Introduction
Traumatic brain injury (TBI) is one of the leading
causes of morbidity and mortality in young adults and children, and
is a leading public health problem worldwide (1). In TBI, neurological impairment is
caused by immediate brain tissue disruption (primary injury) and
post-injury cellular and molecular events (secondary injury) that
exacerbate the primary neurological insult (2). These combined events lead to the
induction of mitochondrial dysfunction and the amplification of
biochemical cell-death signaling cascades, which cause neuronal
cell death and general neurological functional deficits (3,4).
However, the destructive molecular events that follow TBI evolve
over several days, and therefore there is a window of opportunity
during which therapeutic strategies may improve outcome.
Autophagy is a highly regulated process that
involves the degradation of a cell’s cytoplasmic macromolecules and
organelles. In mammalian cells, this catabolic mechanism utilizes
the lysosomal system and has a homeostatic function in normal cell
growth and development, helping to maintain a balance between the
synthesis, degradation and subsequent recycling of cellular
products (5,6). Identification of autophagosomes
remains the ‘gold standard’ method for the detection of autophagy
(7). The first direct evidence
that autophagy is increased following TBI, was reported in a study
by Lai et al, which used a controlled cortical impact model
of TBI to investigate this effect (8). More recently, a study administered a
selective inhibitor of autophagy, 3-methyladenine, in a rat model
of transient focal cerebral ischemia. They identified that this
agent reduced infarct size, as compared with the vehicle treatment,
which had no effect (9). However,
3-methyladenine has other effects, including inhibition of
non-class III PI3-kinases and promotion of glycogen breakdown in
hepatocytes (10).
Chloroquine (CQ) has long been used in the treatment
and prevention of malaria, and less commonly has been employed in
the treatment of autoimmune diseases, due to its immunosuppressive
properties (11,12). Recently, CQ has been recognized as
an inhibitor of autophagy, and thus has been used as a
pharmacological tool to study the role of autophagy in the
laboratory (13,14). CQ has also been used in clinical
trials to evaluate its efficacy as an adjuvant to cancer
therapeutic regimens (15).
Inhibition of lysosome activity by CQ arrests the late stages of
autophagy, including the degradation of the autolysosome, which
prevents the supply of energy to the cell through the autophagy
pathway (16). Recent studies have
revealed that CQ is emerging as a potential therapeutic target in
acute and chronic neurological disorders, including brain ischemia
and Alzheimer’s disease (17,18).
Nevertheless, no studies have examined the potential for CQ to
provide neuroprotection in an animal model of TBI.
This study was designed to investigate the
hypothesis that CQ has neuroprotective effects, via the attenuation
of autophagy and inflammation, in a rat model of TBI.
Materials and methods
Animals
A total of 150 Sprague-Dawley rats (obtained from
Hebei United University Experimental Animal Center, Tangshan,
Hebei, China), weighing 280–320 g, were housed under a 12 h
light/dark cycle with regular food and water supply. All procedures
were performed in accordance with the institutional guidelines for
the care and use of laboratory animals (Hebei Medical University,
Shijiazhuang, China), and conformed to the National Institutes of
Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH
Publication no. 80–23, revised 1996).
Models of TBI
The rat model of TBI was induced using a modified
weight-drop device, as described previously by Marmarou et
al (19). Briefly, rats were
anesthetized with sodium pentobarbital (Nembutal, 60 mg/kg) prior
to surgery. A midline incision was made to expose the skull between
the bregma and lambda suture lines and a steel disc (10 mm in
diameter and 3 mm thickness) was adhered to the skull using dental
acrylic. Following this, rats were placed on a foam mattress
underneath a weight-drop device, where a 450 g weight falls freely
though a vertical tube from 1.5 m onto the steel disk. The
sham-operated animals underwent the same surgical procedure,
however they did not undergo TBI. Following the surgery, the rats
received supporting oxygenation with 95% O2 for no
longer than 2 min and were then returned to their cages. All of the
rats were housed in individual cages and placed on heat pads (37°C)
for 24 h, to maintain normal body temperature during the recovery
period.
Group and drug administration
Rats were randomly assigned to the sham-operated
group (sham, n=30), TBI treated with CQ group (CQ, n=60) and TBI
received only equal volumes of 0.9% saline solution (vehicle,
n=60). CQ was dissolved in 0.9% saline and stored at 4°C. Following
brain injury in the CQ group, CQ was immediately administered as an
intraperitoneal injection (3 mg/kg body weight). All investigations
were blind and the animal codes were revealed only at the end of
the behavioral and histological analyses.
Immunofluorescence
Brain tissues were fixed in 4% paraformaldehyde for
24 h, removed into 30% sucrose solution with 0.1 mol/l
phosphate-buffered saline (PBS; pH 7.4) until sinking to the
bottom. Sections (200 µm apart) from the anterior to
posterior hippocampus (bregma, 1.9–3 mm) were made from the TBI
rats and then embedded in OCT. Frozen sections (15 µm) were
sliced with a frozen slicer, treated with 0.4% Triton X-100 for 10
min and blocked in normal donkey serum for 1 h. For double
labeling, the frozen sections were incubated with a mixture of
rabbit anti-microtubule-associated protein 1 light chain 3 (LC3)
polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA; dilution, 1:100) and mouse anti-neuron-specific nuclear
protein (NeuN) polyclonal antibody (Santa Cruz Biotechnology, Inc.;
dilution, 1:100) overnight at 4°C. The next day, sections were
incubated with a mixture of fluorescein-conjugated anti-rabbit IgG
and anti-mouse IgG (Santa Cruz Biotechnology, Inc.; dilution,
1:1,000) for 2 h at 37°C in the dark. All cell nuclei were
counterstained by 4′,6-diamidino-2-phenylindole (DAPI). Photographs
were taken in a laser scanning confocal microscope (Olympus FV1000;
Olympus Corporation, Tokyo, Japan). Primary antibodies were
replaced with PBS in the negative control group.
Western blot analysis
Briefly, rats were anesthetized and underwent
intracardiac perfusion with 0.1 mol/l PBS (pH 7.4). The hippocampal
region of each rat brain was rapidly isolated, total proteins were
extracted and protein concentration was determined by the BCA
reagent (Beijing Solarbio Science & Technology Co., Ltd,
Beijing, China) method. Samples were subjected to sodium dodecyl
sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Separated
proteins on the gel were transferred onto PVDF membranes (Roche
Diagnostics, Mannheim, Germany). Blots were blocked with 5%
fat-free dry milk for 1 h at room temperature. Following blocking,
the membrane was incubated with the primary antibodies overnight at
4°C, including rabbit anti-LC3 polyclonal antibodies (Santa Cruz
Biotechnology, Inc.; dilution, 1:500), rabbit anti-interleukin
(IL)-1β polyclonal antibody (Santa Cruz Biotechnology, Inc.;
dilution, 1:500), rabbit anti-tumor necrosis factor (TNF)-α
polyclonal antibody (Santa Cruz Biotechnology, Inc.; dilution,
1:500), mouse anti-β-actin monoclonal antibody (Santa Cruz
Biotechnology, Inc.; dilution, 1:500). The antibodies were then
incubated with horseradish peroxidase conjugated anti-rabbit IgG
and anti-mouse IgG (Cell Signaling Technology, Inc., Danvers, MA,
USA; dilution, 1:5,000) for 2 h at room temperature. Following
incubation with a fully titrated second antibody, the immunoblot on
the membrane was visible. After development with an enhanced
chemiluminescence (ECL) detection system, the densitometric signals
were quantified using an imaging program. Immunoreactive bands of
the protein expression were normalized to the intensity of
corresponding bands for β-actin. The western blot analysis results
were analyzed with NIH Image 1.41 software (Bethesda, MD, USA).
Evaluation of brain edema
Brain edema was evaluated by analysis of brain water
content as previously described (20). Rat brains were separated and
weighed immediately with a chemical balance to obtain the wet
weight (WW). Following drying in a desiccating oven for 24 h at
100°C, dry tissues were weighed again to obtain the constant dry
weight (DW). The percentage of water in the tissues was calculated
according to the formula: % brain water = [(WW − DW)/WW] × 100.
Recovery of motor function
The neurobehavioral status of the rats was evaluated
using a set of 10 tasks, collectively termed the Neurologic
Severity Score (NSS), which tests reflexes, alertness, coordination
and motor abilities. One point is awarded for failure to perform a
particular task, thus, a score of ten reflects maximal impairment,
whereas a normal rat scores zero (21). Post-injury, NSS was evaluated at
day 1, 3, 7 and 14. Each animal was assessed by an observer who was
blinded to the type of treatment the animal had received. The
difference between the initial NSS and that at a later time was
calculated for each rat, and this value (∆NSS) reflected the
spontaneous or treatment-induced recovery of motor function.
Morris water maze test
The spatial learning ability was assessed in a
Morris water maze as described previously (22). The Morris water maze consists of a
black circular pool (180 cm diameter, 45 cm high) filled with water
(30 cm depth) at 26°C and virtually divided into four equivalent
quadrants: north (N), west (W), south (S) and east (E). A 2 cm
submerged escape platform (diameter 12 cm, height 28 cm, made
opaque with paint) was placed in the middle of one of the quadrants
equidistant from the sidewall and the center of the pool. Rats were
trained to find the platform prior to TBI or sham operation. For
each trial, the rat was randomly placed into a quadrant start point
(N, S, E or W) facing the wall of the pool and was allowed a
maximum of 60 sec to escape to the platform. Rats that failed to
escape within 90 sec were placed on the platform for a maximum of
20 sec and returned to the cage for a new trial (intertrial
interval, 20 sec). Maze performance was recorded using a video
camera suspended above the maze and interfaced with a video
tracking system (HVS Imaging, Hampton, UK). The average escape
latency of a total of five trials was calculated. This test was
conducted at 3, 7 and 14 days following trauma or sham
operation.
Statistical analysis
All data were presented as the mean ± standard error
(SE). SPSS 16.0 (SPSS Inc., Chicago, IL, USA) was used for
statistical analysis of the data. Statistical analysis was
performed using one-way analysis of variance and followed by the
Student-Newman-Keuls post-hoc test. P<0.05 was considered to
indicate a statistically significant result.
Results
Neurological deficit following TBI
As previously described by Marmarou et al
(19), animals showed no
significant difference in baseline locomotor activity for any
parameters, including horizontal activity, vertical activity, total
distance or stereotypy prior to surgery. Following the induction of
injury, rats exhibited moderate to severe neurological deficits,
including forelimb upon lifting the animal by its tail, decreased
resistance to lateral push and reduced locomotor activity, flexion
of contralateral torso and loss of righting reflex. Any mice not
exhibiting behavioral deficits consistent with the surgery were
excluded from further study.
Treatment with CQ attenuates neurons
autophagy in the hippocampus following TBI
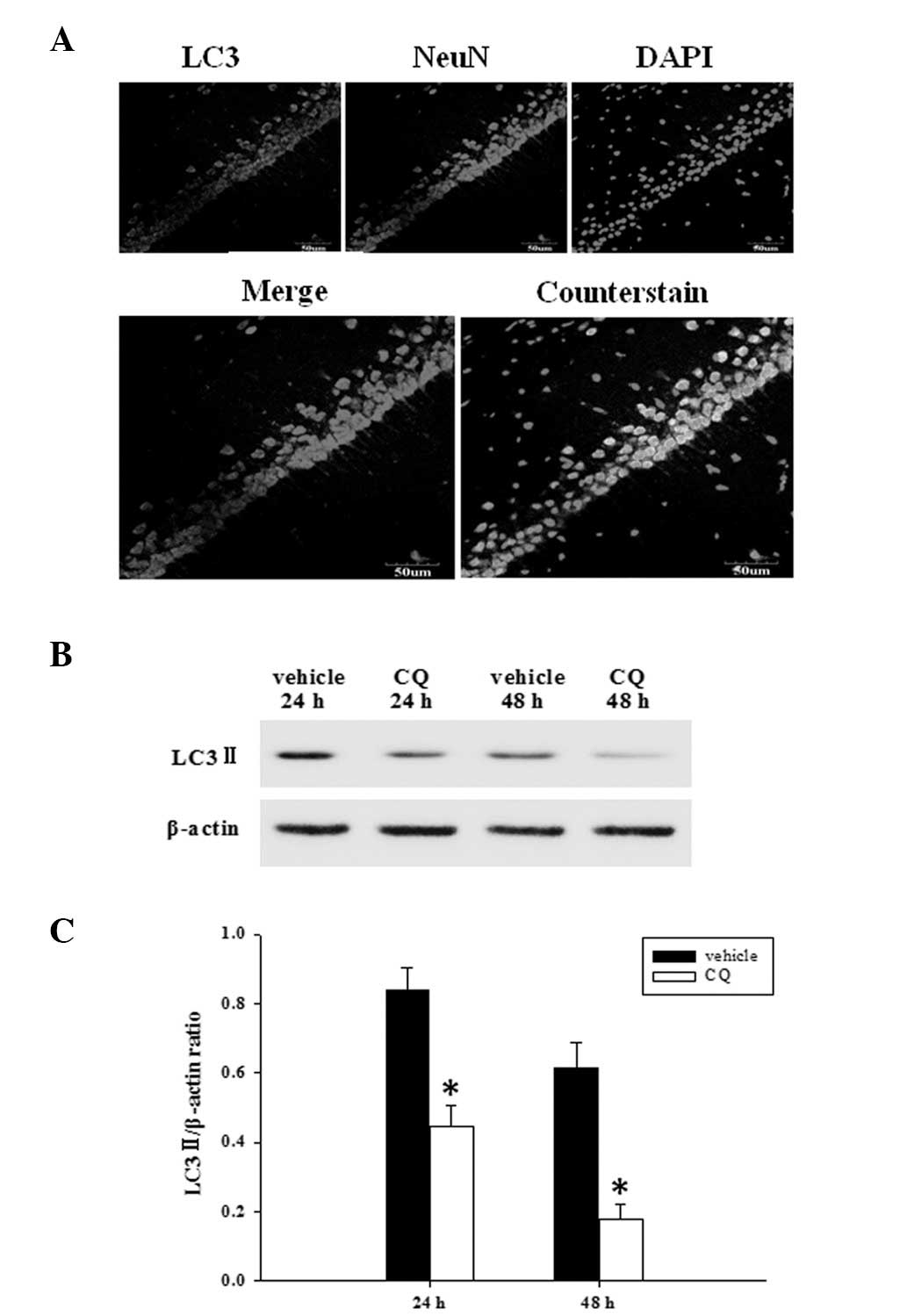
Since it has been recently demonstrated that the
expression of the autophagy marker protein, LC3, was markedly
elevated at 24 h following TBI (8), the co-localization of NeuN and LC3
was followed with immunofluorescent staining at 24 h. As summarized
in Fig. 1A, the majority of
autophagy induced following TBI, occurred in neurons. Then, we
examined whether CQ treatment inhibited the expression of LC3-II,
as determined by western blot analysis (Fig. 1B). As demonstrated in Fig. 1C, at 24 and 48 h following TBI,
administration of CQ significantly attenuated the LC3-II protein
expression in the rat hippocampus compared with the TBI group.
Treatment with CQ attenuates inflammatory
cytokine levels in the hippocampus following TBI
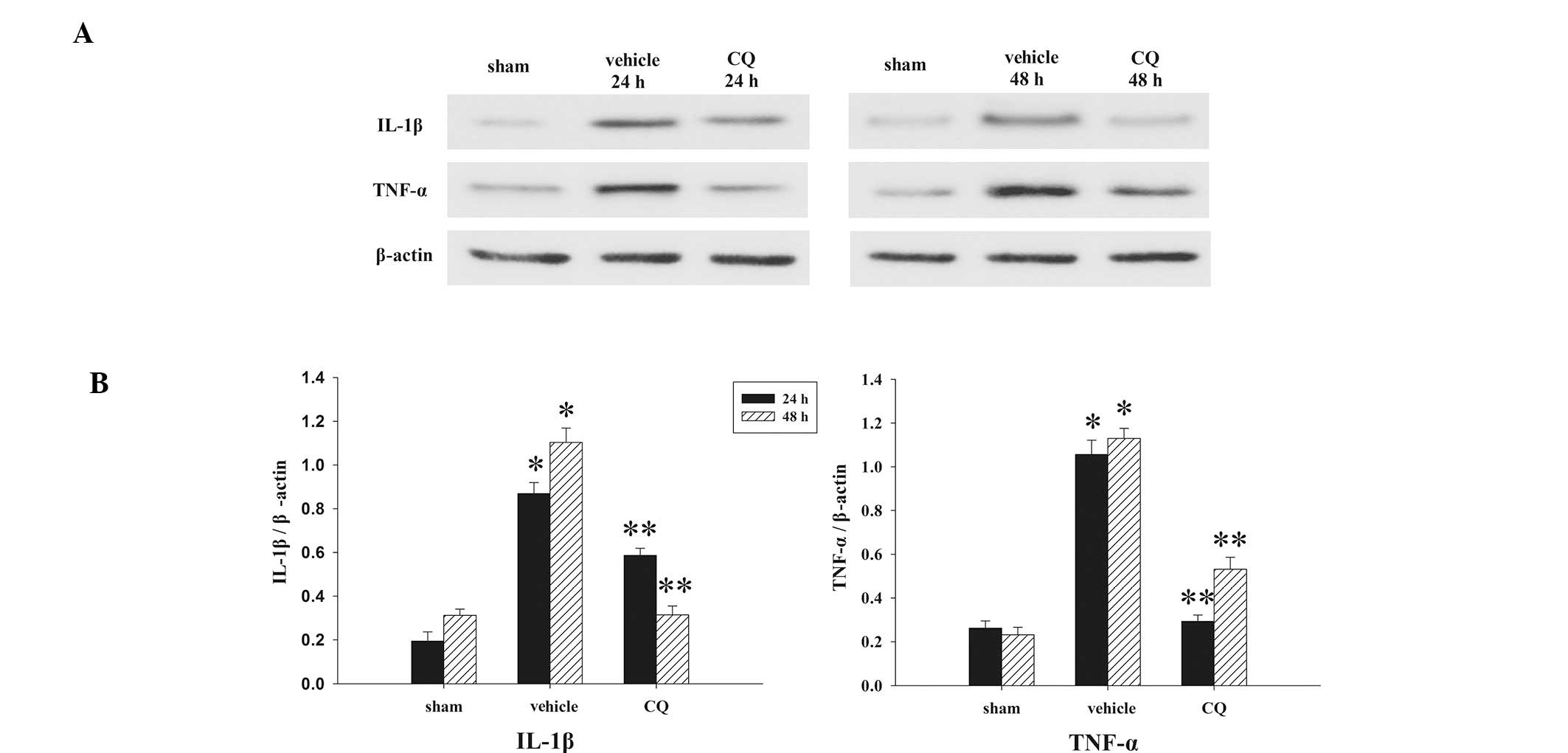
The expression levels of IL-1β and TNF-α in the
hippocampus at 24 and 48 h, were measured by western blot analysis
(Fig. 2A). As revealed in Fig. 2B, the inflammatory cytokine levels
in the sham rat hippocampus at each time point, following the
induction of injury, were consistently presented in a low
background. All measured cytokine levels exhibited significant
increases from different time points in the TBI group. Our results
revealed that administration of CQ produced significant reductions
in the injury-induced upregulation of IL-1β and TNF-α
expression.
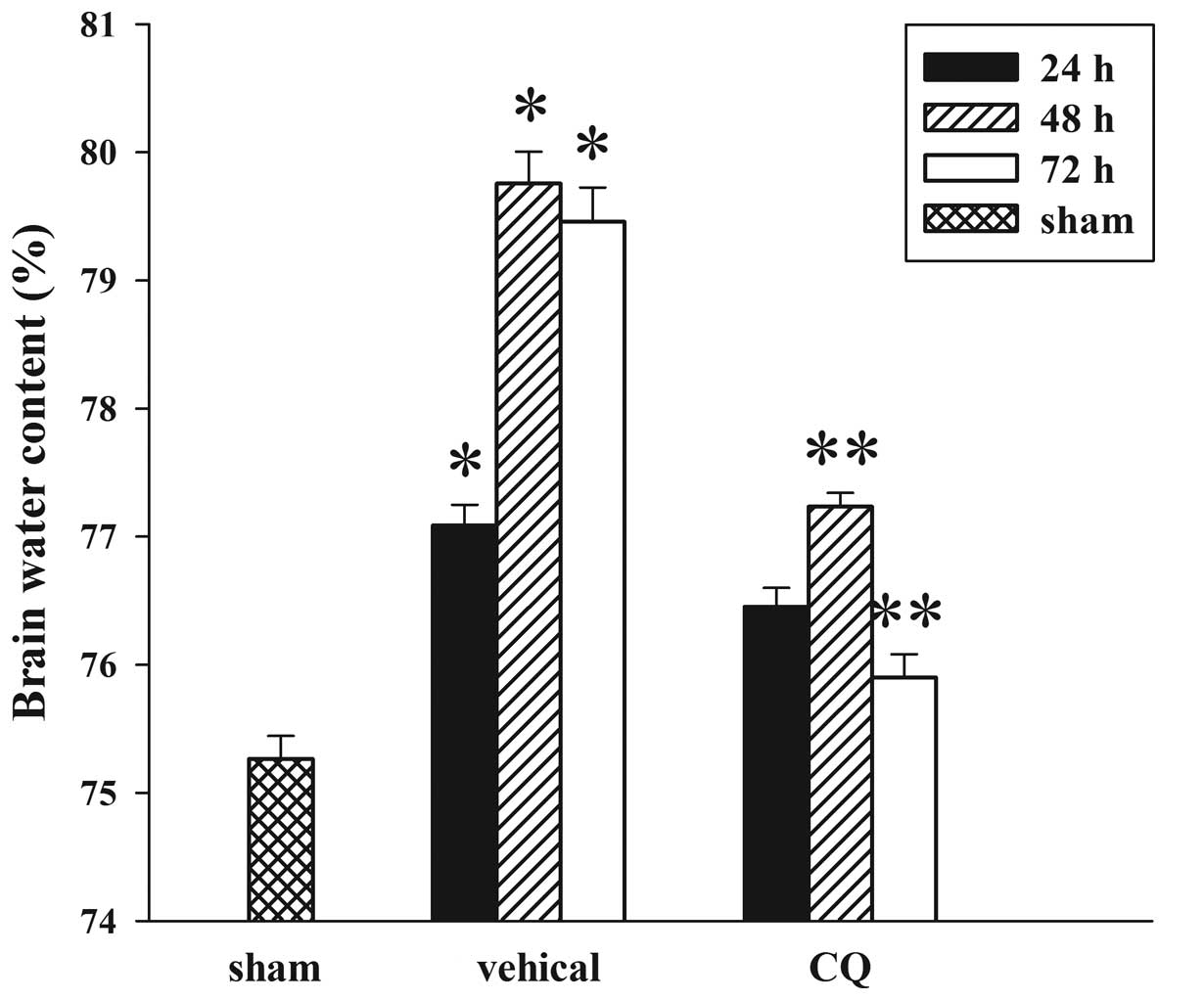
Treatment of CQ attenuates TBI-induced
cerebral edema
The wet-dry weight method was used to evaluate brain
edema. As shown in Fig. 3, CQ
post-injury administration attenuated cerebral edema following TBI.
In the TBI group, brain water content was significantly increased
compared with the sham group at 24, 48 and 72 h after trauma.
Furthermore, tissue water content in the CQ treatment group was
significantly reduced at 48 and 72 h compared with the TBI group at
the same time points.
Treatment of CQ attenuates TBI-induced
motor deficits
Fig. 4 depicts the
temporal changes in functional recovery of the rat, expressed as
∆NSS. It is clear that post-injury administration of CQ
significantly improved the motor function recovery of the trauma
rats between 24 h and 14 days following TBI.
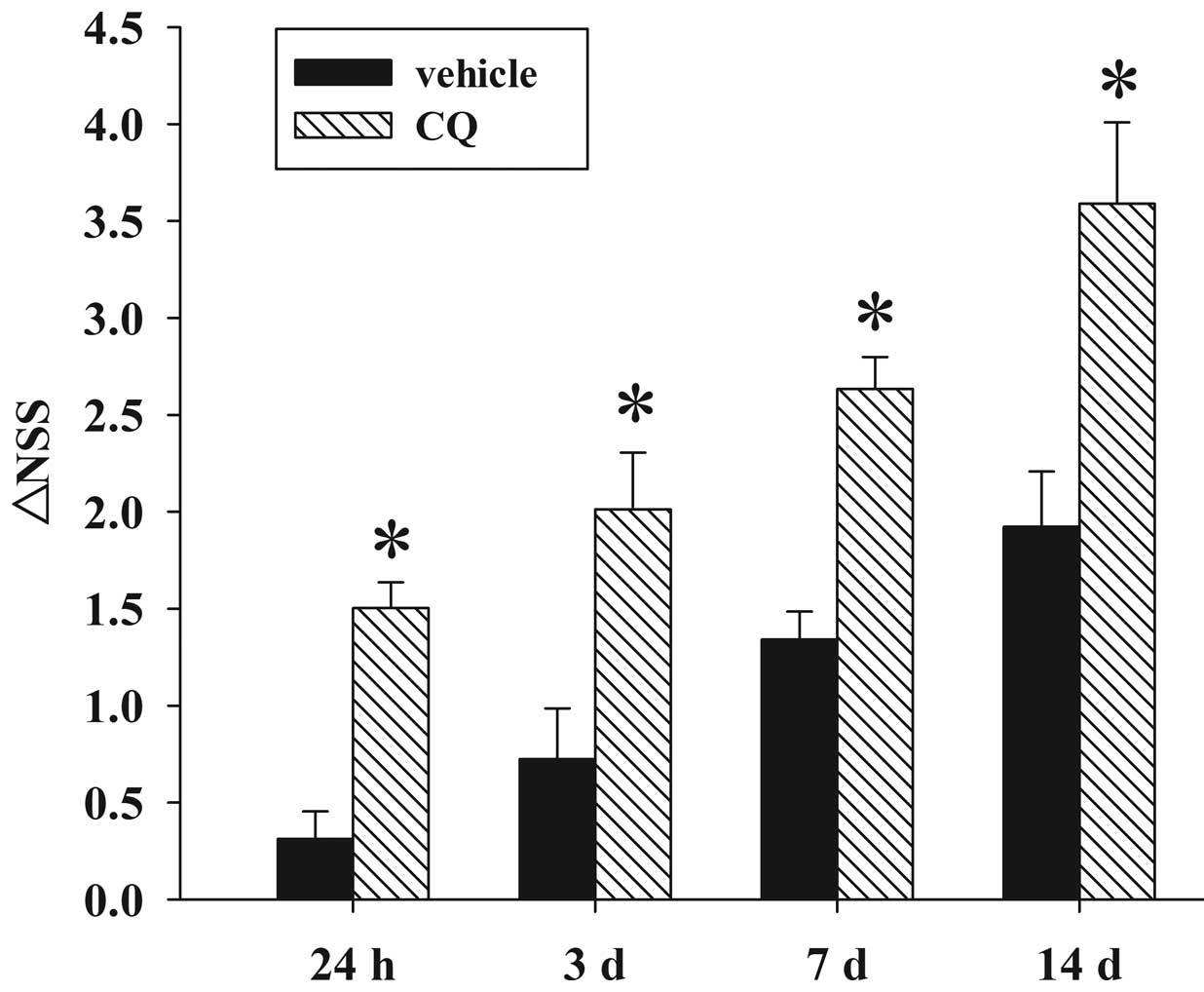 | Figure 4Effect of CQ on TBI-induced motor
deficits. The temporal changes in motor recovery of the rat was
determined at 24 h, 3, 7 and 14 d following TBI and calculated as
∆NSS. Bars represent the mean ± SE (n=5/group). Administration of
CQ significantly improved motor function at 24 h, 3, 7 and 14 d
following TBI (**P<0.05 vs. TBI group), as reflected
by an increase in ∆NSS. TBI, traumatic brain injury; CQ,
chloroquine; SE, standard error; NSS, Neurologic Severity Score; d,
days. |
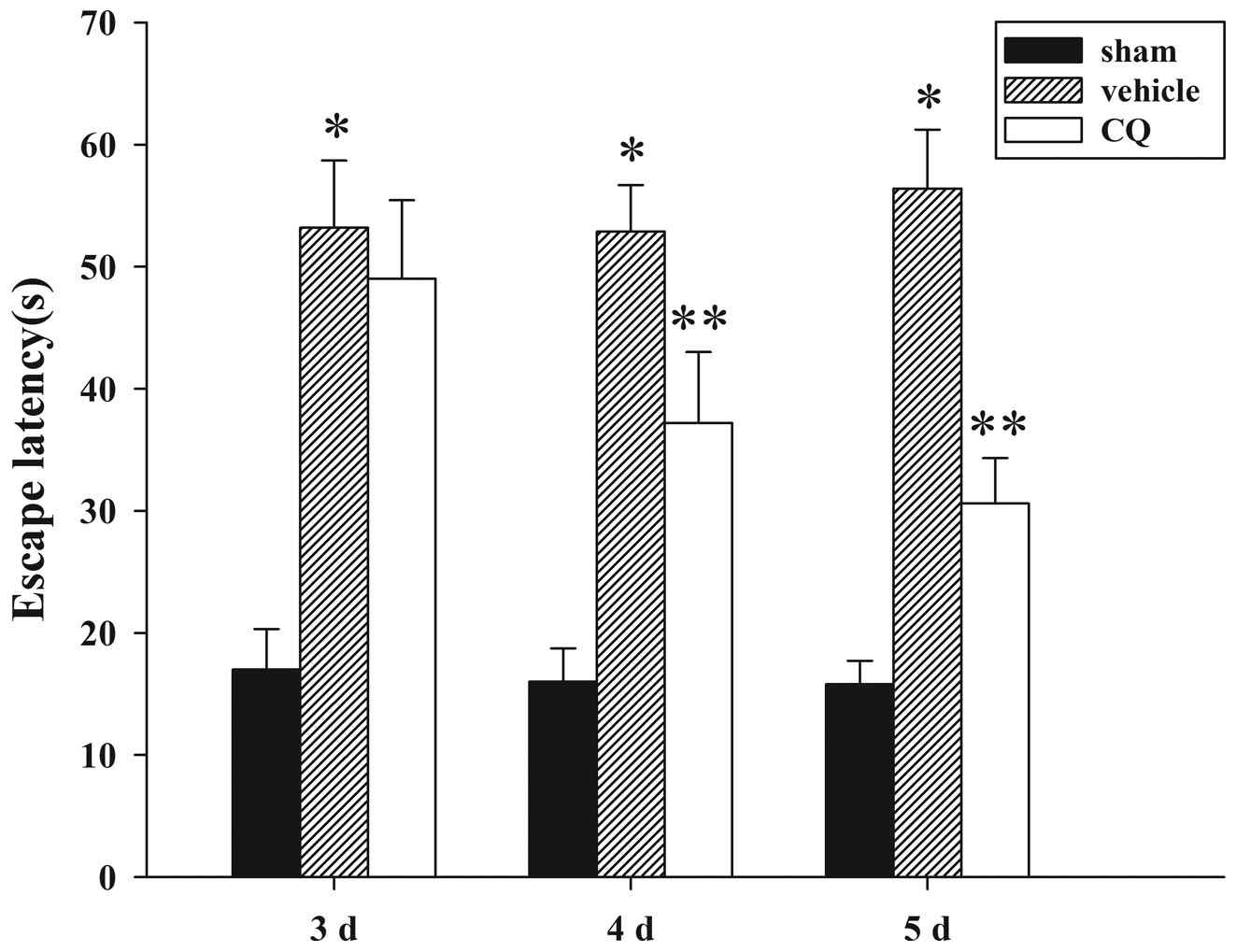
Treatment of CQ attenuates TBI-induced
learning and spatial memory function
Since CQ treatment was able to attenuate brain edema
and improve motor deficits, we next examined whether CQ treatment
could improve spatial learning function, as assessed by the Morris
water maze at day 3, 7 and 14 following TBI or sham operation. As
demonstrated in Fig. 5, TBI caused
a significant spatial learning deficit at 3, 7 and 14 days compared
with the sham group, and CQ treatment significantly reduced the
escape latency at 7 and 14 days compared with the TBI group.
Discussion
TBI is caused by immediate brain tissue disruption
(primary injury) and post-injury cellular and molecular events
(secondary injury). Primary damage is due to mechanical factors and
occurs immediately at the moment of injury. Secondary injury is
delayed and is produced by complex processes that are initiated at
the moment of impact, but does not present clinically for a period
of hours to days following the injury. This delayed
pathophysiological cascade is now believed to result from a
combination of factors, including increases in neurotransmitter
release (23), free radical
overproduction (24), exacerbated
inflammatory response (25) and
subsequent neuronal cell death via apoptosis and autophagy
(8,26).
In the present study, we investigated the efficacy
of the autophagy inhibitor, CQ, as a therapeutic strategy for the
acute treatment of TBI. It was identified that a single injection
of CQ immediately following TBI, is neuroprotective in rats. The
antimalarial drug CQ has been recognized as an autophagy inhibitor
in a variety of disorders, including Alzheimer’s disease and brain
ischemia (15–18). Using a rat model of TBI, we have
confirmed and extended these earlier observations, demonstrating
for the first time, to the best of our knowledge, that post-injury
administration of CQ improves behavioral outcomes and attenuates
secondary cerebral edema following experimentally induced TBI in
rats.
The use of animal models of TBI is essential for
investigating and understanding the complex physiological and
behavioural changes following injury. There are numerous
established animal models of TBI, including rigid indentation,
fluid percussion, impact acceleration, weight-drop and dynamic
cortical deformation (27).
However, head injury is a spontaneous, unpredictable event and no
single animal model is entirely successful in reproducing the
complete spectrum of pathological changes observed following TBI in
humans (27). In this study, we
used Marmarou’s weight-drop model, which is currently the most
commonly used to produce direct focal cortical compression in
vivo, for its advantages of being simple and easy to operate.
Nevertheless, there are limitations associated with this model,
including variation of impact velocity by the falling 450 g steel
column and the possibility of a rebound impact which together may
result in the biomechanical data failing to accurately describe the
resulting brain deformations. Therefore, any mice not exhibiting
moderate to severe neurological deficits consistent with the
surgery, were excluded from further study.
The first study to demonstrated that TBI may
stimulate autophagy was conducted by Diskin et al in 2005
(28). This laboratory later
evaluated the effects of treatment with the autophagy-inducer,
rapamycin, in the closed head injury model. They identified that
following TBI, intraperitoneal injection of rapamycin resulted in
improved neurobehavioral function as determined by an NSS and
increased neuronal survival in the injured region (29). Collectively, these data support the
hypothesis that rapamycin-enhanced autophagy produces beneficial
neurological effects following TBI. By contrast, Lai et al
have demonstrated that systemic administration of the antioxidant,
γ-glutamylcysteine ethyl ester (GCEE), following TBI, resulted in
reduced autophagy as determined by LC3-II formation, which improved
cognitive performance and reduced histological damage (8). Therefore, the role of autophagy after
TBI, whether beneficial or detrimental, remains controversial.
In this study, we demonstrated that post-injury
administration of the autophagy inhibitor, CQ, improved behavioral
outcomes and attenuated secondary cerebral edema following TBI in
rats. Our results are consistent with the findings of Lai et
al, and it is therefore conceivable to hypothesize that the
mechanism underlying the neuroprotective effects of CQ on TBI, is
through the prevention of autophagic neuronal death. Furthermore,
it appears these protective effects may also be explained by CQ’s
anti-inflammatory properties. It has been demonstrated that CQ
inhibits TNF-α, IL-1 and IL-6 production from mononuclear
phagocytes (30,31). In the present study, we observed
that the expression of the inflammatory cytokines, IL-1β and TNF-α,
in the injured rat hippocampus of brain was suppressed by CQ, as
determined by western blot analysis. Hence, we suggest that the
suppression of inflammatory cytokines following TBI may be
associated with the neuroprotective effects of CQ treatment.
It has been hypothesized that CQ, rapamycin and
GCEE, are not specific modulators of autophagy activity, but that
these agents have a variety of other effects on cellular functions
(31–33). Thus, targeting the specific
processes of autophagy requires further investigation.
In summary, this study demonstrated that neuronal
autophagy was inhibited by post-injury treatment of CQ in a rat
model of TBI. Furthermore, CQ attenuates secondary brain edema and
improves cognitive functioning. These findings emphasize that CQ
administered immediately following injury, could be neuroprotective
against the damaging consequences of TBI, and we anticipate that
this study has shed light on the potential use of CQ as a
neuroprotective agent in the treatment of cerebral injuries.
Acknowledgments
This study was supported by the grant from the
Natural Science Foundation of Hebei, China (grant nos. H2012401071
and H2014105079).
Abbreviations:
|
CQ
|
chloroquine
|
|
TBI
|
traumatic brain injury
|
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
NeuN
|
neuron-specific nuclear protein
|
|
DAPI
|
4′,6-diamidino-2-phenylindole
|
|
NSS
|
Neurologic Severity Score
|
|
TNF-α
|
tumor necrosis factor-α
|
|
IL-1
|
interleukin-1
|
References
|
1
|
Thurman DJ, Alverson C, Dunn KA, Guerrero
J and Sniezek JE: Traumatic brain injury in the United States: a
public health perspective. J Head Trauma Rehabil. 14:602–615. 1999.
View Article : Google Scholar
|
|
2
|
Davis AE: Mechanisms of traumatic brain
injury: biomechanical, structural and cellular considerations. Crit
Care Nurs Q. 23:1–13. 2000. View Article : Google Scholar
|
|
3
|
Wang YQ, Wang L, Zhang MY, et al:
Necrostatin-1 suppresses autophagy and apoptosis in mice traumatic
brain injury model. Neurochem Res. 37:1849–1858. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Luo CL, Chen XP, Yang R, et al: Cathepsin
B contributes to traumatic brain injury-induced cell death through
a mitochondria-mediated apoptotic pathway. J Neurosci Res.
88:2847–2858. 2010.PubMed/NCBI
|
|
5
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: a double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mizushima N: Autophagy: process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Galluzzi L, Aaronson SA, Abrams J, et al:
Guidelines for the use and interpretation of assays for monitoring
cell death in higher eukaryotes. Cell Death Differ. 16:1093–1107.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lai Y, Hickey RW, Yaming Chen Y, et al:
Autophagy is increased after traumatic brain injury in mice and is
partially inhibited by the antioxidant gamma-glutamylcysteinyl
ethyl ester. J Cereb Blood Flow Metab. 28:540–550. 2008. View Article : Google Scholar
|
|
9
|
Puyal J, Vaslin A, Mottier V and Clarke
PG: Postischemic treatment of neonatal cerebral ischemia should
target autophagy. Ann Neurol. 66:378–389. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Caro LH, Plomp PJ, Wolvetang EJ, Kerkhof C
and Meijer AJ: 3-Methyladenine, an inhibitor of autophagy, has
multiple effects on metabolism. Eur J Biochem. 175:325–329. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Homewood CA, Warhurst DC, Peters W and
Baggaley VC: Lysosomes, pH and the anti-malarial action of
chloroquine. Nature. 235:50–52. 1972. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Slater AF: Chloroquine: mechanism of drug
action and resistance in Plasmodium falciparum. Pharmacol Ther.
57:203–235. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lopez G, Torres K and Lev D: Autophagy
blockade enhances HDAC inhibitors’ pro-apoptotic effects: potential
implications for the treatment of a therapeutic-resistant
malignancy. Autophagy. 7:440–441. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sotelo J, Briceño E and López-González MA:
Adding chloroquine to conventional treatment for glioblastoma
multiforme: a randomized, double-blind, placebo-controlled trial.
Ann Intern Med. 144:337–343. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kimura T, Takabatake Y, Takahashi A and
Isaka Y: Chloroquine in cancer therapy: a double-edged sword of
autophagy. Cancer Res. 73:3–7. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu C, Gao Y, Barrett J and Hu B:
Autophagy and protein aggregation after brain ischemia. J
Neurochem. 115:68–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang JY, Peng C, Shi H, Wang S, Wang Q
and Wang JZ: Inhibition of autophagy causes tau proteolysis by
activating calpain in rat brain. J Alzheimers Dis. 16:39–47.
2009.PubMed/NCBI
|
|
19
|
Marmarou A, Foda MA, van den Brink W,
Campbell J, Kita H and Demetriadou K: A new model of diffuse brain
injury in rats. J Neurosurg. 80:291–300. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang J, Liu J, Zhou C, et al: Mmp-9
deficiency enhances collagenase-induced intracerebral hemorrhage
and brain injury in mutant mice. J Cereb Blood Flow Metab.
24:1133–1145. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Beni-Adani L, Gozes I, Cohen Y, et al: A
peptide derived from activity-dependent neuroprotective protein
(ADNP) ameliorates injury response in closed head injury in mice. J
Pharmacol Exp Ther. 296:57–63. 2001.
|
|
22
|
Hui-guo L, Kui L, Yan-ning Z and Yong-jian
X: Apocynin attenuate spatial learning deficits and oxidative
responses to intermittent hypoxia. Sleep Med. 11:205–212. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yi JH and Hazell AS: Excitotoxic
mechanisms and the role of astrocytic glutamate transporters in
traumatic brain injury. Neurochem Int. 48:394–403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tyurin VA, Tyurina YY, Borisenko GG, et
al: Oxidative stress following traumatic brain injury in rats. J
Neurochem. 75:2178–2189. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Frugier T, Morganti-Kossmann MC, O’Reilly
D and McLean CA: In situ detection of inflammatory mediators in
post mortem human brain tissue after traumatic injury. J
Neurotrauma. 27:497–507. 2010. View Article : Google Scholar
|
|
26
|
Raghupathi R: Cell death mechanisms
following traumatic brain injury. Brain Pathol. 14:215–222. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
O’Connor WT, Smyth A and Gilchrist MD:
Animal models of traumatic brain injury: a critical evaluation.
Pharmacol Ther. 130:106–113. 2011. View Article : Google Scholar
|
|
28
|
Diskin T, Tal-Or P, Erlich S, et al:
Closed head injury induces upregulation of Beclin 1 at the cortical
site of injury. J Neurotrauma. 22:750–762. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Erlich S, Alexandrovich A, Shohami E and
Pinkas-Kramarski R: Rapamycin is a neuroprotective treatment for
traumatic brain injury. Neurobiol Dis. 26:86–93. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van den Borne BE, Dijkmans BA, de Rooij H,
le Cessie S and Verweij CL: Chloroquine and hydroxychloroquine
equally affect tumor necrosis factor-alpha, interleukin 6, and
interferon-gamma production by peripheral blood mononuclear cells.
J Rheumatol. 24:55–60. 1997.PubMed/NCBI
|
|
31
|
Jang CH, Choi JH, Byun MS and Jue DM:
Chloroquine inhibits production of TNF-alpha, IL-1beta and IL-6
from lipopolysac-charide-stimulated human monocytes/macrophages by
different modes. Rheumatology (Oxford). 45:703–710. 2006.
View Article : Google Scholar
|
|
32
|
Fullerton HJ, Ditelberg JS, Chen SF, et
al: Copper/zinc superoxide dismutase transgenic brain accumulates
hydrogen peroxide after perinatal hypoxia ischemia. Ann Neurol.
44:357–364. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sehgal S, Baker H and Vézina C: Rapamycin
(AY-22,989), a new antifungal antibiotic. II Fermentation,
isolation and characterization. J Antibiot (Tokyo). 28:727–732.
1975. View Article : Google Scholar
|