Introduction
Atherosclerosis is a progressive chronic
inflammatory disease, characterized by the development of plaques
in the artery, which ultimately lead to cardiovascular disease
(1). One of the early events in
atherogenesis is the formation of foam cells, which are macrophages
containing ingested oxidized low-density lipoprotein (LDL)
(2). It has been suggested that
macrophages become foam cells as a consequence of the loss of the
normal balance between cholesterol uptake from lipoproteins and
cholesterol efflux. This has led to investigations to elucidate the
regulation of cholesterol efflux from macrophages (3).
The adenosine triphosphate (ATP)-binding cassette
(ABC) transporters, ABCA1 and ABCG1, are important in the removal
of cholesterol from lipid-laden macrophages as a primary
anti-atherogenic mechanism (4).
ABCA1 promotes cholesterol efflux from foam cells to lipid-free
apolipoprotein A-1 (apoA-1), but not to mature high-density
lipoprotein (HDL) particles, whereas ABCG1 mediates cholesterol
efflux to HDL and not to the lipid-poor apoA-1 (5,6).
Biochemical studies have identified ABCA1 and ABCG1 as direct
transcriptional targets of liver X receptor/retinoid X receptor
(LXR/RXR) (7,8). Peroxisome proliferator-activated
receptor gamma (PPARγ) is also involved in the transcriptional
activation of the ABC transporters in an LXRα-dependent manner
(9). Chawla et al (9) demonstrated that LXRα
activation-induced expression levels of ABCA1 and ABCG1 are
markedly reduced in PPARγ-deficient macrophages, suggesting the
involvement of PPARγ in the LXRα-mediated upregulation of ABC
proteins. PPARγ and LXRα have been demonstrated to exert
anti-atherogenic effects by facilitating the removal of cholesterol
from foam cells (10).
The renin-angiotensin system (RAS) is considered to
be involved in the pathogenesis of atherosclerosis (11,12).
Angiotensin II (Ang II), the most important component of the RAS,
has been found to promote the development of atherosclerosis in
apolipoprotein-E (ApoE)-deficient mice (13). Ang-converting enzyme (ACE) 2 is a
homolog of ACE, which is responsible for generating Ang II from Ang
I. ACE2 efficiently degrades Ang II to form Ang-(1-7), a peptide
exerting actions opposite to those of Ang II (14). Our previous study demonstrated that
Ang-(1-7) ameliorates Ang-II-induced apoptosis in human umbilical
vein endothelial cells (15). A
previous study demonstrated that Ang-(1-7) dose-dependently
inhibits early atherosclerotic lesions and increases plaque
stability by targeting vascular cells in ApoE(−/−) mice
(16). Takata et al
(17) reported that Ang II alters
macrophage cholesterol homeostasis by repressing the expression of
ABCA1, therefore, promoting foam cell formation. However, the
effects of Ang-(1-7) on macrophage cholesterol metabolism remain to
be elucidated. The present study investigated the effects of
Ang-(1-7) on Ang II-stimulated cholesterol efflux and the
associated molecular mechanisms.
Materials and methods
Reagents and antibodies
Ang II, Ang-(1-7), phorbol 12-my ristate 13-acetate
(PMA), apoA-1, SB203580 (a p38 MAPK inhibitor), SP600125 (a JNK
inhibitor) and GW9662 were purchased from Sigma-Aldrich (St. Louis,
MO, USA). Primary mouse anti-human monoclonal antibodies (1:300)
against ABCA1 (sc-53482), ABCG1 (sc-20795), PPARγ (sc-390740) and
LXRα (sc-271064) were purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Primary mouse or rabbit anti-human
monoclonal antibodies (1:300) against total p38 (#9212),
phosphorylated (p-)p38 (#9216), total extracellular
signal-regulated protein kinases 1 and 2 (ERK1/2) (#9102), p-ERK1/2
(#9108), total c-Jun NH2-terminal kinase (JNK) (#9252), p-JNK
(#9255) and β-actin (#4967) were purchased from Cell Signaling
Technology, Inc. (Beverly, MA, USA). Horseradish
peroxidase-conjugated goat anti-mouse or anti-rabbit secondary
antibodies (1:2,000 dilution) were purchased from Santa Cruz
Biotechnology, Inc.
Culture and differentiation of THP-1
cells
Human THP-1 cells were purchased from the Cell Bank
of Chinese Academy of Sciences (Shanghai, China) and cultured in
RPMI-1640 medium, containing 10% fetal bovine serum (FBS), 10
μg/ml streptomycin and 100 U/ml penicillin (Invitrogen Life
Technologies, Carlsbad, CA, USA) at 37°C with 5% CO2. To
differentiate these cells into macrophages, the monocytes were
seeded into 12-well plates at a density of 1×106
cells/well in complete RPMI-1640 medium containing 160 nM PMA and
incubated for 24 h at 37°C.
Drug treatment
The differentiated THP-1 macrophages were washed
three times in phosphate-buffered saline (PBS; Invitrogen Life
Technologies) and labeled by incubation for 48 h at 37°C in the
presence of (3H)cholesterol (0.37×109 Bq/l; GE
Healthcare, Piscataway, NJ, USA) in RPMI-1640 medium, supplemented
with 10% FBS. Following washing three times in PBS, the labeled
cells were treated with Ang II (1 μM) and/or 10 or 100 nM
Ang-(1-7) (18) for 24 h at 37°C.
Unless stated otherwise, 100 nM Ang-(1-7) was used. To investigate
the role of PPARγ in the Ang-(1-7)-mediated induction of ABCA1 and
ABCG1 expression, the THP-1 macrophages were treated with GW9662 (1
μM) for 1 h (19), prior to
incubation for 24 h with 1 μM Ang II with or without 100 nM
Ang-(1-7). To investigate the role of mitogen-activated protein
kinases (MAPKs) in the regulation of PPARγ and LXRα expression,
SB203580 (10 μM) and/or SP600125 (10 μM) (20) were added to the cell culture and
incubated for 1 h at 37°C prior to the addition of 1 μM Ang
II.
Cholesterol efflux assay
Following the treatment described above, the labeled
cells were washed three times in PBS and incubated in serum-free
RPMI-1640 medium containing 10 mg/l apoA-1 as a cholesterol
receptor for 12 h at 37°C. The medium was then collected and
centrifuged at 1,000 × g for 10 min at room temperature to remove
cellular debris. Radioactivity in the medium and cells was analyzed
by liquid scintillation counting (ZX34-LS6500; Beckman Coulter,
Inc., Fullerton, CA, USA). The percentage of cholesterol efflux was
calculated as: Counts/min (cpm) in the medium / (cpm in the cell +
cpm in the medium) × 100.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA was extracted using TRIzol reagent,
according to the manufacturer’s instructions (Invitrogen Life
Technologies). RT was performed using an AMV First Strand cDNA
Synthesis kit (Bio Basic, Inc., Amhurst, NY, USA). qPCR
amplification was performed using an Applied Biosystems StepOnePlus
Real-Time PCR system (Applied Biosystems, Foster City, CA, USA)
using a SYBR green PCR master mix (Applied Biosystems). The primers
used for qPCR are listed in Table
I. Each PCR reaction (20 μl) contained 10 ng cDNA and
ther thermal cyclin conditions were as follows: 95°C for 10 min,
followed by 35 cycles of 95°C for 15 sec and 60°C for 50 sec. As an
internal quantitative control, glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) was amplified in a parallel reaction. All
assays were performed in triplicate and the threshold cycle (Ct)
value was determined as the cycle number at which the fluorescence
signal reached the exponential phase. The relative mRNA expression
levels were normalized against GAPDH and were determined using the
2−ΔΔCt method (21).
 | Table IPrimer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table I
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Sequence (5′–3′) | Primer size (bp) |
|---|
| ABCA1 |
| Forward |
GCTTTCAATCATCCCCTGAA | 20 |
| Reverse |
TGACAGGCTTCACTCCACTG | 20 |
| ABCG1 |
| Forward |
CTCCGGCTTCCTCTTCTTCT | 20 |
| Reverse |
TACACGATGCTGCAGTAGGC | 20 |
| PPARγ |
| Forward |
GAGCCCAAGTTTGAGTTTGC | 20 |
| Reverse |
CTGTGAGGACTCAGGGTGGT | 20 |
| LXRα |
| Forward |
ACGGTGATGCTTCTGGAGAC | 20 |
| Reverse |
AGCAATGAGCAAGGCAAACT | 20 |
| GAPDH |
| Forward |
CGACCACTTTGTCAAGCTCA | 20 |
| Reverse |
AGGGGAGATTCAGTGTGGTG | 20 |
Western blot analysis
Following treatment, the cells were lysed in lysis
buffer (Santa Cruz Biotechnology, Inc.), containing 50 mmol/l Tris
(pH 7.4), 150 mmol/l NaCl, 1% NP-40 and 0.1% sodium dodecyl
sulphate (SDS; Sigma-Aldrich), supplemented with protease and
phosphatase inhibitors, including pepstatin, leupeptin, aprotinin
and phenylmethylsulfonyl fluoride, which were purchased from
Sigma-Aldrich. The protein samples were separated on polyacrylamide
gels (Bio-Rad Laboratories, Inc., Hercules, CA, USA) containing
0.1% SDS, and then transferred onto a nitrocellulose membrane
(Millipore, Bedford, MA, USA). Following blocking for 4 h in a
Tris-buffered solution, containing 5% non-fat dried milk and 0.5%
Tween-20 (Sigma-Aldrich), the membrane was incubated with
individual primary antibodies (1:300; as described above) overnight
at 4°C. The membrane was then washed three times in PBS and
incubated for 1 h with secondary antibodies at room temperature.
The signals were visualized using the enhanced chemiluminescence
detection system (Amersham Biosciences, Little Chalfont, UK) and
developed on X-ray film (GE Healthcare). The band density was
measured using the GelDoc 2000 system equipped with Quantity One
4.6 software (Bio-Rad Laboratories, Inc.) and normalized against
β-actin.
Statistical analysis
The data are expressed as the mean ± standard
deviation. All statistical analyses were performed using SPSS 19.0
software (IBM, Armonk, NY, USA). Statistical differences among
multiple groups were calculated using a one-way analysis of
variance followed by Tukey’s post-hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Ang-(1-7) antagonizes the Ang II-mediated
repression of cholesterol efflux
Compared with the untreated control THP-1
macrophages, treatment with Ang II (1 μM) for 48 h
significantly reduced the rate of cholesterol efflux to apoA-1 by
~50% (7.69±0.48, vs. 14.06±0.56%; P<0.05; Fig. 1),. Notably, treatment with
Ang-(1-7) led to a dose-dependent restoration in cholesterol efflux
to apoA-1. When the cells were treated with 100 nM Ang-(1-7), the
cholesterol efflux rate (12.39±0.50) increased to a level similar
to that observed in the control group.
Ang-(1-7) relieves the Ang II-mediated
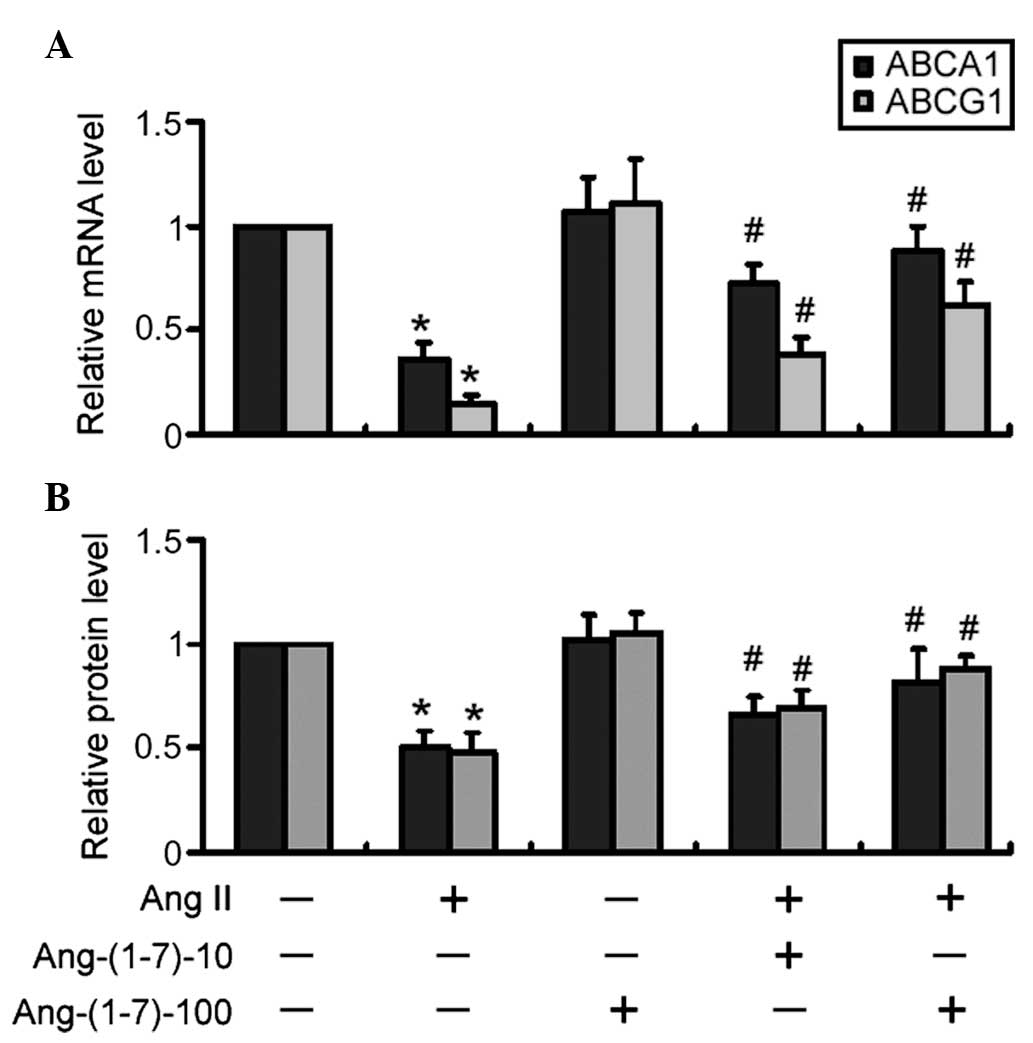
suppression of the gene expression levels of ABCA1 and ABCG1
The mRNA expression levels of ABCA1 and ABCG1 were
significantly (P<0.05) reduced in the Ang II-treated THP-1
macrophages compared with the control cells (Fig. 2A). The co-treatment with Ang-(1-7)
and Ang II significantly increased the mRNA expression levels of
ABCA1 and ABCG1 in a dose-dependent manner (P<0.05), compared
with Ang II only (Fig. 2A). In
addition, western blot analysis revealed that treatment with
Ang-(1-7) significantly (P<0.05) attenuated the Ang II-mediated
reduction in the protein expression levels of ABCA1 and ABCG1 in a
dose-dependent manner (Fig.
2B).
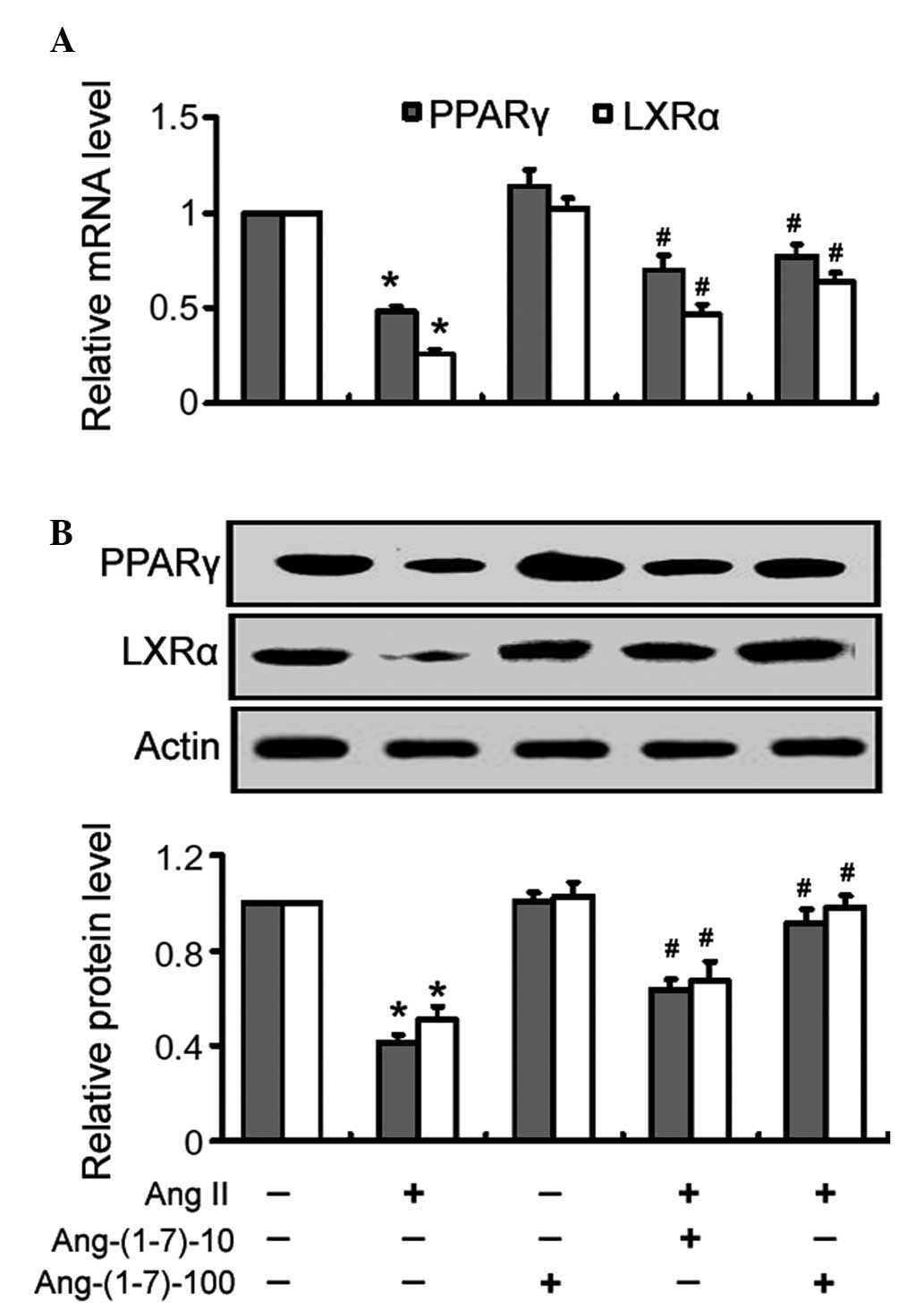
Ang-(1-7) restores the expression levels
of PPARγ and LXRα in Ang II-treated cells
Treatment with Ang II alone significantly
(P<0.05) reduced the mRNA expression levels of PPARγ and LXRα in
the THP-1 macrophages, compared with the control cells (Fig. 3A). Treatment with Ang-(1-7)
inhibited the Ang II-mediated suppression of PPARγ and LXRα
expression in a dose-dependent manner (Fig. 3A). Similarly, the Ang II-mediated
reduction in the protein expression of PPARγ and LXRα was almost
completely inhibited following treatment with Ang-(1-7), as
determined by western blot analysis (Fig. 3B).
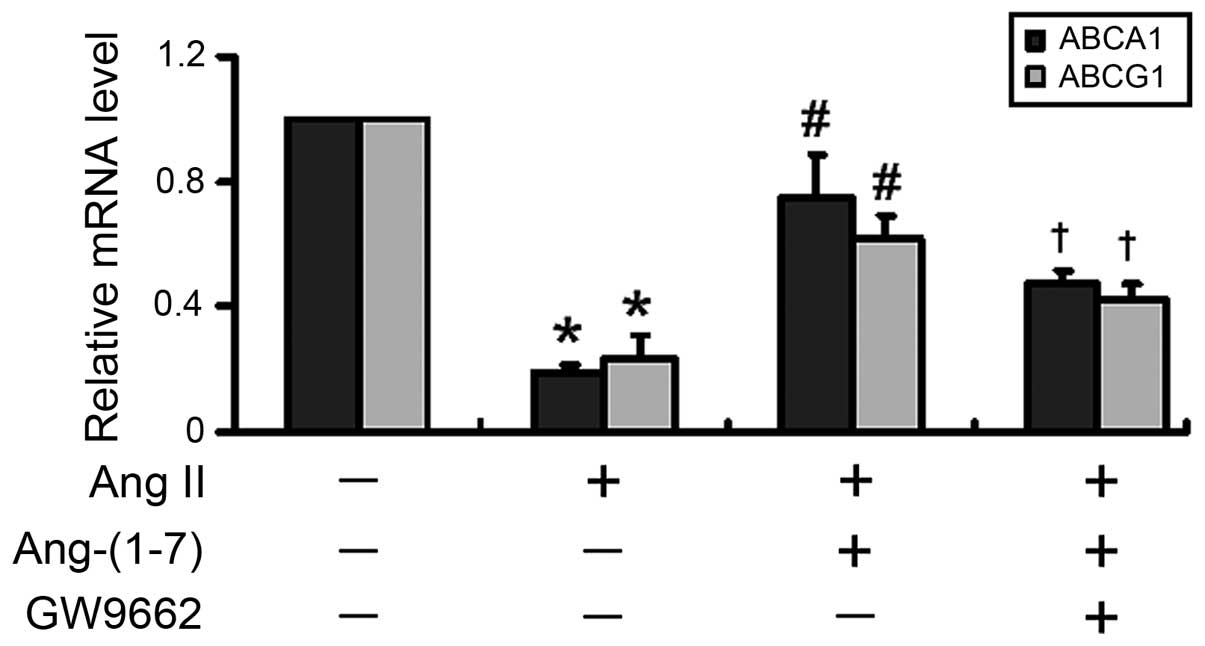
Pharmacological inactivation of PPARγ
attenuates the stimulatory effects of Ang-(1-7) on the mRNA
expression levels of ABCA1 and ABCG1
Subsequently, the present study inhibited the
activity of PPARγ to investigate whether the Ang-(1-7)-mediated
stimulation of ABCA1 and ABCG1 expression was PPARγ-dependent. As
shown in Fig. 4, inhibition of
PPARγ with GW9662, an irreversible and selective PPARγ antagonist,
significantly (P<0.05) eliminated the Ang-(1-7)-mediated
induction of the mRNA expression of ABCA1 and ABCG1.
Inactivation of JNK and p38 MAPKs is
associated with the Ang-(1-7)-mediated induction of PPARγ and LXRα
expression
To elucidate the mechanisms underlying the
Ang-(1-7)-induced expression of PPARγ and LXRα, MAPK signaling
pathways were investigated by western blot analysis. It was
revealed that treatment with Ang-(1-7) significantly (P<0.05)
antagonized the inductive effect of Ang II on the phosphorylation
of JNK and p38 MAPKs, but not ERK1/2 (Fig. 5A). The total protein expression
levels of the MAPKs were not affected by treatment with Ang-(1-7).
Notably, co-incubation with SB203580, a specific inhibitor of p38
MAPK, significantly (P<0.05) restored the expression levels of
PPARγ and LXRα in Ang II-treated THP-1 macrophages (Fig. 5B), which mimicked the indusive
effects of Ang-(1-7). Similarly, inhibition of JNK signaling using
SP600125 significantly (P<0.05) attenuated the inhibition of
PPARγ and LXRα expression by Ang II. Taken together, these results
suggested that inactivation of p38 and JNK MAPK signaling was
involved in the Ang-(1-7)-mediated induction of PPARγ and LXRα
expression.
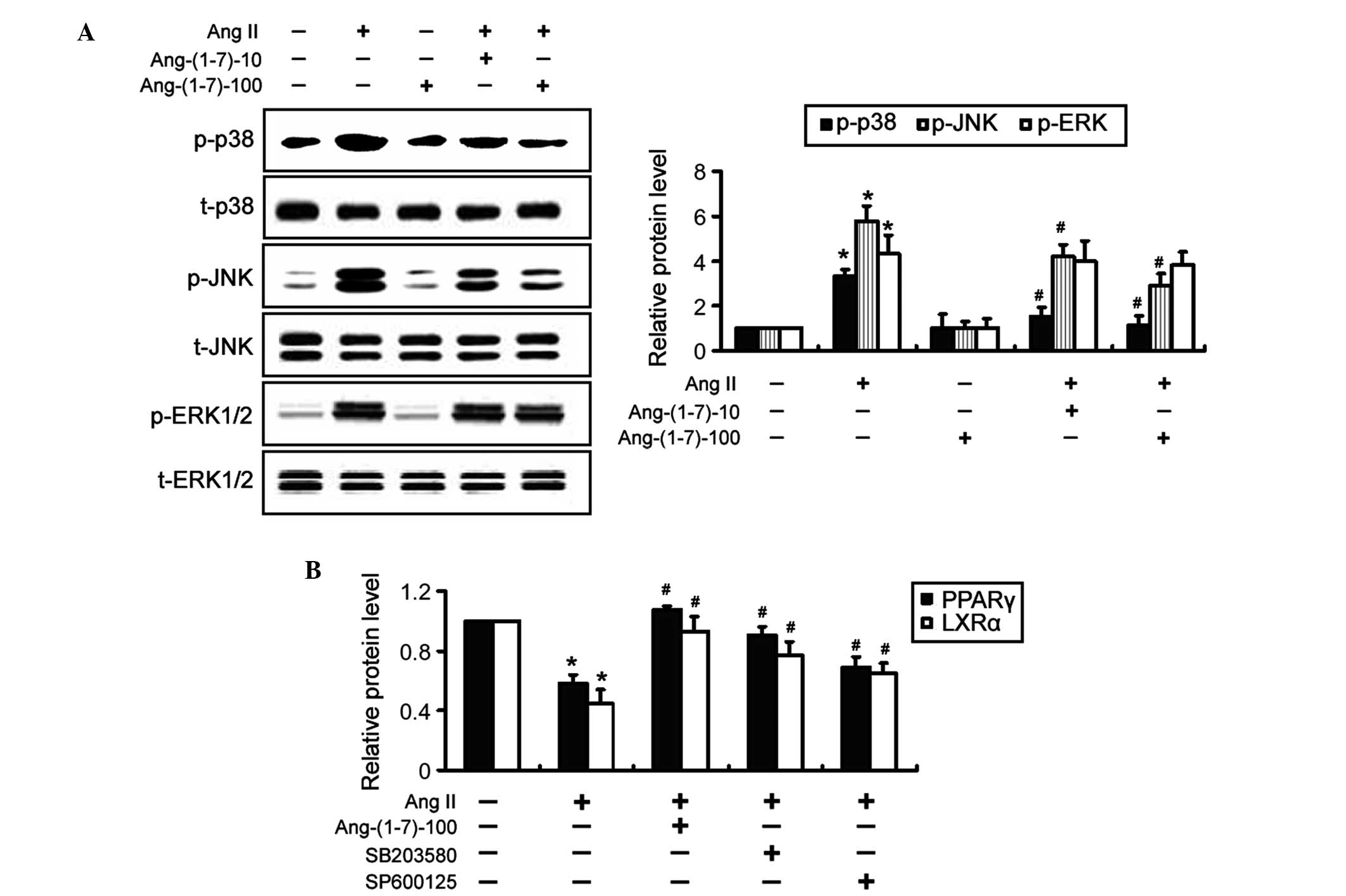 | Figure 5Ang-(1-7)-mediated induction of PPARγ
and LXRα is associated with the suppression of JNK and p38
mitogen-activated protein kinase activation. (A) THP-1 macrophages
were exposed to Ang II and/or Ang-(1-7) for 24 h and the
phosphorylation levels of MAPKs were examined by western blot
analysis. Representative blots of three independent experiments and
densitometric quantification of the blots are shown. (B) THP-1
macrophages were treated with 10 μM SB203580 or SP600125 for
1 h prior to the addition of 1 μM Ang II. Following
treatment, the protein expression levels of PPARγ and LXRα were determined by western
blot analysis. The data are expressed as the mean ± standard
deviation of three independent experiments (*P<0.05,
vs. untreated cells; #P<0.05 vs. Ang II-only cells).
Ang-(1-7)-10 and Ang-(1-7)-100 indicate treatment with 10 or 100 nM
Ang-(1-7), respectively. Ang, angiotensin; p-, phosphory-lated; t-,
total; JNK, c-Jun NH2-terminal kianse; ERK, extracellular
signal-regulated kinase; PPAR, peroxisome proliferator-activated
receptor; LXRα, liver X receptor α. |
Discussion
Ang II ha been implicated in a variety of
cardiovascular diseases, including atherosclerosis, hypertension,
left ventricular hypertrophy, myocardial infarction and heart
failure (22). It has been
demonstrated that Ang II accelerates foam cell formation by
upregulating the expression of acyl-coenzyme A:cholesterol
acyltransferase-1 (ACAT1), which converts intracellular free
cholesterol into cholesterol ester for storage in lipid droplets
(23). In addition, the
upregulation of ACAT1, suppression of ABCA1 expression and
subsequent cholesterol transport is known to mediate the
atherosclerotic effect of Ang II (24). Consistently, the present study
demonstrated that treatment with Ang II resulted in a significant
reduction in cholesterol efflux from the THP-1 foam cells to
apoA-1. This inhibition of cholesterol accumulation was coupled
with the downregulation of ABCA1 and ABCG1. Notably, the presence
of Ang-(1-7) significantly reversed the Ang II-induced repression
of cholesterol efflux. Taken together, these results provided
evidence that Ang-(1-7) and Ang II exhibited counter-regulatory
roles in macrophage cholesterol homeostasis.
The present study revealed that treatment with Ang
II caused a significant decrease in the mRNA expression levels of
ABCA1 and ABCG1, suggesting its importance in transcriptional
regulation. Notably, co-treatment with Ang-(1-7) significantly
restored the expression levels of ABCA1 and ABCG1 in the Ang
II-treated THP-1 macrophages. Since PPARγ and LXRα are the ‘master’
transcription factors, cooperatively regulate the expression levels
of ABCA1 and ABCG1 (8,9), the present study assessed the effects
of Ang-(1-7) on the expression levels of PPARγ and LXRα. It was
found that the Ang II-mediated suppression of t PPARγ and LXRα
expression was significantly reversed following treatment with
Ang-(1-7). The Ang-(1-7)-mediated induction of PPARγ expression has
also been demonstrated in other biological contexts (25,26).
Dhaunsi et al (26)
reported that treatment with Ang-(1-7) prevents the inhibition of
PPARγ expression in diabetes and/or hypertension. The present study
further demonstrated that the pharmacological inhibition of PPARγ
with GW9662 significantly inhibited the Ang-(1-7)-mediated
induction of ABCA1 and ABCG1 mRNA expression. These results
suggested that treatment with Ang-(1-7) facilitated the
PPARγ-dependent transcriptional activation of the ABC genes, which
provided a possible explanation for its promotion of cholesterol
efflux from foam cells.
There is evidence that MAPK signaling is involved in
foam cell formation (27–29). Ren et al (27) demonstrated that oxidized HDL
accelerates foam cell formation via the p38 MAPK-dependent
upregulation of PPARγ and downregulation of cluster of
differentiation 36. Mei et al (29) demonstrated that activation of p38
MAPK promotes cholesterol ester accumulation in macrophages.
Pharmacological inhibition of p38 MAPK signaling, including
treatment with curcumin, has also been revealed to hinder oxidized
LDL-induced foam cell formation (30). In addition to p38 MAPK, activation
of the JNK MAPK is involved in foam cell formation from
macrophages, which are exposed to oxidized LDL (31). In agreement with previous studies,
the present study revealed that the Ang-(1-7)-mediated promotion of
cholesterol efflux in Ang II-treated THP-1 macrophages is coupled
with the inactivation of JNK and p38 MAPK signaling. Additionally,
the inhibition of MAPK signaling using specific pharmacological
inhibitors mimicked the induction of PPARγ and LXRα expression by
Ang-(1-7). These results suggested that the Ang-(1-7)-mediated
restoration of PPARγ and LXRα expression was associated, in part,
with the suppression of MAPK signaling. The inhibition of MAPK
signaling by Ang-(1-7) has also been demonstrated in other
biological settings. Santos et al (32) reported that oral treatment with
Ang-(1-7) prevents obesity and hepatic inflammation in high-fat fed
rats, which is associated with the suppression of the MAPK
signaling pathway. Moon et al (33) revealed that Ang II-induced MAPK
signaling activation is attenuated by Ang-(1-7) in mesangial
cells.
In conclusion, the present study provided the first
evidence, to the best of our knowledge, that Ang-(1-7) facilitated
cholesterol efflux in Ang II-treated THP-1 macrophages, which, in
part, involved the suppression of p38 and JNK signaling and the
induction of PPARγ and LXRα expression. These findings suggested
that Ang-(1-7) may offer therapeutic benefits in preventing the
development of atherosclerosis.
Acknowledgments
This study was published as an abstract in J Am Coll
Cardiol 64: S6, 2014. The present study was supported by grants
from the Scientific and Technological Project of Shanxi Province of
China (no. 20100311098-4), the Doctoral Foundation of The Second
Hospital of Shanxi Medical University of China (no. 20100404), the
Youth Foundation of Shanxi Medical University of China (no.
02201421) and the Youth Foundation of Health and Family Planning
Commission of Shanxi Province of China (no. 2014041).
References
|
1
|
Libby P, Ridker PM and Maseri A:
Inflammation and atherosclerosis. Circulation. 105:1135–1143. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fenyo IM and Gafencu AV: The involvement
of the monocytes/macrophages in chronic inflammation associated
with atherosclerosis. Immunobiology. 218:1376–1384. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu XH, Fu YC, Zhang DW, Yin K and Tang CK:
Foam cells in atherosclerosis. Clin Chim Acta. 424:245–252. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yvan-Charvet L, Wang N and Tall AR: Role
of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and
immune responses. Arterioscler Thromb Vasc Biol. 30:139–143. 2010.
View Article : Google Scholar :
|
|
5
|
Smith JD, Le Goff W, Settle M, Brubaker G,
Waelde C, Horwitz A and Oda MN: ABCA1 mediates concurrent
cholesterol and phospholipid efflux to apolipoprotein A-I. J Lipid
Res. 45:635–644. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Matsuura F, Wang N, Chen W, Jiang XC and
Tall AR: HDL from CETP-deficient subjects shows enhanced ability to
promote cholesterol efflux from macrophages in an apoE- and
ABCG1-dependent pathway. J Clin Invest. 116:1435–1442. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Costet P, Luo Y, Wang N and Tall AR:
Sterol-dependent transactivation of the ABC1 promoter by the liver
X receptor/retinoid X receptor. J Biol Chem. 275:28240–28245.
2000.PubMed/NCBI
|
|
8
|
Kennedy MA, Venkateswaran A, Tarr PT,
Xenarios I, Kudoh J, Shimizu N and Edwards PA: Characterization of
the human ABCG1 gene: liver X receptor activates an internal
promoter that produces a novel transcript encoding an alternative
form of the protein. J Biol Chem. 276:39438–39447. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chawla A, Boisvert WA, Lee CH, et al: A
PPARγ-LXR-ABCA1 pathway in macrophages is involved in cholesterol
efflux and atherogenesis. Mol Cell. 7:161–171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nagy ZS, Czimmerer Z and Nagy L: Nuclear
receptor mediated mechanisms of macrophage cholesterol metabolism.
Mol Cell Endocrinol. 368:85–98. 2013. View Article : Google Scholar
|
|
11
|
Wang Y, Tikellis C, Thomas MC and Golledge
J: Angiotensin converting enzyme 2 and atherosclerosis.
Atherosclerosis. 226:3–8. 2013. View Article : Google Scholar
|
|
12
|
Kawahito H, Yamada H, Irie D, et al:
Periaortic adipose tissue-specific activation of the renin
angiotensin system contributes to atherosclerosis development in
uninephrectomized apoE−/− mice. Am J Physiol Heart Circ Physiol.
305:H667–H675. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fujisaka T, Hoshiga M, Hotchi J, et al:
Angiotensin II promotes aortic valve thickening independent of
elevated blood pressure in apolipoprotein-E deficient mice.
Atherosclerosis. 226:82–87. 2013. View Article : Google Scholar
|
|
14
|
Santos RA, Ferreira AJ, Verano-Braga T and
Bader M: Angiotensin-converting enzyme 2, angiotensin-(1-7) and
Mas: new players of the renin-angiotensin system. J Endocrinol.
216:R1–R17. 2013. View Article : Google Scholar
|
|
15
|
Yang HY, Bian YF, Zhang HP, et al:
Angiotensin-(1-7) treatment ameliorates angiotensin II-induced
HUVEC apoptosis. Clin Exp Pharmacol Physiol. 39:1004–1010. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang JM, Dong M, Meng X, et al:
Angiotensin-(1-7) dose-dependently inhibits atherosclerotic lesion
formation and enhances plaque stability by targeting vascular
cells. Arterioscler Thromb Vasc Biol. 33:1978–1985. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Takata Y, Chu V, Collins AR, et al:
Transcriptional repression of ATP-binding cassette transporter A1
gene in macrophages: a novel atherosclerotic effect of angiotensin
II. Circ Res. 97:e88–e96. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sampaio WO, Souza dos Santos RA,
Faria-Silva R, da Mata Machado LT, Schiffrin EL and Touyz RM:
Angiotensin-(1-7) through receptor Mas mediates endothelial nitric
oxide synthase activation via Akt-dependent pathways. Hypertension.
49:185–192. 2007. View Article : Google Scholar
|
|
19
|
Hwang JS, Kang ES, Ham SA, et al:
Activation of peroxisome proliferator-activated receptor γ by
rosiglitazone inhibits lipopolysaccharide-induced release of high
mobility group box 1. Mediators Inflamm. 2012:3528072012.
View Article : Google Scholar
|
|
20
|
Trasino SE, Kim YS and Wang TT: Ligand,
receptor, and cell type-dependent regulation of ABCA1 and ABCG1
mRNA in prostate cancer epithelial cells. Mol Cancer Ther.
8:1934–1945. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔC(T)) Method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
22
|
Rosenbaugh EG, Savalia KK, Manickam DS and
Zimmerman MC: Antioxidant-based therapies for angiotensin
II-associated cardiovascular diseases. Am J Physiol Regul Integr
Comp Physiol. 304:R917–R928. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kanome T, Watanabe T, Nishio K, Takahashi
K, Hongo S and Miyazaki A: Angiotensin II upregulates
acyl-CoA:cholesterol acyltransferase-1 via the angiotensin II Type
1 receptor in human monocyte-macrophages. Hypertens Res.
31:1801–1810. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Y, Chen Z, Liao Y, et al: Angiotensin
II increases the cholesterol content of foam cells via
down-regulating the expression of ATP-binding cassette transporter
A1. Biochem Biophys Res Commun. 353:650–654. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mario EG, Santos SH, Ferreira AV, Bader M,
Santos RA and Botion LM: Angiotensin-(1-7) Mas-receptor deficiency
decreases peroxisome proliferator-activated receptor γ expression
in adipocytes. Peptides. 33:174–177. 2012. View Article : Google Scholar
|
|
26
|
Dhaunsi GS, Yousif MH, Akhtar S, Chappell
MC, Diz DI and Benter IF: Angiotensin-(1-7) prevents
diabetes-induced attenuation in PPAR-gamma and catalase activities.
Eur J Pharmacol. 638:108–114. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ren J, Jin W and Chen H: oxHDL decreases
the expression of CD36 on human macrophages through PPARgamma and
p38 MAP kinase dependent mechanisms. Mol Cell Biochem. 342:171–181.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rohde E, Schallmoser K, Reinisch A, et al:
Pro-angiogenic induction of myeloid cells for therapeutic
angiogenesis can induce mitogen-activated protein kinase
p38-dependent foam cell formation. Cytotherapy. 13:503–512. 2011.
View Article : Google Scholar :
|
|
29
|
Mei S, Gu H, Ward A, et al: p38
mitogen-activated protein kinase (MAPK) promotes cholesterol ester
accumulation in macrophages through inhibition of macroautophagy. J
Biol Chem. 287:11761–11768. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Min KJ, Um HJ, Cho KH and Kwon TK:
Curcumin inhibits oxLDL-induced CD36 expression and foam cell
formation through the inhibition of p38 MAPK phosphorylation. Food
Chem Toxicol. 58:77–85. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yin R, Dong YG and Li HL: PPARγ
phosphorylation mediated by JNK MAPK: a potential role in
macrophage-derived foam cell formation. Acta Pharmacol Sin.
27:1146–1152. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Santos SH, Andrade JM, Fernandes LR, et
al: Oral Angiotensin-(1-7) prevented obesity and hepatic
inflammation by inhibition of resistin/TLR4/MAPK/NF-κB in rats fed
with high-fat diet. Peptides. 46:47–52. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moon JY, Tanimoto M, Gohda T, et al:
Attenuating effect of angiotensin-(1-7) on angiotensin II-mediated
NAD(P)H oxidase activation in type 2 diabetic nephropathy of
KK-A(y)/Ta mice. Am J Physiol Renal Physiol. 300:F1271–F1282. 2011.
View Article : Google Scholar : PubMed/NCBI
|