apoptosis in SGC7901 human gastric cancer cells. Mol Med Rep.
10:1927–1934. 2014.PubMed/NCBI
|
|
114
|
Chattopadhyay S, Moran RG and Goldman ID:
Pemetrexed Biochemical and cellular pharmacology, mechanisms, and
clinical applications. Mol Cancer Ther. 6:404–417. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Fischer CD: daC osta M and Rothenberg SP:
The heterogeneity and properties of folate binding proteins from
chronic myelogenous leukemia cells. Blood. 46:855–867.
1975.PubMed/NCBI
|
|
116
|
Fischer CD: DaC osta M and Rothenberg SP:
Properties of purified folate-binding proteins from chronic
myelogenous leukemia cells. Biochim Biophys Acta. 543:328–339.
1978. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Barford PA, Blair JA and Malghani MA: The
effect of methotrexate on folate metabolism in the rat. Br J
Cancer. 41:816–820. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Ohbayashi M, Kubota S, Kawase A, Kohyama
N, Kobayashi Y and Yamamoto T: Involvement of
epithelial-mesenchymal transition in methotrexate-induced pulmonary
fibrosis. J Toxicol Sci. 39:319–330. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Shprung T and Gozes I: A novel method for
analyzing mitochondrial movement: inhibition by paclitaxel in a
pheochromocytoma cell model. J Mol Neurosci. 37:254–262. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Foland TB, Dentler WL, Suprenant KA, Gupta
ML Jr and Himes RH: Paclitaxel-induced microtubule stabilization
causes mitotic block and apoptotic-like cell death in a
paclitaxel-sensitive strain of Saccharomyces cerevisiae.
Yeast. 22:971–978. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Salido M, Gonzalez JL and Vilches J: Loss
of mitochondrial membrane potential is inhibited by bombesin in
etoposide-induced apoptosis in PC-3 prostate carcinoma cells. Mol
Cancer Ther. 6:1292–1299. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Mizukami S, Kikuchi K, Higuchi T, Urano Y,
Mashima T, Tsuruo T and Nagano T: Imaging of caspase-3 activation
in HeLa cells stimulated with etoposide using a novel fluorescent
probe. FEBS Lett. 453:356–360. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Matsushima Y, Kanzawa F, Miyazawa N,
Sasaki Y and Saijo N: In vitro antitumor activity of teniposide
against carcinoma of the lung in human tumor clonogenic assay.
Anticancer Res. 6:921–924. 1986.PubMed/NCBI
|
|
124
|
Sánchez-Alcázar JA, Khodjakov A and
Schneider E: Anticancer drugs induce increased mitochondrial
cytochrome c expression that precedes cell death. Cancer
Res. 61:1038–1044. 2001.PubMed/NCBI
|
|
125
|
Uyar D, Takigawa N, Mekhail T, Grabowski
D, Markman M, Lee F, Canetta R, Peck R, Bukowski R and Ganapathi R:
Apoptotic pathways of epothilone BMS 310705. Gynecol Oncol.
91:173–178. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Thomson AER and Robinson MA: Cytocidal
action of colchicine in vitro on lymphocytes in chronic lymphocytic
leukaemia. Lancet. 2:868–870. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Chen XM, Liu J, Wang T and Shang J:
Colchicine-induced apoptosis in human normal liver L-02 cells by
mitochondrial mediated pathways. Toxicol In Vitro. 26:649–655.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Jelínek M, Balušíková K, Schmiedlová M,
Němcová-Fürstová V, Šrámek J, Stančíková J, Zanardi I, Ojima I and
Kovář J: The role of individual caspases in cell death induction by
taxanes in breast cancer cells. Cancer Cell Int. 15:82015.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
André N, Braguer D, Brasseur G, Gonçalves
A, Lemesle-Meunier D, Guise S, Jordan MA and Briand C: Paclitaxel
induces release of cytochrome c from mitochondria isolated
from human neuroblastoma cells'. Cancer Res. 60:5349–5353.
2000.PubMed/NCBI
|
|
130
|
Khawaja NR, Carré M, Kovacic H, Estève MA
and Braguer D: Patupilone-induced apoptosis is mediated by
mitochondrial reactive oxygen species through Bim relocalization to
mitochondria. Mol Pharmacol. 74:1072–1083. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Salim AA, Xiao X, Cho KJ, Piggott AM,
Lacey E, Hancock JF and Capon RJ: Rare Streptomyces sp.
polyketides as modulators of K-Ras localisation. Org Biomol Chem.
12:4872–4878. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Pusceddu S, Indini A and Procopio G:
Everolimus treatment in advanced solid tumors: a personal view.
Future Science. 2015.OA March 20, (Epub ahead of print)
doi:10.4155/fso.14.1. View Article : Google Scholar
|
|
133
|
Chambraud B, Belabes H, Fontaine-Lenoir V,
Fellous A and Baulieu EE: The immunophilin FKBP52 specifically
binds to tubulin and prevents microtubule formation. FASEB J.
21:2787–2797. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Shirane M and Nakayama KI: Inherent
calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and
inhibits apoptosis. Nat Cell Biol. 5:28–37. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Tanaka K, Fujita N, Higashi Y and Ogawa N:
Neuroprotective and antioxidant properties of FKBP-binding
immunophilin ligands are independent on the FKBP12 pathway in human
cells. Neurosci Lett. 330:147–150. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Simon N, Morin C, Urien S, Tillement JP
and Bruguerolle B: Tacrolimus and sirolimus decrease oxidative
phosphorylation of isolated rat kidney mitochondria. Br J
Pharmacol. 138:369–376. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Zini R, Simon N, Morin C, Thiault L and
Tillement JP: Tacrolimus decreases in vitro oxidative
phosphorylation of mitochondria from rat forebrain. Life Sci.
63:357–368. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Zoli W, Ulivi P, Tesei A, Fabbri F,
Rosetti M, Maltoni R, Giunchi DC, Ricotti L, Brigliadori G, Vannini
I, et al: Addition of 5-fluorouracil to doxorubicin-paclitaxel
sequence increases caspase-dependent apoptosis in breast cancer
cell lines. Breast Cancer Res. 7:R681–R689. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Yang Y, Niu X, Zhang Q, Hao L, Ding Y and
Xu H: The efficacy of abraxane on osteosarcoma xenografts in nude
mice and expression of secreted protein, acidic and rich in
cysteine. Am J Med Sci. 344:199–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Saif MW: U.S. Food and Drug Administration
approves paclitaxel protein-bound particles (Abraxane®) in
combination with gemcitabine as first-line treatment of patients
with metastatic pancreatic cancer. JOP. 14:686–688. 2013.PubMed/NCBI
|
|
141
|
Coward P, Lee D, Hull MV and Lehmann JM:
4-Hydroxytamoxifen binds to and deactivates the estrogen-related
receptor gamma. Proc Natl Acad Sci USA. 98:8880–8884. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Richards J, Lim AC, Hay CW, Taylor AE,
Wingate A, Nowakowska K, Pezaro C, Carreira S, Goodall J, Arlt W,
et al: Interactions of abiraterone, eplerenone, and prednisolone
with wild-type and mutant androgen receptor: A rationale for
increasing abiraterone exposure or combining with MDV3100. Cancer
Res. 72:2176–2182. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Darbre PD and King RJB: Differential
effects of steroid hormones on parameters of cell growth. Cancer
Res. 47:2937–2944. 1987.PubMed/NCBI
|
|
144
|
Yates J and King RJB: Correlation of
growth properties and morphology with hormone responsiveness of
mammary tumor cells in culture. Cancer Res. 41:258–262.
1981.PubMed/NCBI
|
|
145
|
Talwar GP, Raina K, Gupta JC, Ray R,
Wadhwa S and Ali MM: A recombinant luteinising-hormone-releasing-
hormone immunogen bioeffective in causing prostatic atrophy.
Vaccine. 22:3713–3721. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Brandes AA, Ermani M, Turazzi S, Scelzi E,
Berti F, Amistà P, Rotilio A, Licata C and Fiorentino MV:
Procarbazine and high-dose tamoxifen as a second-line regimen in
recurrent high-grade gliomas, A phase II study. J Clin Oncol.
17:645–650. 1999.PubMed/NCBI
|
|
147
|
de Bono JS, Logothetis CJ, Molina A,
Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB Jr, Saad F,
et al: COU-AA-301 Investigators: Abiraterone and increased survival
in metastatic prostate cancer. N Engl J Med. 364:1995–2005. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Kosaka T, Miyajima A, Yasumizu Y, Miyazaki
Y, Kikuchi E and Oya M: Limited in vitro efficacy of CYP17A1
inhibition on human castration resistant prostate cancer. Steroids.
92:39–44. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Louderbough JMV, Lopez JI and Schroeder
JA: Matrix hyaluronan alters epidermal growth factor
receptor-dependent cell morphology. Cell Adhes Migr. 4:26–31. 2010.
View Article : Google Scholar
|
|
150
|
Hara F, Aoe M, Doihara H, Taira N, Shien
T, Takahashi H, Yoshitomi S, Tsukuda K, Toyooka S, Ohta T, et al:
Antitumor effect of gefitinib ('Iressa') on esophageal squamous
cell carcinoma cell lines in vitro and in vivo. Cancer Lett.
226:37–47. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Stegmaier K, Corsello SM, Ross KN, Wong
JS, Deangelo DJ and Golub TR: Gefitinib induces myeloid
differentiation of acute myeloid leukemia. Blood. 106:2841–2848.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Augustin A, Lamerz J, Meistermann H,
Golling S, Scheiblich S, Hermann JC, Duchateau-Nguyen G, Tzouros M,
Avila DW, Langen H, et al: Quantitative chemical proteomics
profiling differentiates erlotinib from gefitinib in EGFR wild-type
non-small cell lung carcinoma cell lines. Mol Cancer Ther.
12:520–529. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Huang HL, Chen YC, Huang YC, Yang KC, Pan
H, Shih SP and Chen YJ: Lapatinib induces autophagy, apoptosis and
megakaryocytic differentiation in chronic myelogenous leukemia K562
cells. PLoS One. 6:e290142011. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Diaz R, Nguewa PA, Parrondo R,
Perez-Stable C, Manrique I, Redrado M, Catena R, Collantes M,
Peñuelas I, Díaz-González JA, et al: Antitumor and antiangiogenic
effect of the dual EGFR and HER-2 tyrosine kinase inhibitor
lapatinib in a lung cancer model. BMC Cancer. 10:1882010.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Chen G, Noor A, Kronenberger P, Teugels E,
Umelo IA and De Grève J: Synergistic effect of afatinib with
su11274 in non-small cell lung cancer cells resistant to gefitinib
or erlotinib. PLoS One. 8:e597082013. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Solca F, Dahl G, Zoephel A, Bader G,
Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E and Adolf
GR: Target binding properties and cellular activity of afatinib
(BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp
Ther. 343:342–350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Stoica GE, Kuo A, Powers C, Bowden ET,
Sale EB, Riegel AT and Wellstein A: Midkine binds to anaplastic
lymphoma kinase (ALK) and acts as a growth factor for different
cell types. J Biol Chem. 277:35990–35998. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Sumi Y, Muramatsu H, Hata K, Ueda M and
Muramatsu T: Midkine enhances early stages of collagen gel
contraction. J Biochem. 127:247–251. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Kadomatsu K and Muramatsu T: Midkine and
pleiotrophin in neural development and cancer. Cancer Lett.
204:127–143. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Friboulet L, Li N, Katayama R, Lee CC,
Gainor JF, Crystal AS, Michellys PY, Awad MM, Yanagitani N, Kim S,
et al: The ALK inhibitor ceritinib overcomes crizotinib resistance
in non-small cell lung cancer. Cancer Discov. 4:662–673. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Lisignoli G, Toneguzzi S, Piacentini A,
Cristino S, Grassi F, Cavallo C and Facchini A: CXCL12 (SDF-1) and
CXCL13 (BCA-1) chemokines significantly induce proliferation and
collagen type I expression in osteoblasts from osteoarthritis
patients. J Cell Physiol. 206:78–85. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Kalinina OV, Pfeifer N and Lengauer T:
Modelling binding between CCR5 and CXCR4 receptors and their
ligands suggests the surface electrostatic potential of the
co-receptor to be a key player in the HIV-1 tropism. Retrovirology.
10:1302013. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Massarelli I, Chiellini F, Chiellini E and
Bianucci AM: Three-dimensional models of the oligomeric human
asialoglycoprotein receptor (ASGP-R). Int J Mol Sci. 11:3867–3884.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Fallon RJ and Danaher M: The effect of
staurosporine a protein kinase inhibitor, on asialoglycoprotein
receptor endocytosis. Exp Cell Res. 203:420–426. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Trerè D, Fiume L, De Giorgi LB, Di Stefano
G, Migaldi M and Derenzini M: The asialoglycoprotein receptor in
human hepatocellular carcinomas: Its expression on proliferating
cells. Br J Cancer. 81:404–408. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Becherer U, Guatimosim C and Betz W:
Effects of staurosporine on exocytosis and endocytosis at frog
motor nerve terminals. J Neurosci. 21:782–787. 2001.PubMed/NCBI
|
|
167
|
Belmokhtar CA, Hillion J and
Ségal-Bendirdjian E: Staurosporine induces apoptosis through both
caspase-dependent and caspase-independent mechanisms. Oncogene.
20:3354–3362. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Zhang XD, Gillespie SK and Hersey P:
Staurosporine induces apoptosis of melanoma by both
caspase-dependent and -independent apoptotic pathways. Mol Cancer
Ther. 3:187–197. 2004.PubMed/NCBI
|
|
169
|
Dunai ZA, Imre G, Barna G, Korcsmaros T,
Petak I, Bauer PI and Mihalik R: Staurosporine induces necroptotic
cell death under caspase-compromised conditions in U937 cells. PLoS
One. 7:e419452012. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Chan KC, Knox WF, Gee JM, Morris J,
Nicholson RI, Potten CS and Bundred NJ: Effect of epidermal growth
factor receptor tyrosine kinase inhibition on epithelial
proliferation in normal and premalignant breast. Cancer Res.
62:122–128. 2002.PubMed/NCBI
|
|
171
|
Maity A, Pore N, Lee J, Solomon D and
O'Rourke DM: Epidermal growth factor receptor transcriptionally
up-regulates vascular endothelial growth factor expression in human
glioblastoma cells via a pathway involving phosphatidylinositol
3-kinase and distinct from that induced by hypoxia. Cancer Res.
60:5879–5886. 2000.PubMed/NCBI
|
|
172
|
Ouchi T, Monteiro ANA, August A, Aaronson
SA and Hanafusa H: BRCA1 regulates p53-dependent gene expression.
Proc Natl Acad Sci USA. 95:2302–2306. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Arizti P, Fang L, Park I, Yin Y, Solomon
E, Ouchi T, Aaronson SA and Lee SW: Tumor suppressor p53 is
required to modulate BRCA1 expression. Mol Cell Biol. 20:7450–7459.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Andreassen A, Øyjord T, Hovig E, Holm R,
Flørenes VA, Nesland JM, Myklebost O, Høie J, Bruland OS, Børresen
AL, et al: p53 abnormalities in different subtypes of human
sarcomas. Cancer Res. 53:468–471. 1993.PubMed/NCBI
|
|
175
|
O'Hare T, Pollock R, Stoffregen EP, Keats
JA, Abdullah OM, Moseson EM, Rivera VM, Tang H, Metcalf CA III,
Bohacek RS, et al: Inhibition of wild-type and mutant Bcr-Abl by
AP23464, a potent ATP-based oncogenic protein kinase inhibitor:
Implications for CML. Blood. 104:2532–2539. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Grisolano JL, O'Neal J, Cain J and
Tomasson MH: An activated receptor tyrosine kinase, TEL/PDGFbetaR,
cooperates with AML1/ETO to induce acute myeloid leukemia in mice.
Proc Natl Acad Sci USA. 100:9506–9511. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Aoki M, Nabeshima K, Koga K, Hamasaki M,
Suzumiya J, Tamura K and Iwasaki H: Imatinib mesylate inhibits cell
invasion of malignant peripheral nerve sheath tumor induced by
platelet-derived growth factor-BB. Lab Invest. 87:767–779. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Hercus TR, Thomas D, Guthridge MA, Ekert
PG, King-Scott J, Parker MW and Lopez AF: The
granulocyte-macrophage colony-stimulating factor receptor, linking
its structure to cell signaling and its role in disease. Blood.
114:1289–1298. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Ding W, Shanafelt TD, Lesnick CE,
Erlichman C, Leis JF, Secreto C, Sassoon TR, Call TG, Bowen DA,
Conte M, et al: Akt inhibitor MK2206 selectively targets CLL B-cell
receptor induced cytokines, mobilizes lymphocytes and synergizes
with bendamustine to induce CLL apoptosis. Br J Haematol.
164:146–150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Zhao Y-Y, Tian Y, Zhang J, Xu F, Yang YP,
Huang Y, Zhao HY, Zhang JW, Xue C, Lam MH, et al: Effects of an
oral allosteric AKT inhibitor (MK-2206) on human nasopharyngeal
cancer in vitro and in vivo. Drug Des Devel Ther. 8:1827–1837.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Agarwal E, Chaudhuri A, Leiphrakpam PD,
Haferbier KL, Brattain MG and Chowdhury S: Akt inhibitor MK-2206
promotes anti-tumor activity and cell death by modulation of AIF
and Ezrin in colorectal cancer. BMC Cancer. 14:1452014. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Fu YR, Yi ZJ, Yan YR and Qiu ZY:
Hydroxycamptothecin-induced apoptosis in hepatoma SMMC-7721 cells
and the role of mitochondrial pathway. Mitochondrion. 6:211–217.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Uckun FM, Stewart CF, Reaman G, Chelstrom
LM, Jin J, Chandan-Langlie M, Waddick KG, White J and Evans WE: In
vitro and in vivo activity of topotecan against human B-lineage
acute lymphoblastic leukemia cells. Blood. 85:2817–2828.
1995.PubMed/NCBI
|
|
184
|
Caserini C, Pratesi G, Tortoreto M,
Bedogné B, Carenini N, Supino R, Perego P, Righetti SC and Zunino
F: Apoptosis as a determinant of tumor sensitivity to topotecan in
human ovarian tumors, preclinical in vitro/in vivo studies. Clin
Cancer Res. 3:955–961. 1997.PubMed/NCBI
|
|
185
|
Kim MK, James J and Annunziata CM:
Topotecan synergizes with CHEK1 (CHK1) inhibitor to induce
apoptosis in ovarian cancer cells. BMC Cancer. 15:1962015.
View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Tolis C, Peters GJ, Ferreira CG, Pinedo HM
and Giaccone G: Cell cycle disturbances and apoptosis induced by
topotecan and gemcitabine on human lung cancer cell lines. Eur J
Cancer. 35:796–807. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Szalay K, Rázga Z and Duda E: TNF inhibits
myogenesis and downregulates the expression of myogenic regulatory
factors myoD and myogenin. Eur J Cell Biol. 74:391–398.
1997.PubMed/NCBI
|
|
188
|
Fiers W, Beyaert R, Brouckaert P,
Everaerdt B, Haegeman C, Suffys P, Tavernier J and Vanhaesebroeck
B: TNF Its potential as an antitumour agent. Dev Biol Stand.
69:143–151. 1988.PubMed/NCBI
|
|
189
|
Smith RA and Baglioni C: The active form
of tumor necrosis factor is a trimer. J Biol Chem. 262:6951–6954.
1987.PubMed/NCBI
|
|
190
|
Udagawa N, Takahashi N, Jimi E, Matsuzaki
K, Tsurukai T, Itoh K, Nakagawa N, Yasuda H, Goto M, Tsuda E, et
al: Osteoblasts/stromal cells stimulate osteoclast activation
through expression of osteoclast differentiation factor/RANKL but
not macrophage colony-stimulating factor: Receptor activator of
NF-kappa B ligand. Bone. 25:517–523. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Luan X, Lu Q, Jiang Y, Zhang S, Wang Q,
Yuan H, Zhao W, Wang J and Wang X: Crystal structure of human RANKL
complexed with its decoy receptor osteoprotegerin. J Immunol.
189:245–252. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Ciusani E, Croci D, Gelati M, Calatozzolo
C, Sciacca F, Fumagalli L, Balzarotti M, Fariselli L, Boiardi A and
Salmaggi A: In vitro effects of topotecan and ionizing radiation on
TRAIL/Apo2L-mediated apoptosis in malignant glioma. J Neurooncol.
71:19–25. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Chen S, Gomez SP, McCarley D and
Mainwaring MG: Topotecan-induced topoisomerase IIalpha expression
increases the sensitivity of the CML cell line K562 to subsequent
etoposide plus mitoxantrone treatment. Cancer Chemother Pharmacol.
49:347–355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Naldini L, Weidner KM, Vigna E, Gaudino G,
Bardelli A, Ponzetto C, Narsimhan RP, Hartmann G, Zarnegar R,
Michalopoulos GK, et al: Scatter factor and hepatocyte growth
factor are indistinguishable ligands for the MET receptor. EMBO J.
10:2867–2878. 1991.PubMed/NCBI
|
|
195
|
Stamos J, Lazarus RA, Yao X, Kirchhofer D
and Wiesmann C: Crystal structure of the HGF beta-chain in complex
with the Sema domain of the Met receptor. EMBO J. 23:2325–2335.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Paulson AK, Linklater ES, Berghuis BD, App
CA, Oostendorp LD, Paulson JE, Pettinga JE and Melnik MK: VandeW
oude GF and Graveel CR: MET and ERBB2 are coexpressed in
ERBB2+ breast cancer and contribute to innate
resistance. Mol Cancer Res. 11:1112–1121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Xie Q, Su Y, Dykema K, Johnson J, Koeman
J, De Giorgi V, Huang A, Schlegel R, Essenburg C, Kang L, et al:
Overexpression of HGF promotes HBV-induced hepatocellular carcinoma
progression and is an effective indicator for Met-targeting
therapy. Genes Cancer. 4:247–260. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Simonetti S, Molina MA, Queralt C, de
Aguirre I, Mayo C, Bertran-Alamillo J, Sanchez JJ, Gonzalez-Larriba
JL, Jimenez U, Isla D, et al: Detection of EGFR mutations with
mutation-specific antibodies in stage IV non-small-cell lung
cancer. J Transl Med. 8:1352010. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Maseki S, Ijichi K, Tanaka H, Fujii M,
Hasegawa Y, Ogawa T, Murakami S, Kondo E and Nakanishi H:
Acquisition of EMT phenotype in the gefitinib-resistant cells of a
head and neck squamous cell carcinoma cell line through
Akt/GSK-3β/snail signalling pathway. Br J Cancer. 106:1196–1204.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Stahtea XN, Roussidis AE, Kanakis I,
Tzanakakis GN, Chalkiadakis G, Mavroudis D, Kletsas D and Karamanos
NK: Imatinib inhibits colorectal cancer cell growth and suppresses
stromal-induced growth stimulation, MT1-MMP expression and pro-MMP2
activation. Int J Cancer. 121:2808–2814. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Hirai H, Sootome H, Nakatsuru Y, Miyama K,
Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, et al:
MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy
by standard chemotherapeutic agents or molecular targeted drugs in
vitro and in vivo. Mol Cancer Ther. 9:1956–1967. 2010. View Article : Google Scholar : PubMed/NCBI
|