Introduction
Liver fibrosis is the essential pathophysiological
consequence of chronic liver injury and is characterized by the
excessive accumulation of extracellular matrix (ECM) proteins,
particularly collagens (1). Liver
fibrosis has traditionally been regarded as an irreversible
process. However, mounting clinical evidence has indicated that
even advanced fibrosis is reversible (2). The activation of hepatic stellate
cells (HSCs), the primary source of the ECM, that is characterized
by the expression of α-smooth muscle actin (α-SMA), is the pivotal
process in liver fibrosis (1).
Therefore, the inhibition of the accumulation of activated HSCs by
modulating either their activation and/or proliferation or
promoting their apoptosis is one strategy to regress liver fibrosis
(1).
microRNAs (miRNAs) are a group of small,
evolutionarily conserved, non-coding, naturally occurring RNA
molecules, which post-transcriptionally modulate gene expression
and determine cell fate by regulating multiple gene products and
cellular pathways (3,4). Deregulation of miRNAs has been
consistently associated with a number of different human
malignancies, including diseases of the liver (5,6).
Several miRNAs have been identified that may be involved in the
process of liver fibrosis and hepatocellular carcinoma (7,8).
The miR-34 family was first identified as direct
transcriptional targets of p53, and is composed of miR-34a, miR34b
and miR34c (9). The miR-34 family
members target numerous genes, including cyclin-dependent kinases 1
and 4, B-cell lymphoma 2, cAMP-response element binding protein
(CREB), forkhead box protein P1 (Foxp1), to modulate the cell
cycle, differentiation, proliferation and apoptosis (10). Lower miR-34 expression in cancer
has been reported by several groups, suggesting its possible
involvement in oncogenesis as a tumor suppressor (4,11–13).
However, elevated miR-34 expression has been reported in activated
HSCs, in rats with induced hepatic fibrosis or liver tumors and in
patients with liver diseases (14–18).
It has been reported that miR-34 family members may be involved in
the process of liver fibrosis by targeting acyl-CoA synthetase
long-chain family member 1 (ACSL1) (15). However, whether other cellular
factors or proteins are involved remains elusive.
Peroxisome proliferator-activated receptor γ (PPARγ)
belongs to a superfamily of nuclear receptors controlling the
transcriptions of numerous different genes. PPARγ is an important
anti-fibrotic factor and is involved in the maintenance of HSCs in
a quiescent phenotype (19).
Several studies have reported that PPARγ antagonists or activators
impeded the HSCs activation during live fibrosis (20–22).
PPARγ is now considered to be a promising therapeutic target for
antifibrotic chemotherapy (23).
The present study, utilizing bioinformatics and a
reporter assay, investigated whether the PPARγ gene was a target
protein of miR-34a/c. Expression of PPARγ in activated or
miR-34a/c-silenced HSCs was assessed in human and rat cell models
in vitro, in order to elucidate whether the miR-34 family
promoted liver fibrosis by targeting PPARγ.
Materials and methods
Cells and reagents
The immortalized human HSC line LX-2 (Institute of
Cell and Molecule Biology, Central South University, Changsha,
China), a generous gift from Dr. Tan (24), was maintained in Dulbecco’s
modified Eagle’s medium (DMEM; Invitrogen Life Technologies,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS;
Gibco BRL, Grand Island, NY, USA), 100 μg/ml streptomycin (Amresco,
Solon, OH, USA) and 100 U/ml penicillin (Amresco) and incubated at
37°C with 5% CO2. Prior to the start of the experiments,
cells were tested by PCR for mycoplasma, using commercially
available primers, and were shown to be mycoplasma-negative. The
cells were activated by administration of transforming growth
factor β1 (TGF-β1; Sigma-Aldrich, St. Louis, MO, USA). Human
Embryonic Kidney (HEK; Cell Center of Shanghai Institutes for
Biological Sciences, Chinese Academy of Sciences, Shanghai, China)
293 cells were maintained in DMEM supplemented with 10% FBS and
antibiotics at 37°C in 5% CO2. The mimics and inhibitors
of miR34a and miR-34c, and the respective RNA controls, were
obtained from Shanghai JIMA Pharmacy Technology Co., Ltd.
(Shanghai, China).
Bioinformatics approaches
To search for miRNA-34 family target genes, the
online miRNA database miRanda (http://www.microrna.org), Targetscan (http://www.targetscan.org) and Pictar (http://pictar.bio.nyu.edu) were used.
Isolation and identification of rat
HSCs
Primary HSCs were isolated from normal male Sprague
Dawley rats aged between five and six months (weighing 400–500 g;
Shanghai Laboratory Animal Center of Chinese Academy of Sciences,
Shanghai, China) by in situ perfusion and density-gradient
centrifugation, as previously described (25). The rats were maintained at 25°C on
a 12/12 h light/dark cycle, with ad libitum access to rodent
chow and water. The rats received humane care according to the
Guide for the Care and Use of Laboratory Animals of the Chinese
Academy of Sciences. The study was approved by the ethics committee
of Yiwu Central Hospital. All institutional and national guidelines
for the care and use of laboratory animals were followed. Isolated
HSCs were suspended in DMEM supplemented with 10% FBS, and
penicillin and streptomycin at a cell density of 5×105
cells/ml, seeded in culture flasks and cultured at 37°C in 5%
CO2.
Indirect immunofluorescence assay
(IFA)
The cells seeded on glass coverslips or 96-well
plates were washed with phosphate-buffered saline (PBS) and fixed
with 4% paraformaldehyde for 20 min at room temperature (RT). After
being washed three times with PBS, the cells were incubated in
blocking buffer [PBS containing 3% bovine serum albumin (BSA), 0.3%
Triton™ X-100 and 10% FBS] for at least 30 min and then in binding
buffer (PBS containing 3% BSA and 0.3% Triton X-100) with
monoclonal antibodies (mAbs) against α-SMA (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) at a dilution of 1:100
for 1 h at RT. Following three washes with PBS, the cells were
incubated with fluorescein isothiocyanate-conjugated goat
anti-mouse immunoglobulin (Ig)G (Thermo Fisher Scientific, San
Jose, CA, USA) at a 1:100 dilution with binding buffer for 1 h at
RT. The cell nuclei were stained with DAPI (Sigma-Aldrich). The
stained samples were then examined with a Leica TCS SPII confocal
microscope (Leica Microsystems, Wetzlar, Germany).
Total RNA extraction and quantitative
polymerase chain reaction (qPCR) analysis
For general PCR, total RNA was extracted with TRIzol
(Invitrogen Life Technologies) from HEK 293 cells and cDNA was
synthesized using moloney murine leukemia virus reverse
transcriptase (Promega Corp., Madison, WI, USA). For miRNA
detection, total RNA was prepared by using the mirVana™ miRNA
Isolation kit (Ambion, Austin, TX, USA) according to the
manufacturer’s instructions. First-strand cDNA was synthesized
using the Taqman miRNA RT kit (Applied Biosystems, Foster City, CA,
USA). For the detection of the miRNA levels by qPCR,
TaqMan® microRNA assay (Applied Biosystems) was used to
quantify the relative expression levels of miR-34a (assay ID:
000426), miR-34c (assay ID: 000428), and U6 (assay ID: 001973) as
an internal control, in an Applied Biosystems 7500 Detection system
(Applied Biosystems). The relative amount of miRNAs was normalized
against U6 small nuclear RNA, and the fold change for each miRNA
was calculated using the 2−ΔΔCt method (26). The relative miRNA expression was
calculated from three different experiments.
Vector construction
The 3′-untranslated region (UTR) fragments of the
indicated target mRNAs containing putative miR-34a and miR-34c
binding sites were amplified by PCR from the cDNA of HEK293 cells.
The amplified fragments were cloned into the XbaI sites of
pGL3-promoter vector (Promega Corp.) downstream of the luciferase
coding region to generate reporter vector pGL-UTRs. The vector
pGL-PPARγ-mut with mutated binding sites was mutated with the
QuikChange II XL Site-Directed Mutagenesis kit (Stratagene, La
Jolla, CA, USA) according to the manufacturer’s instructions. The
oligonucleotides used for cloning are demonstrated in Table I. All of the primers were
synthesized by Sangong Biotech Co., Ltd. (Shanghai, China). The
constructed clones were confirmed by sequencing (Sangong Biotech
Co., Ltd.).
 | Table IPrimers used in this study. |
Table I
Primers used in this study.
| Primer | Sequence
(5′-3′) |
|---|
| LEF1-F |
GCAGGTCTAGAGAAACATGGTGGAA |
| LEF1-R |
GCAGGTCTAGACTGGGGTGCTGATG |
| Wnt2B-F |
GCAGGTCTAGATGGGAAGGAGTTGTC |
| Wnt2B-R |
GCAGGTCTAGAGGAGTGTTCTAGGG |
| PPARγ-F |
GCAGGTCTAGAGGACTTGTACTAGCAG |
| PPARγ-R |
GCAGGTCTAGAGGTGTCAGATTTTCCC |
| PPARγ-mut1 |
GAGTCCTGAGCCTGTCGCAACATTTCC |
| PPARγ-mut2 |
GGAAATGTTGCGACAGGCTCAGGACTC |
| AREG-F |
GCAGGTCTAGAAACAGAAAGAAGAA |
| AREG-R |
GCAGGTCTAGAAATAGCATAAAAGTG |
| LDHA-F |
GCAGGTCTAGACCTTGCATTTTGGGA |
| LDHA-R |
GCAGGTCTAGAGGAAGAATTATGCAC |
| ACSL1-F |
GCAGGTCTAGATTTCAGGTCGCAGATAG |
| ACSL1-R |
GCAGGTCTAGACTGGTCCGCTTGTTG |
| FABP3-F |
GCAGGTCTAGACACCACATTGCCTCATT |
| FABP3-R |
GCAGGTCTAGACAAGCCTGGGTTCTGT |
| XBP1-F |
GCAGGTCTAGAGGGCGCCTGCGTCGG |
| XBP1-R |
GCAGGTCTAGACGGGGTGTTCTGGCC |
Luciferase reporter assays
The HEK293 cells were plated in 24-well plates the
day prior to transfection. The cells were co-transfected with
internal control pRL-TK (Promega Corp.), reporter vectors and
mimics of miR-34a/c or negative control probes (Shanghai JIMA
Pharmacy Technology Co., Ltd.) using Lipofectamine® 2000
(Invitrogen Life Technologies) according to the manufacturer’s
instructions. A total of 48 h later, the cells were harvested and
applied to the luciferase measurement with a Dual-Luciferase
Reporter Assay System (Promega Corp.) using a GloMax®
20/20 detection system (Promega Corp.). Values are represented as
the number of relative light units (RLU).
Transient transfection
rHSCs or LX-2 cells were seeded into six- or 96-well
plates the day prior to transfection. The cells were transfected
with inhibitors of miR-34a/c or negative control miRNAs (Shanghai
JIMA Pharmacy Technology Co., Ltd.) using Lipofectamine RNAiMAX
Transfection Reagent (Invitrogen Life Technologies). Following 6 h
of culture with the transfection mix, the cells were cultured in
normal culture medium (rHSCs) or medium containing 5 ng/ml TGF-β1
(LX-2 cells). A total of 48 h later, the cells were harvested and
subjected to western blot analysis.
Western blot analysis
The cells were harvested in lysis buffer [50 mM
Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium
deoxycholate, 0.1% SDS and protease inhibitor cocktail (Roche
Diagnostics, Basel, Switzerland)] were first quantified by the
bicinchoninic acid method (Pierce Biotechnology, Inc., Rockford,
IL, USA) and then denatured by boiling for 5 min. A total of 30 μg
of protein per sample were separated by 10% SDS-PAGE and
transferred to a polyvinylidene difluoride membrane (Millipore,
Billerica, MA, USA). The membranes were blocked with tris-buffered
saline (TBS; 20 mM Tris-HCl (pH 7.4), 150 mM NaCl) containing 5%
milk for 1 h at RT. Next, the membranes were incubated with
monoclonal mouse antibodies against α-SMA (1:100 dilution;
sc-53142), against PPARγ (1:100 dilution; sc-7273) or against GAPDH
(1:100 dilution; sc-365062) (all Santa Cruz Biotechnology, Inc.)
for 1 h at 37°C, followed by three time washes with TBS-T (TBS
containing 0.1% (v/v) Tween-20) buffer. Next, the membranes were
incubated with horseradish peroxidase-conjugated goat anti-mouse or
anti-rabbit IgG at a dilution of 1:10,000 in TBS for 1 h at RT,
followed by three washes with TBS-T. The proteins were then
detected using the Supersignal® West Pico
chemiluminescent substrate (Pierce Biotechnology, Inc.) on an
AlphaEase® FC Imaging System (Alpha Innotech
Corporation, San Leandro, CA, USA).
Statistical analysis
Values are presented as the mean ± standard
deviation. The software GraphPad Prism 5.0 (GraphPad Software,
Inc., La Jolla, CA, USA) was used for all statistical analysis and
graphical illustrations. The statistical significance was analyzed
using Student t-test. P<0.01 was considered to indicate a
statistically significant difference.
Results
PPARγ is a target protein of miR-34a and
miR-34c
To identify the potential target mRNAs of miR-34a
and miR-34c, which contain the same ‘seed region’, three different
prediction tools (miRanda, Targetscan and Pictar) were used. Of all
the predicted genes, eight genes were selected for confirmation.
The criteria for target selection was not only concerning the
binding possibility, but also concerning the potential functions in
liver fibrosis. Therefore, the selected genes encoded proteins
either correlated with certain important signaling pathways
[lymphoid enhancer factor 1 (LEF1), Wnt-2B, PPARγ, amphiregulin
(AREG)] (23,27–29)
or associated with lipid metabolism [lactate dehydrogenase (LDHA),
ACSL-1, fatty acid binding protein 3 (FABP3), X box binding protein
1 (XBP1)] (30,31).
HEK293 cells were co-transfected with reporter
vectors pGL-UTRs bearing the predicted binding sites and mimics of
miR-34a or miR-34c together with the internal control pRL-TK. The
increased expression of miR-34a or miR-34c by mimics transfection
was confirmed by qPCR at 48 h following transfection (Fig. 1A), and a concentration of 20 nM
mimics was selected for the following experiments. The results
demonstrated that the overexpression of miR-34a/34c significantly
decreased the relative luciferase activity in HEK293 cells
transfected with UTRs of LEF1, PPARγ, AREG, LDHA, ACSL-1 and XBP1.
LEF1, LDHA and ACSL-1 have been reported to be targeted by
miR-34a/34c previously (15,32).
Since PPARγ was reported to be involved in the maintenance of a
quiescent HSCs phenotype and the activation of HSCs resulted in the
loss of PPARγ (19,33), PPARγ was selected for further
confirmation. The reporter vector bearing the mutated UTR of PPARγ
was constructed (Fig. 1C). It was
identified that the overexpression of miR-34a/34c significantly
decreased the relative luciferase activity in HEK293 cells
transfected with pGL-PPARγ (Fig.
1D), while it caused no apparent relative luciferase activity
changes in HEK293 cells transfected with pGL-PPARγmut (Fig. 1E). These results indicated that
PPARγ was a direct target gene of miR-34a and miR-34c.
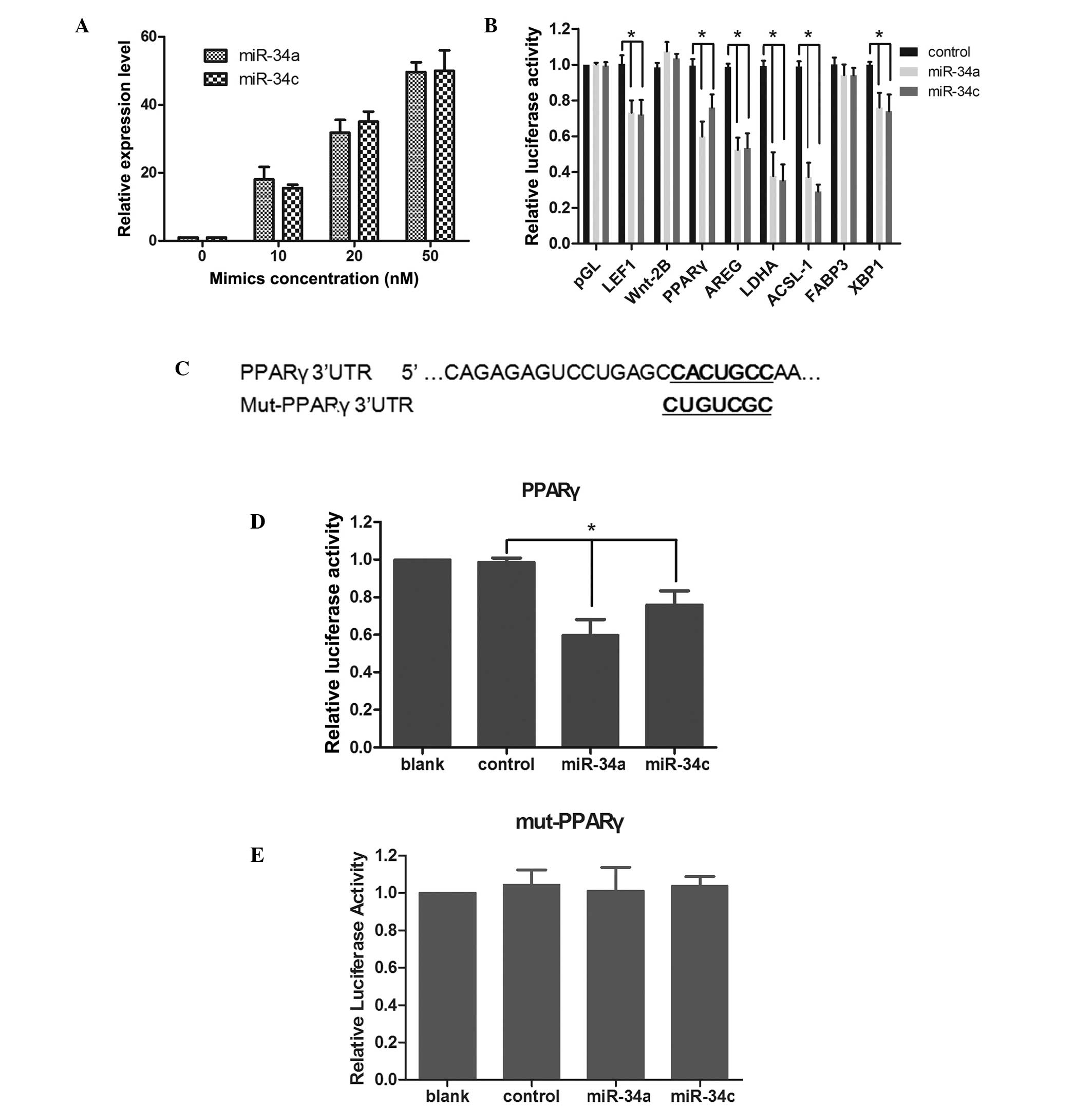 | Figure 1PPARγ is a direct target protein of
miR-34a and miR-34c. Mimics of miR-34a and miR-34c were introduced
into HEK-293 cells and upregulation of miR-34a and miR-34c was
confirmed by quantitative polymerase chain reaction. (A) A
concentration of 20 nM mimics was selected for the following
assays. (B) Luciferase reporter assay was used to confirm the
direct interaction between miR-34a/miR-34c and target mRNAs. HEK293
cells were co-transfected with mimics, pRL-TK together with
reporter vector pGL-UTRs, and the relative luciferase activity was
detected. (C) The predicted miR-34a/miR-34c binding site on the
PPARγ mRNA 3′-UTR and mutated binding site are demonstrated as bold
and underlined. (D and E) Mutation luciferase reporter assay was
used to further confirm the direct interaction between
miR-34a/miR-34c and PPARγ. HEK293 cells were co-transfected with
mimics, pRL-TK together with (D) wild reporter vector pGL-PPARγ (E)
and mutant reporter vector pGL-PPARγ-mut. Relative luciferase
activity was detected. Data are represented as the mean ± standard
deviation from triplicate independent experiments.
*P<0.01, for comparison between the miR-34a/c
transfected cells and the control miRNA transfected cells. UTR,
untranslated region; Mut, mutation; LEF, lymphoid enhancer factor;
AREG, amphiregulin; LDHA, lactate dehydrogenase; FABP, fatty acid
binding protein; XBP, X box binding protein PPARγ,
proliferator-activated receptor γ; HEK293, human embryonic kidney
293; miRNA, microRNA. |
Expression of miR-34a/c and PPARγ during
the activation of HSCs
The upregulation of miR-34 family members and
downregulation of PPARγ have been previously reported during liver
fibrosis (15,23). Since the activation of HSCs is the
pivotal process in liver fibrosis, the expression of miR-34a/c and
PPARγ was detected during the activation of HSCs. Rat HSCs and
human HSCs (LX-2) were used in the studies.
Firstly, primary rat HSCs were isolated, and
~2×108 HSCs were harvested from each rat. The fraction
of freshly isolated living HSCs was ≤90%, as defined by trypan blue
staining. The morphology and growth characteristics of the freshly
isolated cells were observed with an inverted phase contrast
microscope. The cells were small and round with the quiescent
phenotype when freshly isolated; however, they demonstrated a weak
adhesive growth pattern following culture for two days and
presented a wall-adhesive growth pattern following culture for ten
days (Fig. 2Aa–c). IFA and western
blot analysis of α-SMA were applied for demonstrating the activated
phenotype of HSCs. The IFA results demonstrated that the cells
expressed no or little α-SMA on day 2 (Fig. 2Ad) and expressed abundant α-SMA on
day 10 (Fig. 2Ad). Western blot
analysis also demonstrated that the expression of α-SMA was
increased progressively with the time in culture, and reached the
highest expression at day 10 (Fig.
2B). These results indicated that following culture for ten
days, HSCs had been highly activated.
Next, the expression of miR-34a and miR-34c in the
rat HSCs was detected by qPCR. As demonstrated in Fig. 2C, the expression of miR-34a and
miR-34c in rat HSCs revealed a significant ~15-fold and 5-fold
increase, respectively, following culture for seven days, and
peaked with the highest expression at day 10, with an increase of
~20-fold and 10-fold, respectively. The expression of miR-34a was
always marginally higher than that of miR-34c.
Next, the expression of PPARγ was detected by
western blot analysis. As demonstrated in Fig. 2B, PPARγ decreased progressively
with the time in culture and demonstrated a negative correlation
with the expression of miR-34a/c and α-SMA.
The expression of miR-34a/c and PPARγ was also
analyzed during the activation of human HSC LX-2 cells. TGF-β1 is
one of the critical factors for the activation of HSC during
chronic inflammation (34). LX-2
cells were treated with TGF-β1 for the indicated time (from 0 to 48
h) in 5 ng/ml or in the indicated concentration (from 0 to 5 ng/ml)
for 48 h. The upregulated expression of α-SMA in both conditions
indicated the activation of LX-2 by TGF-β1 stimulation (Fig. 2D). In addition, the expression of
PPARγ, miR-34a and miR-34c demonstrated changes that were
consistent with the results in the rat HSCs during the activation
of LX-2 (Fig. 2D and E). In
conclusion, during activation of HSCs, miR-34a and miR-34c were
upregulated and PPARγ was downregulated, and the expression of
PPARγ was negatively correlated with the expression of miR-34a and
miR-34c and the degree of HSC activation.
Inhibitors of miR-34a and miR-34c
upregulate the expression of PPARγ and downregulate the expression
of α-SMA in human HSCs
To further investigate the association between the
miR-34 family and PPARγ and their effect on the activation of HSCs,
activated HSCs were transfected with miR-34a and miR-34c inhibitors
alone or together, and the expression of PPARγ and α-SMA was
detected. As demonstrated in Fig.
3A, PPARγ was upregulated following transfection with miR34a
and miR34c alone or together, compared with the negative control
miRNA in TGF-β1-stimulated LX-2 cells, while α-SMA was
downregulated in HSCs transfected with miR-34a/c compared with
negative control microRNA, which suggested that activation of HSCs
was reversed. Furthermore, application of the miR34a inhibitor
resulted in higher expressional regulation than the miR34c
inhibitor did, and the two inhibitors in combination induced the
highest expressional regulation. However, in the activated rat
HSCs, no expression change was observed (Fig. 3B). These results indicated that the
miR-34a family accelerated the activation of human HSCs by
modulating PPARγ, and the decreased expression of PPARγ may have
resulted from differential regulatory mechanisms in rat and human
HSCs.
Discussion
The miR-34 family is transcriptionally controlled by
p53 tumor suppressor protein and regulates a plethora of target
proteins, which are involved in cell cycle, apoptosis,
differentiation and cellular development (10). The downregulation of miR-34a has
been previously identified in numerous cancer types (12,13,35),
and therefore, miR-34a is considered to be a tumor suppressor.
However, upregulation of miR-34 has been found in numerous liver
diseases from fatty liver disease to hepatocellular carcinoma
(36–38). It was reported that miR-34 family
members may target ACSL1, which has a central role in lipid
metabolism and fatty acid metabolism in the liver, and impairs the
lipid metabolism in the liver, resulting in the development of
hepatic fibrosis (15). Whether
other factors are involved in this process has yet to be
elucidated.
In the present study, using a bioinformatics
approach, eight proteins were selected for experimental
confirmation. Among them, LEF1, Wnt-2B, PPARγ and AREG have been
reported to be correlated with several signaling pathways,
including Wnt and PPAR (23,27–29),
which may have an important role during liver fibrosis, while LDHA,
ACSL-1, FABP3 and XBP1 were reported to be associated with lipid
metabolism (30,31). ACSL-1 is also used as a positive
control since it has been reported to be the target of miR-34a and
miR-34c (15). The reporter assay
indicated that LEF1, PPARγ, AREG, LDHA, ACSL-1 and XBP1 are
possible target genes of miR-34a and miR-34c. Among these, PPARγ
has been reported to be potently involved in liver fibrosis
(23). Therefore, PPARγ was
selected for further analysis in the present study. The mutation
reporter assay confirmed that PPARγ is the direct target of miR-34a
and miR-34c.
PPARγ is a ligand-activated nuclear transcription
factor that belongs to the nuclear hormone receptor superfamily.
PPARγ has a key role in HSC biology and is involved in the
maintenance of a quiescent HSC phenotype (19). PPARγ receptors were found to have
anti-proliferative and anti-fibrotic effects on activated HSCs, as
well as to induce HSC apoptosis through a mechanism involving an
extrinsic apoptosis pathway (20,23).
It has been reported previously that miR-34 family
members are upregulated and PPARγ is downregulated during liver
fibrosis (15,23). Considering the hallmark of liver
fibrosis is the activation of HSCs, the present study next detected
the expression of miR-34 family members and PPARγ during the
activation of HSCs. The results demonstrated that miR-34 family
member expression was increased, while PPARγ expression was reduced
during the activation of HSCs both in rats and humans, which is
consistent with previous studies (15,23).
Furthermore, the expression of PPARγ was negatively correlated with
the expression of miR-34 family members and the degree of
activation.
The present study further examined the association
of miR-34 and PPARγ in activated HSCs by using miR-34 inhibitors.
The results from the human HSCs demonstrated that PPARγ was
upregulated and α-SMA was downregulated when the cells were
transfected with miR-34 and miR-34c inhibitors alone or in
combination, indicating that miR-34a and miR-34c inhibitors may
decrease the activation of HSCs by upregulating the expression of
PPARγ. The miR-34 family may exhibit profibrotic effects by
targeting PPARγ. However, no expression changes of PPARγ and α-SMA
were observed in rat HSCs transfected with miR-34 and miR-34c
inhibitors alone or in combination. Further bioinformatics analysis
demonstrated that the binding site of human PPARγ mRNA is located
in a poorly conserved region in mammals, and there were no
predicted binding sites for miR-34a and miR-34c on the 3′UTR of rat
PPARγ mRNA. This is not in accordance with the general observation
that binding sites are always conserved in species, while the
results from the mutation reporter assay confirmed this
interaction. These data suggested that although the expression of
miR-34 family members, PPARγ and α-SMA demonstrated a similar
expression pattern in rat HSCs and in human HSCs, the regulation
mechanism in the two cell lines may be contrasting. It has been
previously reported that TGF-β1 inhibits the expression of PPARγ in
activated rat HSCs through the β-catenin pathway (39). TGF-β1 is one important cytokine
expressed following liver injury and is the most important cytokine
stimulating fibrogenesis in HSCs (1). It may be possible that in rat HSCs,
the inhibition of PPARγ is mainly the result of the upregulation of
TGF-β1 during the activation of HSCs, while in human HSCs, the
inhibition of PPARγ results from the combination effect of
upregulation of TGF-β1 and miR-34a/c.
For the first time, to the best of our knowledge,
the present study identified and confirmed PPARγ to be a target
gene of the miR-34 family. The regulation of the miR-34 family is
negatively associated with PPARγ in activated HSCs. These data
suggested that miR-34 family members may be involved in liver
fibrosis by targeting PPARγ.
References
|
1
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moreira RK: Hepatic stellate cells and
liver fibrosis. Arch Pathol Lab Med. 131:1728–1734. 2007.PubMed/NCBI
|
|
3
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wiggins JF, Ruffino L, Kelnar K, et al:
Development of a lung cancer therapeutic based on the tumor
suppressor microRNA-34. Cancer Res. 70:5923–5930. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Raisch J, Darfeuille-Michaud A and Nguyen
HT: Role of microRNAs in the immune system, inflammation and
cancer. World J Gastroenterol. 19:2985–2996. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vettori S, Gay S and Distler O: Role of
MicroRNAs in Fibrosis. Open Rheumatol J. 6:130–139. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Noetel A, Kwiecinski M, Elfimova N, Huang
J and Odenthal M: microRNA are central players in anti- and
profibrotic gene regulation during liver fibrosis. Front Physiol.
3:492012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Katayama Y, Maeda M, Miyaguchi K, et al:
Identification of pathogenesis-related microRNAs in hepatocellular
carcinoma by expression profiling. Oncol Lett. 4:817–823.
2012.PubMed/NCBI
|
|
9
|
He L, He X, Lim LP, et al: A microRNA
component of the p53 tumour suppressor network. Nature.
447:1130–1134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen F and Hu SJ: Effect of microRNA-34a
in cell cycle, differentiation, and apoptosis: a review. J Biochem
Mol Toxicol. 26:79–86. 2012. View Article : Google Scholar
|
|
11
|
Trang P, Wiggins JF, Daige CL, et al:
Systemic delivery of tumor suppressor microRNA mimics using a
neutral lipid emulsion inhibits lung tumors in mice. Mol Ther.
19:1116–1122. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pang RT, Leung CO, Lee CL, et al:
MicroRNA-34a is a tumor suppressor in choriocarcinoma via
regulation of Delta-like1. BMC Cancer. 13:252013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yan K, Gao J, Yang T, et al: MicroRNA-34a
inhibits the proliferation and metastasis of osteosarcoma cells
both in vitro and in vivo. PLoS One. 7:e337782012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pogribny IP, Starlard-Davenport A,
Tryndyak VP, et al: Difference in expression of hepatic microRNAs
miR-29c, miR-34a, miR-155, and miR-200b is associated with
strain-specific susceptibility to dietary nonalcoholic
steatohepatitis in mice. Lab Invest. 90:1437–1446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li WQ, Chen C, Xu MD, et al: The
rno-miR-34 family is upregulated and targets ACSL1 in
dimethylnitrosamine-induced hepatic fibrosis in rats. FEBS J.
278:1522–1532. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo CJ, Pan Q, Li DG, Sun H and Liu BW:
miR-15b and miR-16 are implicated in activation of the rat hepatic
stellate cell: An essential role for apoptosis. J Hepatol.
50:766–778. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murakami Y, Toyoda H, Tanaka M, et al: The
progression of liver fibrosis is related with overexpression of the
miR-199 and 200 families. PLoS One. 6:e160812011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kota J, Chivukula RR, O’Donnell KA, et al:
Therapeutic microRNA delivery suppresses tumorigenesis in a murine
liver cancer model. Cell. 137:1005–1017. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hazra S, Xiong S, Wang J, et al:
Peroxisome proliferator-activated receptor gamma induces a
phenotypic switch from activated to quiescent hepatic stellate
cells. J Biol Chem. 279:11392–11401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sharvit E, Abramovitch S, Reif S and Bruck
R: Amplified inhibition of stellate cell activation pathways by
PPAR-γ, RAR and RXR agonists. PLoS One. 8:e765412013. View Article : Google Scholar
|
|
21
|
Attia YM, Elalkamy EF, Hammam OA, Mahmoud
SS and El-Khatib AS: Telmisartan, an AT1 receptor blocker and a
PPAR gamma activator, alleviates liver fibrosis induced
experimentally by Schistosoma mansoni infection. Parasit Vectors.
6:1992013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bruck R, Weiss S, Aeed H, Pines M, Halpern
Z and Zvibel I: Additive inhibitory effect of experimentally
induced hepatic cirrhosis by agonists of peroxisome proliferator
activator receptor gamma and retinoic acid receptor. Dig Dis Sci.
54:292–299. 2009. View Article : Google Scholar
|
|
23
|
Zhang F, Kong D, Lu Y and Zheng S:
Peroxisome proliferator-activated receptor-γ as a therapeutic
target for hepatic fibrosis: from bench to bedside. Cell Mol Life
Sci. 70:259–276. 2012. View Article : Google Scholar
|
|
24
|
Guo GH, Tan DM, Zhu PA and Liu F:
Hepatitis B virus X protein promotes proliferation and upregulates
TGF-beta1 and CTGF in human hepatic stellate cell line, LX-2.
Hepatobiliary Pancreat Dis Int. 8:59–64. 2009.PubMed/NCBI
|
|
25
|
Weiskirchen R and Gressner AM: Isolation
and culture of hepatic stellate cells. Methods Mol Med. 117:99–113.
2005.PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
27
|
Mao CD and Byers SW: Cell-context
dependent TCF/LEF expression and function: alternative tales of
repression, de-repression and activation potentials. Crit Rev
Eukaryot Gene Expr. 21:207–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cha YH, Kim NH, Park C, Lee I, Kim HS and
Yook JI: MiRNA-34 intrinsically links p53 tumor suppressor and Wnt
signaling. Cell Cycle. 11:1273–1281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Busser B, Sancey L, Brambilla E, Coll JL
and Hurbin A: The multiple roles of amphiregulin in human cancer.
Biochim Biophys Acta. 1816:119–131. 2011.PubMed/NCBI
|
|
30
|
Lee AH, Scapa EF, Cohen DE and Glimcher
LH: Regulation of hepatic lipogenesis by the transcription factor
XBP1. Science. 320:1492–1496. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hanhoff T, Lücke C and Spener F: Insights
into binding of fatty acids by fatty acid binding proteins. Mol
Cell Biochem. 239:45–54. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kaller M, Liffers ST, Oeljeklaus S, et al:
Genome-wide characterization of miR-34a induced changes in protein
and mRNA expression by a combined pulsed SILAC and microarray
analysis. Mol Cell Proteomics. 10:M111.010462. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Marra F, Efsen E, Romanelli RG, et al:
Ligands of peroxisome proliferator-activated receptor gamma
modulate profibrogenic and proinflammatory actions in hepatic
stellate cells. Gastroenterology. 119:466–78. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Inagaki Y and Okazaki I: Emerging insights
into transforming growth factor beta Smad signal in hepatic
fibrogenesis. Gut. 56:284–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang S, Li Y, Gao J, et al: MicroRNA-34
suppresses breast cancer invasion and metastasis by directly
targeting Fra-1. Oncogene. 32:4294–4303. 2013. View Article : Google Scholar
|
|
36
|
Castro RE, Ferreira DM, Afonso MB, et al:
miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat
liver and activated by disease severity in human non-alcoholic
fatty liver disease. J Hepatol. 58:119–125. 2013. View Article : Google Scholar
|
|
37
|
Meng F, Glaser SS, Francis H, et al:
Epigenetic regulation of miR-34a expression in alcoholic liver
injury. Am J Pathol. 181:804–817. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pineau P, Volinia S, McJunkin K, et al:
miR-221 overexpression contributes to liver tumorigenesis. Proc
Natl Acad Sci USA. 107:264–269. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qian J, Niu M, Zhai X, Zhou Q and Zhou Y:
β-Catenin pathway is required for TGF-beta1 inhibition of PPARγ
expression in cultured hepatic stellate cells. Pharmacol Res.
66:219–225. 2012. View Article : Google Scholar : PubMed/NCBI
|
















