Introduction
Hepatocellular carcinoma (HCC) is one of the most
common and life-threatening types of human malignant tumor
(1). The inadequate effects of
conventional chemotherapy and resistance to drugs present a
challenge to HCC treatment. In particular, advanced HCC cells
respond poorly to the induction of apoptosis by chemotherapeutic
agents due to reprogramming of cellular apoptotic machinery
(2). Thus, there is a requirement
to examine potential targets of the cellular apoptotic machinery to
develop novel and potent therapeutic drugs for the treatment of
HCC.
Tumor necrosis factor-related apoptosis-induced
ligand (TRAIL), a type II transmembrane protein from the tumor
necrosis factor family, triggers the extrinsic pathway of apoptosis
by binding to the cell surface receptors death receptor (DR) 4 and
5 (3). These receptors then form
homomeric and heteromeric complexes and stimulate the recruitment
of Fas-associated death domain (FADD) and caspase-8, which
self-activate and initiate downstream caspase cleavage events
(4). TRAIL is promising as a novel
therapeutic agent due to its high potential for the selective
induction of apoptosis (5).
However, several studies have indicated that HCC cells are
relatively refractory to TRAIL and that TRAIL alone is unable to
induce apoptotic cell death in these cells (6). A number of factors may be responsible
for the resistance of HCC cells to TRAIL. TRAIL-induced apoptosis
is mediated by caspase-8 (7) and
the resistance of HCC cells to TRAIL is correlated with a
downregulation in the activity of caspase-8, which disturbs
apoptotic signals in the cancer cells (8). Notably, the activity of caspase-8 is
inhibited by its inhibitory protein, the cellular FLICE-like
inhibitory protein (c-FLIP) in chemoresistant cells (9). c-FLIP is expressed at higher levels
in TRAIL-resistant HCC cells compared with TRAIL-sensitive cells
(10). Furthermore, the
downregulation of c-FLIP renders highly resistant HCC cells
sensitive to TRAIL treatment (11). Therefore, novel therapeutic
strategies to eliminate the c-FLIP-mediated chemoresistance of HCC
cells are required, and the identification of novel therapeutic
compounds with anti-HCC activity, particularly those derived from
naturally occurring materials, is necessary.
Rocaglamide is isolated from the genus Aglaia
(family Meliaceae) (12). A number
of species from this genus are used in traditional medicine to
treat coughs, injuries, asthma and inflammatory skin diseases.
Rocaglamide has also been observed to possess anticancer properties
in leukemia (13,14). However, the detailed mechanisms
underlying the anticancer activities of rocaglamide in solid tumors
remain to be elucidated.
The aim of the present study was to investigate
whether rocaglamide sensitized resistant HCC cells to TRAIL-induced
death, by regulation of caspase-8/c-FLIP in vitro.
Furthermore, the efficacy of rocaglamide in TRAIL-resistant
Huh-7-derived tumor xenografts was determined.
Materials and methods
Cell culture and reagents
HepG2 and Huh-7 cells were obtained from the
Shanghai Cell Collection (Shanghai, China) and cultured in
Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL, Carlsbad, CA,
USA) supplemented with 10% fetal bovine serum (FBS; Gibco-BRL), 2
mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. The
cells were cultured at 37°C in 5% CO2. Rocaglamide
(>98% pure) was procured from Enzo Life Sciences (Lörrach,
Germany). TRAIL was purchased from PeproTech, Inc. (Rocky Hill, NJ,
USA) and all chemicals were purchased from Sigma (St. Louis, MO,
USA), unless indicated otherwise.
Treatment of cells with rocaglamide
and/or TRAIL
For the investigation of time-dependence, the cells
(70% confluent) were treated with rocaglamide (100 nM) and/or TRAIL
(100 ng/ml) for different time periods (0–24 h). The cells were
then harvested for cell viability analysis. For the investigation
of dose-dependence, the cells (70% confluent) were pretreated with
rocaglamide (0–100 nM) for 12 h in DMEM, supplemented with 10% FBS,
2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin, and
were then treated with TRAIL (0–100 ng/ml) for an additional 12 h.
To investigate treatment with a combination of rocaglamide and
TRAIL, the cells were pretreated with either rocaglamide or
dimethyl sulfoxide (DMSO) for 12 h, followed by 12 h incubation in
the presence of TRAIL. Subsequently, 24 h after rocaglamide or DMSO
treatment, the cells were harvested and the cell lysates were
prepared, according to previous methods (15) and stored at -80°C for later
use.
Cell viability assay
HepG2 and Huh-7 cells (1×104/well) were
seeded in 96-well plates in complete culture medium and incubated
for 24 h. The cells were then exposed to 100 nM rocaglamide and/or
100 ng/ml TRAIL for 24 h. The control cells were treated with DMSO
at a concentration equal to that used for the drug-treated cells.
The complete culture medium was then removed and MTT (200 μl, 0.5
mg/ml in 10% FBS-containing DMEM) was added to each well and the
plate was incubated for 2 h at 37°C in a humidified incubator. The
solution was then removed from the wells and 200 μl DMSO was added
to each well prior to agitation. The absorbance at 570 nm was read
using a microplate reader (Bio-Tek ELx800; BioTek Instruments Inc.,
Winooski, VT, USA). The value for the vehicle-treated cells was
considered to indicate 100% viability. Furthermore, a crystal
violet assay was carried out. Briefly, the cells
(1.0×105/ml) were seeded in a 12-well plate for 12 h,
and treated with TRAIL (0–100 ng/ml) and/or RocA(1–100 nM) for 12
h. The treated cells were washed with phosphate-buffered saline
(PBS), fixed with 4% paraformaldehyde for 15 min, and stained using
crystal violet (Sigma; cat no. C3886) for a further 30 min.
Western blot analysis
Western blot analysis was performed, as described
previously (7) using the
following: mouse anti-c-FLIP monoclonal antibody (NF6; cat.no
ALX-804-428; Alexis Biochemicals, San Diego, CA, USA), mouse
anti-caspase-8 monoclonal antibody (cat.no 9746; Cell Signaling
Technology, Inc. Danvers, MA, USA), mouse anti-DR4 monoclonal
antibody (cat.no ab13890; Abcam, Cambridge, UK), rabbit anti-DR5
polyclonal antibody (cat.no ab47179; Abcam), Pro-Apoptosis Bcl-2
Family Antibody Sampler kit (cat.no 9942; Cell Signaling
Technology, Inc.) and Apoptosis Western Blot Cocktail (cat.no
ab136812; Abcam). An enhanced chemiluminescence detection reagent
(SuperSignal West Pico) was used for detection (Pierce
Biotechnologies, Rockford, IL, USA) and α-tubulin and β-actin were
used as loading controls. All western blots were representative of
at least three independent experiments.
Transfection of c-FLIP
Small interfering (si)RNA control or siRNA FLIP
(Santa Cruz Biotechnology) of high purity were delivered into the
HepG2 cells in the 12-well plate using Lipofectamine 2000
(Invitrogen Life Technologies, Carlsbad, CA, USA) according to the
manufacturer’s instructions. Briefly, 2 μl Lipofectamine 2000 was
added to 50 nmol/l siRNA in a final volume of 100 μl culture medium
and the solution was added to the washed cells, which were then
incubated at 37°C for 4 h. The transfection mixture was then
removed and the cells were incubated with fresh complete medium for
an additional 36 h. Finally, the cells were exposed to TRAIL or
DMSO for 12 h for subsequent experiments.
Detection of apoptosis and necrosis using
flow cytometry
The HepG2 and Huh-7 cells were seeded and cultured
in each well of a 12-well plate for 24 h and then pretreated with
either rocaglamide or DMSO for 12 h, followed by 12 h incubation in
the presence of TRAIL. An annexin V/propidium iodide (PI) apoptosis
detection kit (eBioscience, Inc., San Diego, CA, USA) was used for
the detection of apoptotic and necrotic cells. The cells
(5×105) were harvested and resuspended in 400 μl 1×
binding buffer with 4 μl FITC annexin V and 4 μl PI. Flow
cytometric analysis was then performed using a Becton-Dickinson
FACSCalibur™ system; BD Biosciences, Franklin Lakes, NJ, USA) and
FlowJo 7.6.3 software (Tree Star, Inc., Ashland, OR, USA).
Tumorigenicity studies in severe combined
immunodeficient (SCID) mice
The Huh-7 cells (3×106), suspended in 100
μl mix (equal volumes of DMEM and Matrigel), were implanted
subcutaneously into the right flank of 10 female SCID mice
(6-week-old) and then randomly divided into two equal groups, one
of which received an intraperitoneal injection of rocaglamide (2.5
mg/kg in 80 μl olive oil; n=5) and the other, used as a vehicle
control, received olive oil alone (n=5). These treatments were
performed once daily for 32 days and the tumor volumes and body
weights of the animals were measured twice a week. The tumor
volumes (mm3) were calculated using the following
formula: Tumor volume = LS2 / 2, where L is the longest
diameter and S is the shortest. At the end of the experiments, the
mice were sacrificed and tumor samples were harvested, fixed in
formalin and embedded in paraffin as tissue sections for
immunohistochemical analysis. The animal experiments were performed
in accordance with the relevant institutional and national
regulations and the research procedures were approved by Tongji
Medical College (Wuhan, China). The SCID mice were purchased from
Beijing HFK Bioscience, Co., Ltd. (Beijing, China).
Immunohistochemistry and terminal
deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL)
staining
Immunohistochemical staining was performed using
either 1:1 hematoxylin and 0.5% eosin (H&E) or an anti-cleaved
caspase-3 antibody (Cell Signaling Technology, Inc.), as previously
described (16). Apoptosis was
assessed by TUNEL (In situ Cell Death Detection kit; Roche
Diagnostics, Basel, Switzerland) according to the manufacturer’s
instructions. The primary antibody was omitted in the first step to
establish a negative control, which yielded negative results. The
percentage of positive cells was independently determined by two
examiners. At least five random fields of each section were
visualized (magnification ×400) and analyzed using a Nikon OPTIPHOT
150 microscope (Nikon, Tokyo, Japan) connected to a SPOT Insight
charge-coupled device camera (Diagnostic Instruments, Inc.,
Sterling Heights, MI, USA). The values are expressed as the mean ±
standard deviation (SD).
Statistical analysis
Where indicated, the data are expressed as the mean
± SD from at least three independent experiments. Statistical
analysis of the data was conducted using Student’s t-test.
P<0.01 was considered to indicate a statistically significant
difference.
Results
Rocaglamide enhances TRAIL-induced
apoptosis in resistant HCC cells
To investigate whether rocaglamide enhanced
TRAIL-mediated apoptosis in HCC cells in the present study, HepG2
and Huh-7 cells, which are highly chemoresistant to TRAIL (17), were selected. The effect of
rocaglamide on TRAIL-induced cytotoxicity was then examined using
an MTT assay. Treatment with rocaglamide or TRAIL alone was
minimally cytotoxic to the HepG2 and Huh-7 cells; however,
pretreatment of the cells with rocaglamide enhanced the cytotoxic
effect of TRAIL in a time-dependent manner in the two cell lines
(Fig. 1B). Furthermore, the effect
of rocaglamide on TRAIL-induced cell death was demonstrated by
observing the morphological signs of apoptosis. Although
rocaglamide and TRAIL alone did not induce morphological signs of
cell death, rocaglamide markedly enhanced the effect of
TRAIL-induced apoptosis (Fig. 1C).
Cell apoptosis was also confirmed using annexin V/PI staining and
flow cytometry in the HepG2 and Huh-7 cells. Treatment with
rocaglamide alone led to apoptosis in ~9% HepG2 and 11% Huh-7 cells
and treatment with TRAIL induced apoptosis in ~16% HepG2 and 17%
Huh-7 cells. However, the combination of rocaglamide and TRAIL
induced apoptosis in ~55% HepG2 and 57% Huh-7 cells (Fig. 1D and E), which is evidently more
than an additive effect. A similar result was obtained by
measurement of cell viability using crystal violet staining
(Fig. 1F). Taken together, the
data from the present study indicate that rocaglamide has the
potential to sensitize highly chemoresistant HepG2 and Huh-7 cells
to TRAIL-based therapy.
Rocaglamide promotes TRAIL-induced
caspase-dependent apoptotic cell death
TRAIL-induced apoptosis is mediated by activation of
the caspase cascade (18). In
particular, the cleavage of caspase-8 is an essential step in the
TRAIL-mediated caspase activation cascade (19). Therefore, the present study
investigated whether the cleavage of caspase-8 was triggered in
TRAIL-resistant cell lines following treatment with rocaglamide or
TRAIL alone. The results revealed that modest reductions in the
level of the procaspase-8 protein occurred in the
rocaglamide-treated and TRAIL-treated HepG2 and Huh-7 cells. An
increase in the level of active-caspase-8 was also observed in
these cells (Fig. 2A; lanes 2 and
3 vs. lane 1). However, combined treatment with rocaglamide and
TRAIL significantly augmented the TRAIL-induced cleavage/activation
of caspases-8 (Fig. 2A; lane 4 vs.
lanes 1, 2 and 3). Notably, treatment with rocaglamide alone did
not affect the expression levels of pro-caspase-3 compared with the
control (Fig. 2B; lane 2 vs. lane
1). Furthermore, treatment with TRAIL alone resulted in a small
reduction in the level of pro-caspase-3 and a small increase in the
cleavage of poly ADP ribose polymerase (PARP) and activated
caspase-3 substrates in the HepG2 and Huh-7 cells (Fig. 2B; lane 3 vs. lanes 1 and 2).
However, combined treatment with rocaglamide and TRAIL resulted in
significantly increased activity of the TRAIL-induced
cleavage/activation of pro-caspase-3 and cleavage of PARP (Fig. 2B; lane 4 vs. lanes 1, 2 and 3),
indicating that combined treatment induced apoptotic death in the
hepG2 and Huh-7 cells, at least partly through a caspase-dependent
pathway. Thus, these results clearly suggest that rocaglamide
sensitizes TRAIL-resistant HCC cells to TRAIL-induced cell death
through the enhancement of caspase activity.
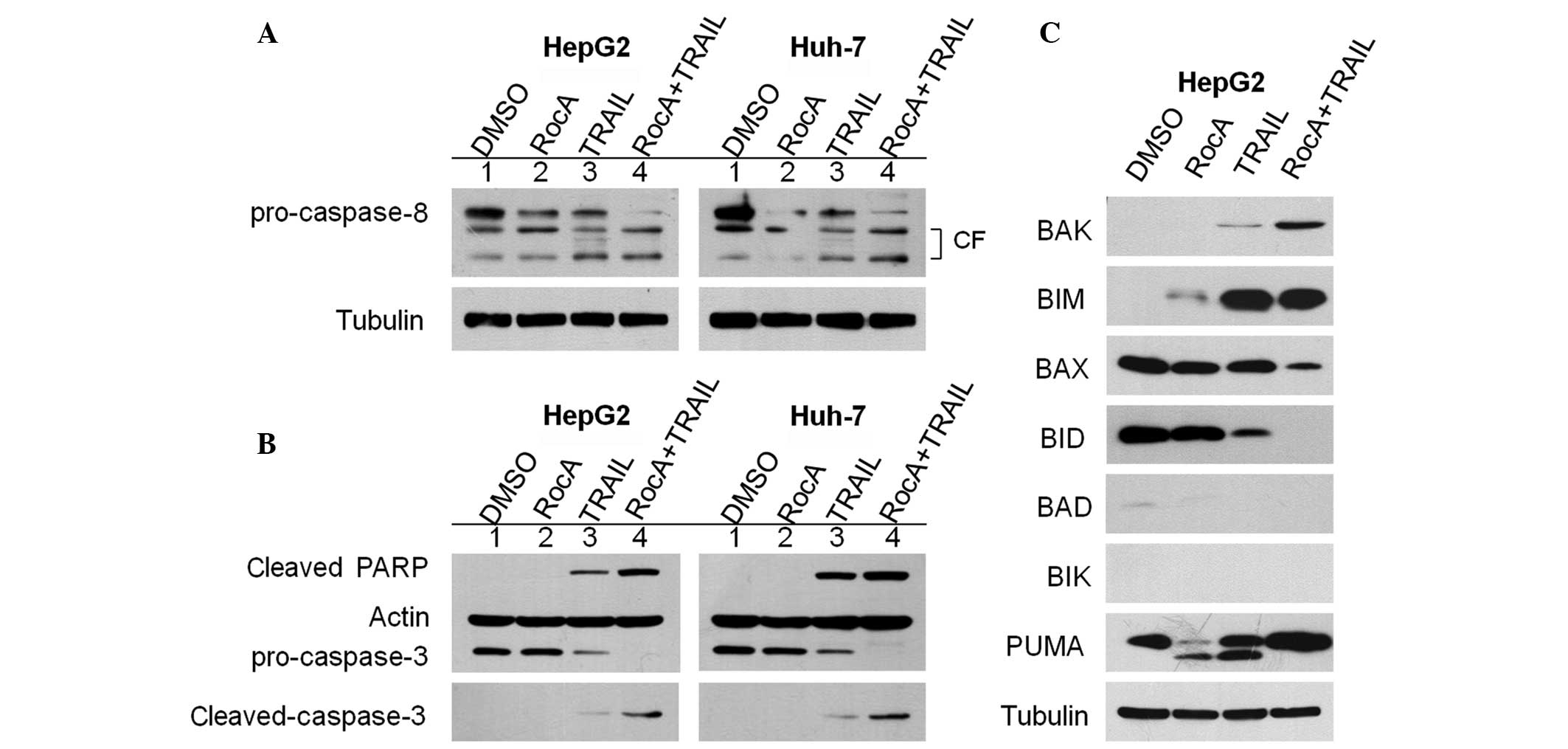 | Figure 2RocA enhances caspase activity
triggered by TRAIL. (A and B) HepG2 and Huh-7 cells were pretreated
with RocA (100 nM) for 12 h and then exposed to TRAIL (100 ng/ml)
for 12 h. Cell lysates were subjected to western blot analysis
using the antibodies indicated. (C) HepG2 cells were pretreated
with RocA (100 nM) for 12 h and TRAIL (100 ng/ml) for 12 h. The
cells were lysed and western blot analysis was performed using the
antibodies indicated. The control contained cells treated with DMSO
only. Results are representative of three independent experiments.
RocA, rocaglamide; CF, cleaved (active) form; TRAIL, tumor necrosis
factor-related apoptosis-inducing ligand; DMSO, dimethyl sulfoxide;
PARP, poly ADP ribose polymerase; BAK, Bcl-2 homologous antagonist
killer; BIM, Bcl-2-interacting mediator of cell death; BAX,
Bcl-2-associated X protein; BID, BH3-interacting domain death
agonist; BAD, Bcl-2-associated death promoter; BIK,
Bcl-2-interacting killer; PUMA, p53-upregulated modulator of
apoptosis. |
As pro-apoptotic B-cell lymphoma 2 (Bcl-2) family
proteins, including BH3-interacting domain death agonist (BID),
Bcl-2-associated X protein (BAX) and p53-upregulated modulator of
apoptosis (PUMA), are able to sensitize cancer cells to
TRAIL-induced apoptosis (20), the
present study investigated the expression levels of these proteins.
In the HepG2 and Huh-7 cells treated with the rocaglamide/TRAIL
combination, the protein levels of pro-apoptotic Bcl-2-interacting
mediator of cell death (BIM), Bcl-2 homologous antagonist killer
(BAK) and PUMA were significantly increased, whereas the protein
levels of other pro-apoptotic proteins, including BID and BAX were
reduced (Fig. 2C), indicating the
possible inhibition of protein synthesis by rocaglamide (21). Therefore, these results suggest
that the upregulation of the BIM, BAK and PUMA proteins is
associated with the rocaglamide-mediated sensitization of HepG2 and
Huh-7 cells to TRAIL-induced apoptosis. Overall, these results
indicate that rocaglamide substantially increases the apoptotic
potential of TRAIL in HCC cells through extrinsic and intrinsic
pathways.
Rocaglamide sensitizes TRAIL-induced
apoptosis via c-FLIP downregulation
c-FLIP, which is highly homologous to caspase-8 but
is catalytically inactive, is able to bind to caspase-8 and the
FADD and inhibit TRAIL-induced apoptosis, thus disrupting the
death-inducing signaling complex (DISC) (22). Notably, HCC tumors exhibit high
resistance to TRAIL due to the overexpression of c-FLIP (23). Following the observation that
rocaglamide is able to overcome TRAIL-resistance in the HCC cells
and activate caspase-8, the present study investigated whether this
effect correlated with c-FLIP. Therefore, the effect of rocaglamide
on the levels of c-FLIP, which is highly expressed in HCC cells,
was examined (Fig. 3A). Notably,
the resulting data also suggest that rocaglamide treatment resulted
in downregulation of the c-FLIP protein in the HepG2 and Huh-7
cells (Fig. 3A). The resulting
effect of rocaglamide on the levels of c-FLIP was consistent with
the effects of rocaglamide on caspase-dependent apoptosis.
TRAIL triggers the extrinsic pathway of apoptosis by
binding to cognate DR4 or DR5 (3),
which results in the formation of receptor homotrimers and the
propagation of a proapoptotic signal through caspase-8, which then
self-activates and initiates downstream caspase events (24). In addition, the levels of DR4 or
DR5 correlate with the sensitivity of TRAIL-mediated cell death
(25). Therefore, the present
study investigated the effect of rocaglamide on DR4 and DR5. No
significant changes in the levels of DR4 or DR5 were observed in
the rocaglamide-treated HepG2 and Huh-7 cells (Fig. 3B).
Knockdown of c-FLIP mimics the effect of
rocaglamide in overcoming TRAIL-resistance in HepG2 cells
In order to determine the effect of downregulation
of c-FLIP on the rocaglamide-induced effect, the present study also
examined whether the apoptosis-inducing effects of rocaglamide were
also exerted in c-FLIP-knockdown HepG2 cells. siRNA was used to
specifically downregulate c-FLIP and an siRNA vector served as a
control. This treatment resulted in a marked suppression in the
levels of c-FLIP of ~70% (Fig.
4A). The cell viability, evaluated by crystal violet staining,
indicated that the c-FLIP-silenced HepG2 cells exhibited an
increased sensitivity to TRAIL treatment compared with the that of
the control (Fig 4B). In addition,
the morphological signs of cell death were significantly increased
following treatment with TRAIL alone in the c-FLIP-silenced HepG2
cells (Fig. 4C). Furthermore,
apoptosis was analyzed using annexin V/PI staining by flow
cytometry in the c-FLIP-knockdown HepG2 cells. The results revealed
that the induced rate of apoptosis was ~8% by siRNA-FLIP, 15% by
TRAIL and 56% by siRNA-FLIP combined with TRAIL (Fig. 4D and E), suggesting that the
downregulation of c-FLIP in the HepG2 cells mimicked the effect of
rocaglamide on TRAIL-mediated apoptosis in the TRAIL-resistant HCC
cells.
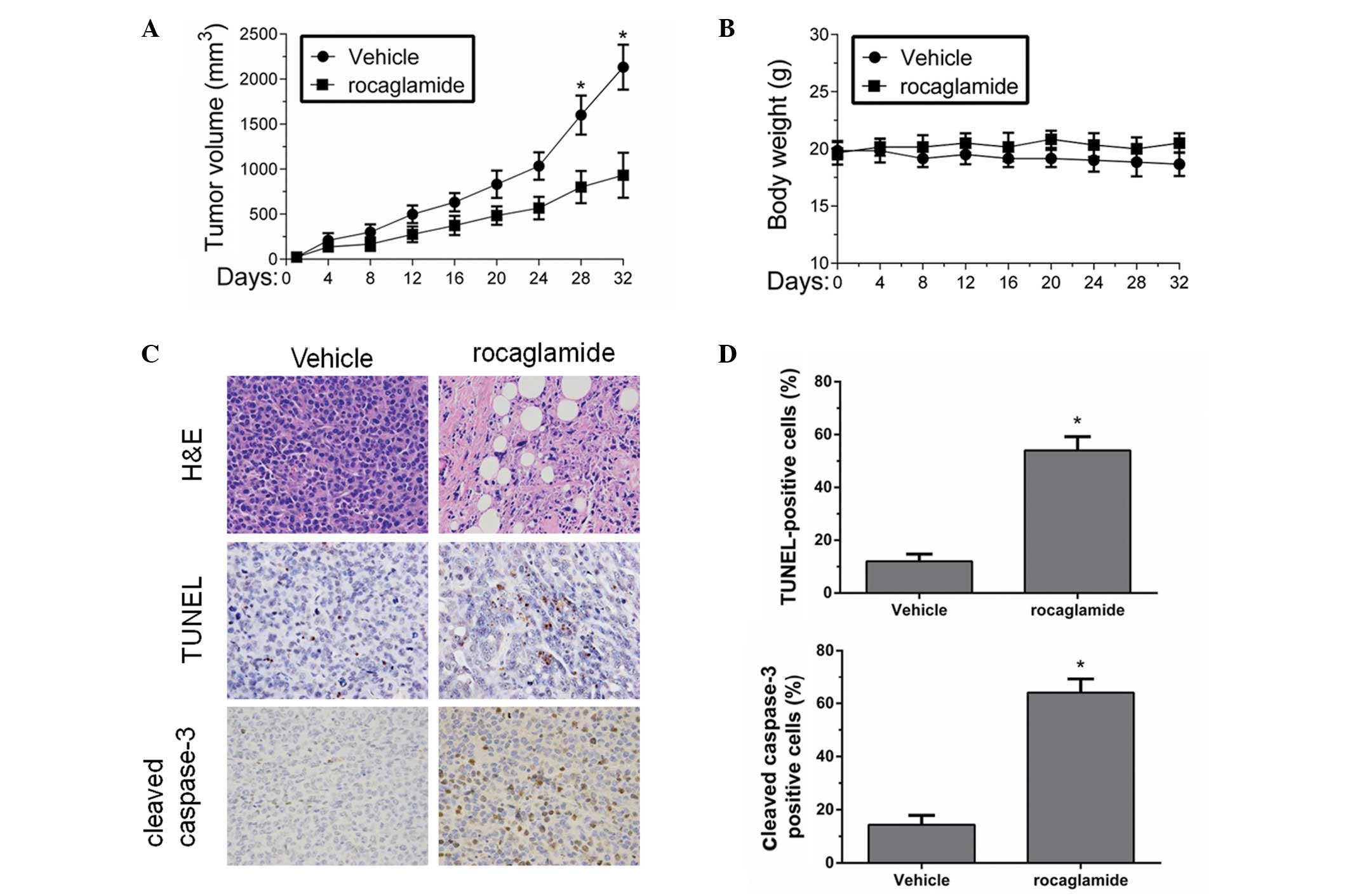
Rocaglamide induces apoptosis of tumor
cells in SCID mice models
To confirm whether the synergistic effect of
rocaglamide and TRAIL in resistant cell lines had potentially
relevant clinical implications, the present study investigated the
in vivo effect of rocaglamide and TRAIL on the growth of HCC
xenograft tumors. As natural killer cells produce TRAIL in
vivo (26,27), rocaglamide alone was administered
in the in vivo study. The Huh-7 cells were subcutaneously
injected into the right flanks of SCID mice and, when tumors were
visible, the mice were matched for tumor volumes and were assigned
to a control group and a rocaglamide-treated group. Tumor volumes
in the rocaglamide-treated group were ~45±12% compared with the
control group (Fig. 5A).
Rocaglamide significantly suppressed tumor growth compared with
that in the control group. Notably, treatment with rocaglamide did
not lead to any reduction in body weight and no apparent signs of
toxicity were observed in the mice during the treatment (Fig. 5B), suggesting that rocaglamide is
generally tolerated well in vivo.
The present study further investigated the effect of
rocaglamide on apoptosis in vivo by examining tumor tissues
harvested from the control and rocaglamide-treated mice using
H&E, TUNEL and cleaved caspase-3 staining. In the untreated
controls, staining with H&E revealed a compact mass of
epithelial cells, whereas following rocaglamide treatment, the
tumor appearance was that of loose epithelial cell aggregates with
increased interspersed mesenchymal cells (Fig. 5C, top panels). In addition, the
TUNEL assays demonstrated a three-fold increase in the percentage
of apoptotic cells in the rocaglamide treatment group compared with
the untreated controls (Fig. 5C,
middle panels and D, upper panel). Furthermore, the cleaved
caspase-3 staining confirmed a five-fold increase in apoptosis in
the tumor sections from the group treated with rocaglamide,
relative to the untreated control (Fig. 5C, bottom panels and D, lower
panel). Therefore, the in vivo investigation suggests that
rocaglamide is an effective drug, which has the potential to
inhibit the growth of HCC cell-derived tumors in vivo.
Discussion
HCC is a significant cause of mortality worldwide
due to its poor prognosis (28).
The ability to evade apoptosis is a characteristic property of HCC
cells, which results mainly from the lack of response to apoptotic
stimuli (29). HCC cells have
acquired drug resistance to cell death and are frequently
refractory to classical chemotherapy (30). Thus, new strategies are required to
address the resistance of HCC to apoptosis in order to improve the
poor prognosis. Considerable attention has been directed towards
investigating the effect of triggering apoptosis in HCC cells using
natural products that stimulate DR-mediated apoptosis (31). In the present study, rocaglamide, a
naturally occurring product, was demonstrated to sensitize
TRAIL-resistant HCC cells to apoptosis through the suppression of
c-FLIP in both in vitro and in vivo conditions.
As the type of cancer cell in HCC may affect the
response of the cell to therapeutic agents, the robustly
TRAIL-resistant HCC cell lines, HepG2 and Huh-7, were selected
(16). Furthermore, the effect of
rocaglamide on HepG2 and Huh-7 cells was examined, as was their
sensitivity to TRAIL. Notably, the HepG2 and Huh-7 cells, were
minimally responsive to treatment with TRAIL alone, indicating
complete resistance to TRAIL. Treatment of the TRAIL-resistant
HepG2 and Huh-7 cells with rocaglamide resulted in dose-dependent
growth inhibition. Thus, these data provide evidence that
rocaglamide has the potential to inhibit the proliferation of HCC
cells and lead to their elimination.
TRAIL-based therapy offers promising therapeutic
potential due to its specificity for cancer cells without evident
adverse effects on normal cells (5,32).
However, resistance to TRAIL-mediated therapy has been observed in
HCC cells, which indicates that treatment with TRAIL alone may be
ineffective in treating HCC. A noteworthy observation from the
present study is that the HepG2 and Huh-7 cells, which are highly
TRAIL-resistant, increased in sensitivity to TRAIL following
pretreatment with rocaglamide, supporting the possibility of
investigating rocaglamide as an adjuvant to combination therapy for
HCC patients. In addition, rocaglamide triggered the process of
apoptosis in the presence of TRAIL at nanomolar concentrations,
suggesting the efficacy of rocaglamide may be potent and
non-toxic.
The detailed mechanisms of resistance to TRAIL in
HCC cells remain to be elucidated. The regulation of DISC-engaged
molecules, including c-FLIP and caspase-8, has been observed to
contribute to the sensitivity of TRAIL-mediated apoptosis in cancer
cells (9,33,34).
In addition, TRAIL resistance correlates with accelerated
degradation of caspase-8 protein in cancer cells (8,35).
The present study demonstrated that rocaglamide significantly
activates caspase-8 in HCC cells. Furthermore, the
rocaglamide-induced caspase-8 activation was found to be
accompanied by increased cleavage of the apoptotic marker for
caspase-3, PARP, in vitro. These findings are consistent
with previous studies indicating that the activation of caspanse-8
induces apoptosis and sensitizes cancer cells to TRAIL (36,37).
c-FLIP inhibits the apoptotic signaling cascade by
preventing the recruitment and activation of caspase-8 at the DISC
(38), demonstrating that an
elevated intracellular level of c-FLIP confers resistance against
proapoptotic stimuli in tumor cells (39). In addition, ectopic expression of
c-FLIP inhibits the release of active caspase-8 fragments from the
DISC, resulting in disruption of the DISC complex (40). By contrast, downregulation of
c-FLIP results in the sensitization of chemoresistant tumor cells
(41). Collectively, these
findings suggest that c-FLIP may be a novel therapeutic drug target
for HCC. The present study provides evidence indicating that
treatment with rocaglamide significantly reduces the protein
expression level of c-FLIP in HepG2 and Huh-7 cells at nanomolar
concentrations. Furthermore, combined treatment with rocaglamide
and TRAIL significantly induced apoptosis, suggesting that
rocaglamide induces sensitivity to TRAIL by the suppression of
c-FLIP in HepG2 cells. Notably, siRNA was used to downregulate
c-FLIP in the HepG2 cells and revealed that the downregulation of
c-FLIP in HepG2 cells mimicked the effect of rocaglamide on
TRAIL-mediated apoptosis in TRAIL-resistant HCC cells.
Targeting the transcriptional activation of c-FLIP
is considered to be a promising approach for the downregulation of
c-FLIP expression in cancer cells (42). However, agents directly inhibiting
FLIP at the mRNA and protein levels remain to be elucidated.
Subsequent studies are planned to determine whether rocaglamide
treatment attenuates the transcriptional activation of c-FLIP are
required, and to examine whether the effect of rocaglamide on
c-FLIP occurs at the transcriptional level. Consistent with
previous studies, the results of the present study suggest that the
downregulation of the transcriptional activation of c-FLIP
sensitizes HCC cells to TRAIL. However, the detailed mechanisms
involved in regulating the expression of c-FLIP remain to be
elucidated.
The present study demonstrated that rocaglamide, a
naturally occurring product, sensitized chemoresistant HCC cells to
TRAIL-mediated apoptosis by decreasing the expression of c-FLIP and
activating caspase-8 in vitro. Furthermore, rocaglamide
markedly inhibited the growth of tumors derived from Huh-7 cells
in vivo in a xenograft mice model. Thus, these findings
provide significant evidence for the development of rocaglamide as
a novel therapeutic agent for use as an adjuvant to TRAIL in the
treatment of HCC.
Acknowledgements
The present study wa supported by the National
Natural Science Foundation of China (grant nos. 81370581, 81000290,
30972796).
References
|
1
|
Arzumanyan A, Reis HM and Feitelson MA:
Pathogenic mechanisms in HBV- and HCV-associated hepathocellular
carcinoma. Nat Rev Cancer. 13:123–135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang K and Lin B: Inhibitor of apoptosis
proteins (IAPs) as regulatory factors of hepatic apoptosis. Cell
Signal. 25:1970–1980. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kischkel FC, Lawrence DA, Chuntharapai A,
Schow P, Kim KJ and Ashkenazi A: Apo2L/TRAIL-dependent recruitment
of endogenous FADD and caspase-8 to death receptors 4 and 5.
Immunity. 12:611–620. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reed JC: Apoptosis-targeted therapies for
cancer. Cancer Cell. 3:17–22. 2003. View Article : Google Scholar
|
|
5
|
Hall MA and Cleveland JL: Clearing the
TRAIL for cancer therapy. Cancer Cell. 12:4–6. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Herr I, Schemmer P and Büchler MW: On the
TRAIL to therapeutic intervention in liver disease. Hepatology.
46:266–274. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dickens LS, Boyd RS, Jukes-Jones R, Hughes
MA, Robinson GL, Fairall L, Schwabe JW, Cain K and Macfarlane M: A
death effector domain chain DISC model reveals a crucial role for
caspase-8 chain assembly in mediating apoptotic cell death. Mol
Cell. 47:291–305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang L, Zhu H, Teraishi F, Davis JJ, Guo
W, Fan Z and Fang B: Accelerated degradation of caspase-8 protein
correlates with TRAIL resistance in a DLD1 human colon cancer line.
Neoplasia. 7:594–602. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haag C, Stadel D, Zhou S, Bachem MG,
Möller P, Debatin KM and Fulda S: Identification of c-FLIP(L) and
c-FLIP(S) as critical regulators of death receptor-induced
apoptosis in pancreatic cancer cells. Gut. 60:225–237. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Du X, Bao G, He X, Zhao H, Yu F, Qiao Q,
Lu J and Ma Q: Expression and biological significance of c-FLIP in
human hepatocellular carcinoma. J Exp Clin Cancer Res. 28:242009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lai LJ and Ho TC: Pigment
epithelial-derived factor inhibits c-FLIP expression and assists
ciglitazone-induced apoptosis in hepatocellular carcinoma.
Anticancer Res. 31:1173–1180. 2011.PubMed/NCBI
|
|
12
|
Kim S, Salim AA, Swanson SM and Kinghorn
AD: Potential of cyclopenta[b]benzofurans from Aglaia
species in cancer chemotherapy. Anticancer Agents Med Chem.
6:319–345. 2006.
|
|
13
|
Lucas DM, Edwards RB, Lozanski G, West DA,
Shin JD, Vargo MA, Davis ME, Rozewski DM, Johnson AJ, Su BN, et al:
The novel plant-derived agent silvestrol has B-cell selective
activity in chronic lymphocytic leukemia and acute lymphoblastic
leukemia in vitro and in vivo. Blood. 113:4656–4666. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Giaisi M, Köhler R, Fulda S, Krammer PH
and Li-Weber M: Rocaglamide and a XIAP inhibitor cooperatively
sensitize TRAIL-mediated apoptosis in Hodgkin’s lymphomas. Int J
Cancer. 131:1003–1008. 2012.PubMed/NCBI
|
|
15
|
Luan Z, He Y, Alattar M, Chen Z and He F:
Targeting the prohibitin scaffold-CRAF kinase interaction in
RAS-ERK-driven pancreatic ductal adenocarcinoma. Mol Cancer.
13:382014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kunzi-Rapp K, Genze F, Küfer R, Reich E,
Hautmann RE and Gschwend JE: Chorioallantoic membrane assay:
vascularized 3-dimensional cell culture system for human prostate
cancer cells as an animal substitute model. J Urol. 166:1502–1507.
2001. View Article : Google Scholar
|
|
17
|
Chen Q, Lou W, Shen J, Ma L, Yang Z, Liu
L, Luo J and Qian C: Potent antitumor activity in experimental
hepatocellular carcinoma by adenovirus-mediated coexpression of
TRAIL and shRNA against COX-2. Clin Cancer Res. 16:3696–3705. 2010.
View Article : Google Scholar
|
|
18
|
Jin Z, Li Y, Pitti R, Lawrence D, Pham VC,
Lill JR and Ashkenazi A: Cullin3-based polyubiquitination and
p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis
signaling. Cell. 137:721–735. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Häcker S, Dittrich A, Mohr A, Schweitzer
T, Rutkowski S, Krauss J, Debatin KM and Fulda S: Histone
deacetylase inhibitors cooperate with IFN-gamma to restore
caspase-8 expression and overcome TRAIL resistance in cancers with
silencing of caspase-8. Oncogene. 28:3097–3110. 2009.PubMed/NCBI
|
|
20
|
Unterkircher T, Cristofanon S, Vellanki
SH, Nonnenmacher L, Karpel-Massler G, Wirtz CR, Debatin KM and
Fulda S: Bortezomib primes glioblastoma, including glioblastoma
stem cells, for TRAIL by increasing tBid stability and
mitochondrial apoptosis. Clin Cancer Res. 17:4019–4030. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sadlish H, Galicia-Vazquez G, Paris CG,
Aust T, Bhullar B, Chang L, Helliwell SB, Hoepfner D, Knapp B,
Riedl R, Roggo S, Schuierer S, Studer C, Porco JA Jr, Pelletier J
and Movva NR: Evidence for a functionally relevant rocaglamide
binding site on the eIF4A-RNA complex. ACS Chem Biol. 8:1519–1527.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Irmler M, Thome M, Hahne M, Schneider P,
Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C,
Rimoldi D, French LE and Tschopp J: Inhibition of death receptors
signals by cellular FLIP. Nature. 338:190–195. 1997.
|
|
23
|
Kim JY, Kim EH, Park SS, Lim JH, Kwon TK
and Choi KS: Quercetin sensitizes human hepatoma cells to
TRAIL-induced apoptosis via Sp1-mediated DR5 up-regulation and
proteasome-mediated c-FLIPS down-regulation. J Cell Biochem.
105:1386–1398. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bao Q and Shi Y: Apoptosome: a platform
for the activation of initiator caspases. Cell Death Differ.
14:56–65. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang G and Wang X, Yu H, Wei S, Williams
N, Holmes DL, Halfmann R, Naidoo J, Wang L, Li L, Chen S, Harran P,
Lei X and Wang X: Small-molecule activation of the TRAIL receptor
DR5 in human cancer cells. Nat Chem Biol. 9:84–89. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Smyth MJ, Cretney E, Takeda K, Wiltrout
RH, Sedger LM, Kayagaki N, Yagita H and Okumura K: Tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL) contributes to
interferon gamma-dependent natural killer cell protection from
tumor metastasis. J Exp Med. 193:661–670. 2001. View Article : Google Scholar
|
|
27
|
Hayakawa Y, Screpanti V, Yagita H,
Grandien A, Ljunggren HG, Smyth MJ and Chambers BJ: NK cell TRAIL
eliminates immature dendritic cells in vivo and limits dendritic
cell vaccination efficacy. J Immunol. 172:123–129. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Villanueva A, Hernandez-Gea V and Llovet
JM: Medical therapies for hepatocellular carcinoma: a critical view
of the evidence. Nat Rev Gastroenterol Hepatol. 10:34–42. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nojiri K, Sugimoto K, Shiraki K, Tameda M,
Inagaki Y, Ogura S, Kasai C, Kusagawa S, Yoneda M, Yamamoto N,
Takei Y, Nobori T and Ito M: Sorafenib and TRAIL have synergistic
effect on hepatocellular carcinoma. Int J Oncol. 42:101–108.
2013.PubMed/NCBI
|
|
30
|
Schattenberg JM, Schuchmann M and Galle
PR: Cell death and hepatocarcinogenesis: Dysregulation of apoptosis
signaling pathways. J Gastroenterol Hepatol. 26:213–219. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Z, Zhou X and Li J, Liu X, Chen Z,
Shen G, Guan T, Ye N, Wei X, Huang N, Yang L, Wei Y and Li J:
Suppression of hepatoma tumor growth by systemic administration of
the phytotoxin gelonin driven by the survivin promoter. Neoplasma.
60:469–479. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kelley SK and Ashkenazi A: Targeting death
receptors in cancer with Apo2L/TRAIL. Curr Opin Pharmacol.
4:333–339. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sayers TJ, Brooks AD, Koh CY, Ma W, Seki
N, Raziuddin A, Blazar BR, Zhang X, Elliott PJ and Murphy WJ: The
proteasome inhibitor PS-341 sensitizes neoplastic cells to
TRAIL-mediated apoptosis by reducing levels of c-FLIP. Blood.
102:303–310. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ganten TM, Haas TL, Sykora J, Stahl H,
Sprick MR, Fas SC, Krueger A, Weigand MA, Grosse-Wilde A, Stremmel
W, Krammer PH and Walczak H: Enhanced caspase-8 recruitment to and
activation at the DISC is critical for sensitization of human
hepatocellular carcinoma cells to TRAIL-induced apoptotic by
chemotherapeutic drugs. Cell Death Differ. 11(Suppl 1): S86–S96.
2004. View Article : Google Scholar
|
|
35
|
Qi L, Bellail AC, Rossi MR, Zhang Z, Pang
H, Hunter S, Cohen C, Moreno CS, Olson JJ, Li S and Hao C:
Heterogeneity of primary glioblastoma cells in the expression of
caspase-8 and the response to TRAIL-induced apoptosis. Apoptosis.
16:1150–1164. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kaminskyy VO, Surova OV, Vaculova A and
Zhivotovsky B: Combined inhibition of DNA methyltransferase and
histone deacetylase restores caspase-8 expression and sensitizes
SCLC cells to TRAIL. Carcinogenesis. 32:1450–1458. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu L, Yim H, Choi JH, Kim ST, Jin Y and
Lee SK: ATM kinase promotes both caspase-8 and caspase-9 activation
during TNF-α-induced apoptosis of HeLa cells. FEBS Lett.
588:929–935. 2014.PubMed/NCBI
|
|
38
|
Feoktistova M, Geserick P, Kellert B,
Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Häcker G
and Leverkus M: cIAPs block Ripoptosome formation, a RIP1/caspase-8
containing intracellular cell death complex differentially
regulated by cFLIP isoforms. Mol Cell. 43:449–463. 2011. View Article : Google Scholar
|
|
39
|
Safa AR, Day TW and Wu CH: Cellular
FLICE-like inhibitory protein (c-FLIP): a novel target for cancer
therapy. Curr Cancer Drug Targets. 8:37–46. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kataoka T: The caspase-8 modulator c-FLIP.
Crit Rev Immunol. 25:31–58. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cheung HH, Mahoney DJ, Lacasse EC and
Korneluk RG: Down-regulation of c-FLIP enhances death of cancer
cells by smac mimetic compound. Cancer Res. 69:7729–7738. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shirley S and Micheau O: Targeting c-FLIP
in cancer. Cancer Lett. 332:141–150. 2013. View Article : Google Scholar : PubMed/NCBI
|