Introduction
Obstructive sleep apnea syndrome (OSAS) is a
prevalent disorder, affecting 4% of adults (1). Patients with OSAS manifest with
repetitive episodes of transient oxygen de-saturation during sleep,
resulting in progressive multisystem damage (2). It has been recently reported that
OSAS is an independent risk factor for a number of cerebral
vascular disorders and is associated with Alzheimer's disease.
Surgical treatment may only partially improve cognitive function in
patients with OSAS. Thus, OSAS-induced damage to the brain may be
irreversible (3,4). Although the pathophysiological basis
of cerebral complications in OSAS is likely to be multifactorial,
including sympathetic excitation, cerebral vascular contraction,
inflammation, oxidative stress and a disorder of the metabolism
(5–8), the precise underlying mechanisms
remain to be elucidated and require further investigation.
The mitogen-activated protein kinases (MAPKs)
comprise a family of ubiquitous proline-directed
protein-serine/threonine kinases, which are essential in the
sequential transduction of biological signals from the cell
membrane to the nucleus (9). In
mammalian cells, there are three well-defined sub-groups of MAPKs:
Extracellular signal-regulated kinases (ERKs, including ERK1 and
ERK2 isoforms), the c-Jun N-terminal kinases (JNKs, including JNK1,
JNK2 and JNK3 isoforms), and the P38MAPKs, including P38-α, -β, -γ
and -δ isoforms. Studies have demonstrated that in mammalian cells,
MAPKs can be activated by a variety of stimuli, and in turn, the
activated MAPKs may typically phosphorylate a number of downstream
substrates, including c-Raf-1, MAPK kinase, ERK-1, and c-Fos, which
regulate a number of genes involved in neuronal apoptosis,
including B-cell lymphoma 2 (Bcl-2) and caspase-3 (10–12).
Studies have demonstrated that sustained cellular hypoxia is
associated with the activation of a MAPK pathway mediated by the
transcription factor hypoxia-inducible factor-1, vascular
endothelial growth factor and inducible nitric oxide synthase
(13–15). These factors mediate an adaptive
response to hypoxia, and are directed toward increasing tissue
perfusion and oxygenation to overcome the initial hypoxic insult.
It is known that intermittent episodes of hypoxia, particularly the
associated episodes of intermittent re-oxygenation, are an
important factor for OSAS-associated cerebral injury. Previous
studies by our group have revealed that intermittent episodes of
hypoxia changed the degree of JNK activation in the cortex and
hippocampus in a rat model of intermittent hypoxia of differing
degrees (16). Additional study is
required to elucidate the response of the hippocampus, a major
component of the brain for memory, to hypoxia by examining the
expression of key MAPKs, including ERK1/2, P38MAPK and JNK.
The present study assessed the effects of
intermittent hypoxia on MAPKs and the expression of apoptotic genes
in the hippocampus of a rat model, which may represent a critical
mechanism for OSAS-associated brain damage in humans.
Materials and methods
Animal model of hypoxia
The protocol of the present study was approved by
the ethics committee of North China University of Science and
Technology (Tangshan, China). A total of 60 male Sprague-Dawley
rats (weight, 170±10 g; age, 8 weeks; Beijing Experimental Animal
Center, Chinese Academy of Science, Beijing, China) were selected
as a model in the present study. The rats were housed in
polycarbonate cages with compressed fiber bedding at 21–25°C and
40–60% relative humidity. Food and water were available ad
libitum in the cage. The animals were randomly divided into
three groups, consisting of control, continued hypoxia and
intermittent hypoxia groups, and rats in these three groups were
further assigned to 2nd, 4th, 6th and 8th week sub-groups, each
including five rats. The corresponding control sub-groups contained
the same quantity of rats.
The animals in the intermittent hypoxia group were
kept in a hypoxia chamber with cycled changes of the hypoxic
conditions (2 min) for 8 h daily between 8:00 am and 4:00 pm. The
chamber was filled with nitrogen and compressed air (Gas Tech
Hexagonal Co., Ltd., Tianjin, China), and in each cycle, for the
first 30 sec, the oxygen concentration inside was lowered to 10%
and maintained for 50 sec, followed by an increase in the oxygen
concentration to 21% for 40 sec. Animals in the continued hypoxia
group were kept in the hypoxia chamber, which was filled with
nitrogen and compressed air and a continuously maintained oxygen
concentration of 10%, for 8 h between 8:00 am and 4:00 pm. The
animals in the control group were kept in the hypoxia by filed with
air (21% oxygen) chamber for 8 h between 8:00 am and 4:00 pm. The
change of the oxygen concentration in the chamber was measured
using an oxygen monitor and the oxygen concentration was maintained
within the range of required concentrations ± 0.5%. Blood (0.1 ml)
was extracted from the arteria carotis using a micro-injector, a
total of 12 times in one cycle (with measurements performed every
10 sec). Blood gas values were measured using a blood gas analyzer
(AVL OMNI automatic blood analyzer; Roche Diagnostics, Basel,
Switzerland).
Tissue preparation
A total of five rats in the control or treatment
groups were decapitated under anesthesia (10% chloral hydrate; 40
mg/kg i.p). Sections of brain hippocampal tissues were separated
and fixed with 4% paraformaldehyde solution for histological
detection and immunohistochemical analysis. Another section of
hippocampal tissue was rapidly frozen in liquid nitrogen. The
frozen tissue samples were homogenized in 1:10 (w/v) ice-cold
homogenization buffer A [10 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.9, 0.5 mM
MgCl2, 10 mM KCl, 0.1 mM EDTA, 0.1 mM ethylene glycol
tetraacetic acid, 50 mM NaF, 5 mM dithiothreitol, 10 mM
β-glycerophosphate, 1 mM sodium orthovanadate, 1% NP-40 and
proteinase inhibitors; 1 mM each of benzamidine, bisnitrophenyl
phosphate and phenylmethylsulfonyl fluoride, and 5 µg/ml
each of aprotinin, leupeptin and pepstatin A; Xinran Biological
Technology Co. Ltd., Shanghai, China], followed by centrifugation
at 1,000 × g for 15 min at 4°C. The supernatants, as cytosolic
components, were collected and the protein concentration was
determined.
Histological analysis with hematoxylin
and eosin (H&E) staining
Post-fixed hippocampal brain tissues were embedded
in paraffin and cut into 5-µm coronal sections using a
microtome. Paraffin-embedded brain sections were de-paraffinized
with xylene and re-hydrated using an ethanol gradient (100–70% v/v)
(both from Tianjin Sheng Winton Chemical Co., Ltd., Tianjin,
China), followed by washing with water. The sections were stained
with 0.1% (w/v) H&E (Nanjing Aoduofuni Biotechnology Co., Ltd.,
Nanjing, China), and examined using light microscopy (Olympus BX53;
Olympus, Tokyo, Japan). The number of surviving hippocampal CA1
pyramidal cells per 1 mm length was used to calculate the neuronal
density.
Western blot analysis
Protein samples (20 µg each) were separated
using 10 or 7.5% SDS-PAGE (Sigma-Aldrich, St. Louis, MO, USA) and
electrotransferred onto nitrocellulose membranes (Bio-Rad
Laboratories, Hercules, CA, USA) according to a previously
described method (16). Following
blocking with 3% bovine serum albumin for 3 h, the membranes were
probed with the following primary antibodies: phosphorylated
(p)-ERKl/2 rabbit anti-mouse polyclonal antibody (cat. no. SC7383),
p-P38MAPK monoclonal antibody (cat. no. bs-547612), p-JNK (cat. no.
elr-0011876) and rabbit polyclonal anti-β-actin (cat. no. 4970P)
(all 1:1,000 dilution; Cell Signaling Technology, Inc., Danvers,
MA, USA). Furthermore, Bcl-2 rabbit polyclonal anti-mouse antibody
and Bcl-2-associated X protein (Bax) polyclonal rabbit anti-mouse
antibody were used (both from Wuhan Boster Biological Engineering
Co., Ltd., Wuhan, China). Membrane-bound antibodies were further
detected using alkaline phosphatase-conjugated goat anti-mouse or
rabbit immunoglobulin (Ig) G (1:10,000 dilution, Sigma-Aldrich).
The immunoreactivity was assessed using a NBT/BCIP assay kit
(Kexing Biological Technology Co., Ltd., Shanghai, China). The band
densities on the membrane were measured using an image analyzer
(Lab Works Software version 17.0; UVP Inc., Upland, CA, USA) and
normalized to the internal control.
Immunohistochemical analysis
Coronal sections of the tissue samples were blocked
in 5% normal goat serum and then incubated in mouse anti-rat Bax
(1:250) or Bcl-2 (1:200) antibody (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) overnight at 4°C. An equivalent dilution of rat
IgG was used as the primary antibody for the negative control.
Subsequently, the sections were incubated in biotinylated rabbit
anti-mouse secondary antibody (1:500 dilution) for 90 min at room
temperature followed by incubation with an avidin-biotin complex
for 90 min. Finally, the sections were developed with stable
3,3′-diaminobenzidine (DAB color kit; Wuhan Boster Biological
Engineering Co., Ltd.) and the nuclei were counter-stained with
hematoxylin. Brown-stained positive cells were examined
microscopically. Quantitative analysis of positive cells in the
hippocampal CA1 region was performed on five slices of each
specimen using Motic-6.0 image acquisition and image analysis
system (magnification, ×200; Motic Med 6.0 digital medical image
analysis system; Beijing Aeronautics and Astronautics University,
Beijing, China). The ratio of positive cells to the total cell
number was calculated.
Malondialdehyde (MDA) and superoxide
dismutase (SOD) analysis
The frozen hippocampal tissue samples were
homogenized in PBS and centrifuged (12,000 ×g for 15 min). The MDA
content and the activities of SOD were detected, respectively
according to the kit specification. MDA, a lipid peroxide
degradation product, was condensed with thiobarbituric acid to form
a red product, which was detected at 532 nm using the MDA test kit
from Jiancheng Bioengineering Co., Ltd., Nanjing, China. A standard
curve was included in the experiment and the content of MDA was
expressed as nmol/mg total tissue protein. SOD activity was
determined using an enzyme kit (Ransod; Randox Laboratories, Inc.,
Crumlin, UK). The method employs xanthine and xanthine oxidase to
produce superoxide radicals that react with
2-(4-iodophenyl)-3-(4-nitrophenyl)-5-phenyltetrazolium chloride to
form a red formazan dye. The SOD activity was measured
photometrically by the degree of inhibition of this reaction at 550
nm at 37°C and expressed as U/mg total tissue protein. A total of 1
unit of SOD inhibits the rate of increase in absorbance at 550 nm
by 50% under the conditions of the assay. The SOD activity was
determined from the percentage inhibition of the test sample
according to an SOD standard curve.
Statistical analysis
All values are expressed as the mean ± standard
deviation. Comparisons between groups were made using one-way
analysis of variance and Newman-Keuls test. P<0.05 was
considered to indicate a statistically significant difference. SPSS
16.0 (SPSS, Inc., Chicago, IL, USA) was used for analysis.
Results
Blood gas parameters
The blood PO2 of rats in the control
group (21% O2) was maintained in a range between 98 and
102 mmHg; The lowest blood oxygen PO2 of rats in the
intermittent hypoxia group (10% O2) reached 48.8 mmHg.
In addition, the lowest blood oxygen PO2 of rats in the
continued hypoxia group (10% O2) was maintained in a
range between 37.4 and 39.6 mmHg.
Intermittent hypoxia causes neuronal cell
loss in the hippocampal CA1 region
To examine the effects of intermittent hypoxia, as
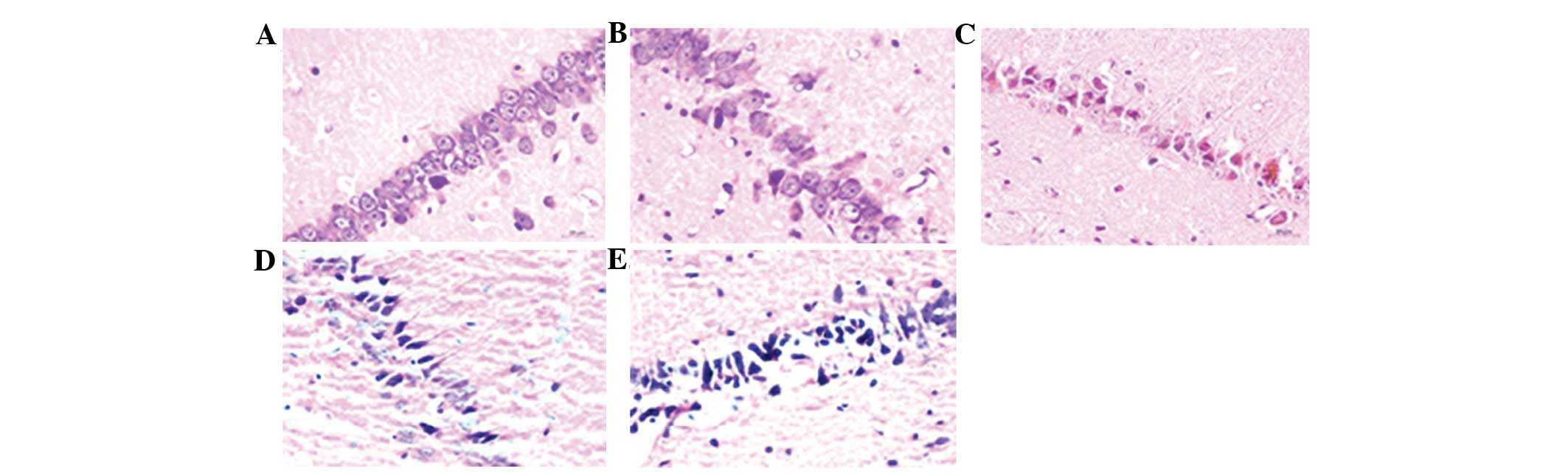
observed in OSAS, on neuronal loss, H&E staining was performed
to examine the survival status of CA1 pyramidal neurons (Fig. 1). The normal cells exhibited
round-shaped and pale-stained nuclei. Shrunken cells with pyknotic
nuclei following ischemia were counted as dead cells. The number of
surviving neurons in the hypoxic groups were significantly lower
than that in the control group (P<0.05) (Table I). In addition, intermittent
hypoxia led to a significant neuronal degeneration and the number
of surviving neurons in the intermittent hypoxia group was
significantly lower than that in the continued hypoxia group
(P<0.05).
 | Table IRate of surviving nerve cells in the
hippocampal CA1 region of rats in various groups (% of total cells
counted). |
Table I
Rate of surviving nerve cells in the
hippocampal CA1 region of rats in various groups (% of total cells
counted).
| Group | Rate of survival of
nerve cells (%)
|
|---|
| Week 2 | Week 4 | Week 6 | Week 8 |
|---|
| Control | 99.4±06 | 99.3±0.7 | 99.5±0.5 | 99.4±0.6 |
| Continued | 98.4±5.6 | 91.6±10.8a | 87.2±19.6a | 84.5±22.7a |
| Intermittent | 91.50±16.48a,b | 82.75±15.80a,b | 74.75±24.70a,b | 70.70±26.10a,b |
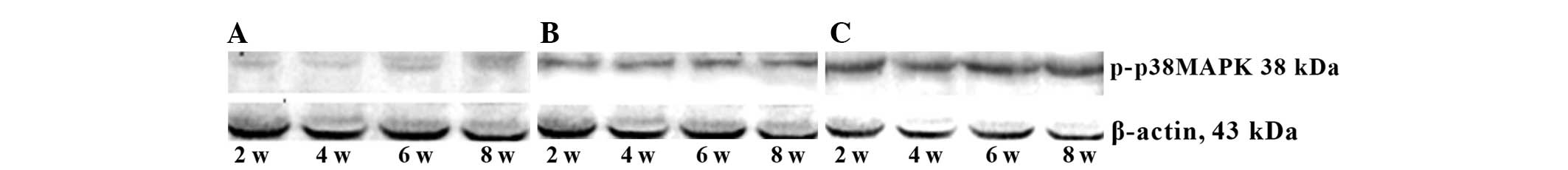
MAPK phosphorylation is increased
following intermittent hypoxia
Western blot analysis revealed that levels of
phosphorylated ERK1/2, P38MAPK and JNK in the continued hypoxia
group were significantly higher than those in the control group
(P<0.05), and levels of phosphorylated ERK1/2 were increased
from the 2nd week and reached a peak at the 4th week, then declined
after hypoxia, while the levels of phosphorylated P38MAPK and JNK
were gradually increased and peaked at the 8th week after hypoxia.
By contrast, the levels of phosphorylated ERK1/2, P38MAPK and JNK
in the intermittent hypoxia group were all markedly and constantly
increased from the 2nd week to the 8th week after hypoxia. In
addition, the levels of phosphorylated ERK1/2, P38MAPK and JNK in
the intermittent hypoxia group were significantly higher than those
in the continued hypoxia group at all time-points (P<0.05;
Figs. 2Figure 3–4; Tables
II–IV).
| Table IIPhospho-ERK1/2 expression in the
hippocampal regions of rats in various groups. |
Table II
Phospho-ERK1/2 expression in the
hippocampal regions of rats in various groups.
| Group | Phospho-ERK1/2
expression (band densities)
|
|---|
| Week 2 | Week 4 | Week 6 | Week 8 |
|---|
| Control | 0.52±0.15 | 0.48±0.12 | 0.64±0.18 | 0.44±0.18 |
| Continued |
2.49±0.96a | 5.84±1.17a | 3.58±1.02a | 1.96±0.82a |
| Intermittent | 4.68±1.56a,b | 6.10±1.12a,b | 7.86±1.56a,b | 9.78±3.41a,b |
| Table IVPhospho-JNK expression in the
hippocampal regions of rats in various groups. |
Table IV
Phospho-JNK expression in the
hippocampal regions of rats in various groups.
| Group | Phospho-JNK
expression (band densities)
|
|---|
| Week 2 | Week 4 | Week 6 | Week 8 |
|---|
| Control | 0.68±0.22 | 0.64±0.20 | 0.66±0.20 | 0.68±0.24 |
| Continued | 1.12±0.28a | 1.18±0.26a | 3.40±0.42a | 4.98±0.56a |
| Intermittent | 1.82±0.25a,b | 3.07±0.19a,b | 4.75±0.32a,b | 8.72±1.40a,b |
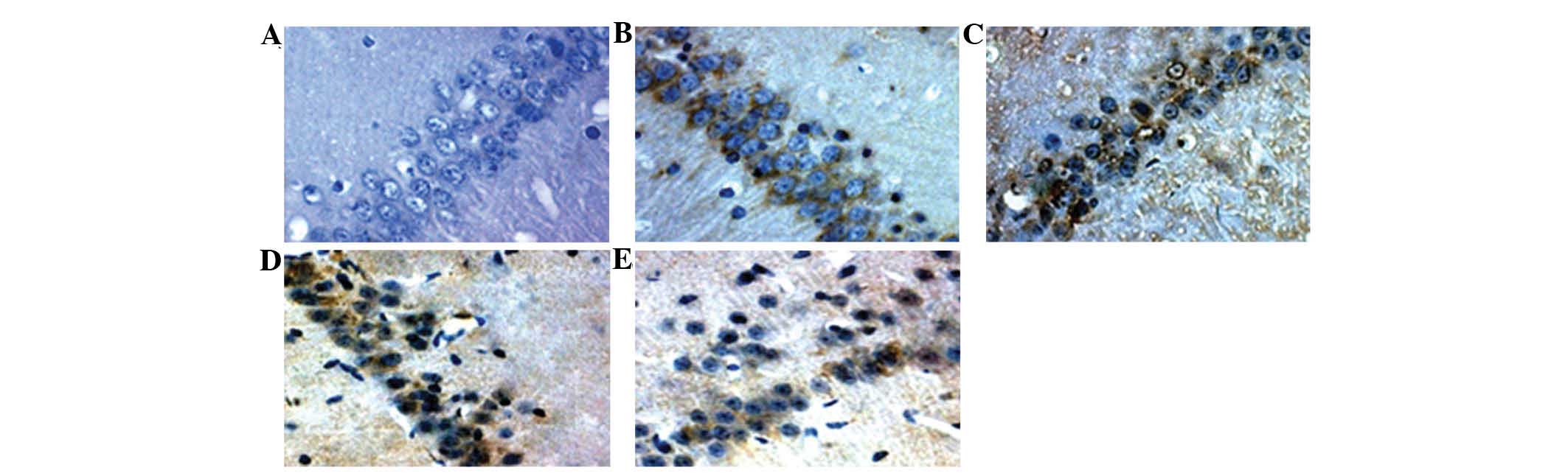
Bax and Bcl-2 are increased following
intermittent hypoxia
To verify the downstream effects of MAPK activation
following intermittent hypoxia in the rat model of OSAS,
immunohistochemical assays were performed to identify the
expression of two members of the Bcl-2 family, namely the
pro-apoptotic protein Bax and the anti-apoptotic protein Bcl-2. The
results revealed an increase in Bcl-2 expression at the 2nd, 4th
and 6th week, but a decease at the 8th week in the continued
hypoxia group in comparison to that in the control group. By
contrast, Bcl-2 expression was increased at the 2nd and 4th week,
but deceased at the 6th and 8th week in the intermittent hypoxia
group. In addition, the levels of Bcl-2 in the intermittent hypoxia
group were significantly higher than those in the continued hypoxia
group at the high end of the range (33.84±8.32 at the 4th week in
the intermittent hypoxia group vs. 30.96±9.66 at the 6th week in
the continued hypoxia group) and at the low end of the range
(9.24±2.42 the intermittent group vs. 14.36±4.46 in the continued
group at the 8th week) levels (Table
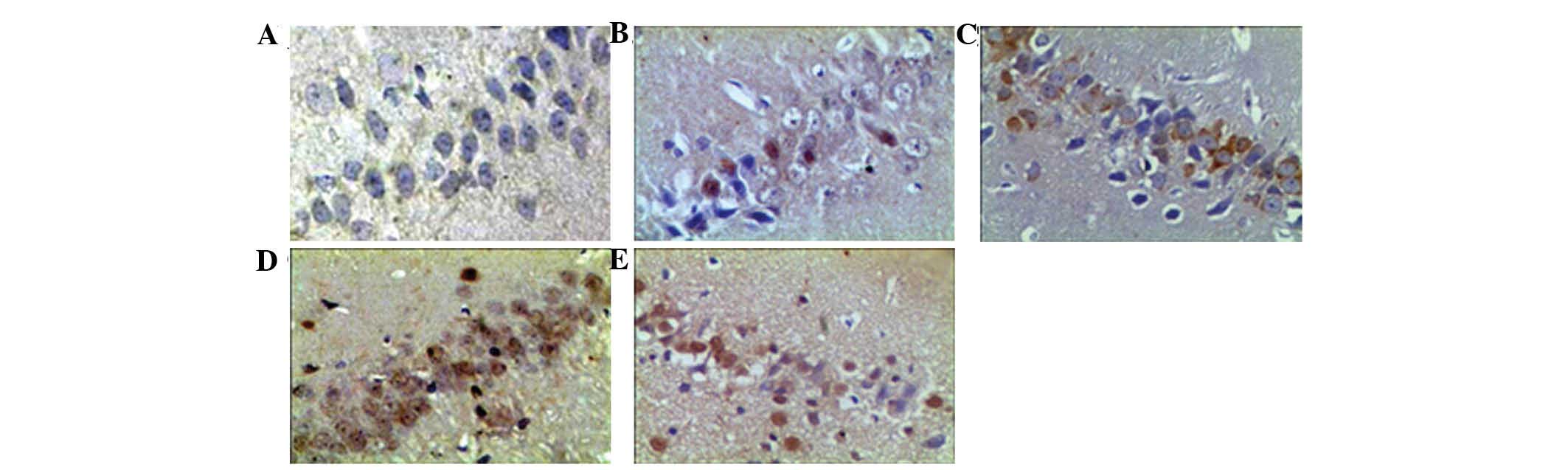
V and Fig. 5). Bax expression
in the continued hypoxia group was significantly higher than that
in the control group (P<0.05) at the 6th and 8th week, whereas
its expression in the intermittent hypoxia group was increased at
all experimental time-points (Table
VI and Fig. 6). The Bcl-2/Bax
ratio was markedly lower in the intermittent hypoxia group than
that in the continued hypoxia group (P<0.05; Table VII.
| Table VRate of Bcl-2-positive cells in the
hippocampal CA1 region of rats in response to hypoxia. |
Table V
Rate of Bcl-2-positive cells in the
hippocampal CA1 region of rats in response to hypoxia.
| Group | Rate of
Bcl-2-positive cells (%)
|
|---|
| Week 2 | Week 4 | Week 6 | Week 8 |
|---|
| Control | 5.56±1.10 | 5.58±1.12 | 5.52±1.11 | 5.28±1.10 |
| Continued | 16.54±3.96a | 24.64±5.74a | 30.96±8.32a | 14.36±4.60a |
| Intermittent | 24.86±6.38a,b | 33.84±9.66a,b | 20.78±5.36a,b | 9.24±2.42a,b |
| Table VIRate of Bax-positive cells in the
hippocampal CA1 region of rats in response to hypoxia. |
Table VI
Rate of Bax-positive cells in the
hippocampal CA1 region of rats in response to hypoxia.
| Group | Rate of
Bax-positive cells
|
|---|
| Week 2 | Week 4 | Week 6 | Week 8 |
|---|
| Control | 2.78±0.82 | 2.66±0.76 | 2.64±0.78 | 2.70±0.82 |
| Continued | 4.14±1.56 | 4.08±1.64 | 9.72±2.80a | 22.68±6.94a |
| Intermittent | 11.44±3.24a,b | 18.80±4.60a,b | 26.64±8.26a,b | 32.72±9.24a,b |
| Table VIIRatio of Bcl-2/Bax-positive cells in
the hippocampal CA1 region of rats in response to hypoxia. |
Table VII
Ratio of Bcl-2/Bax-positive cells in
the hippocampal CA1 region of rats in response to hypoxia.
| Group | Rate of positive
cells (fold)
|
|---|
| Week 2 | Week 4 | Week 6 | Week 8 |
|---|
| Control | 2.0 | 2.1 | 2.1 | 2.0 |
| Continued | 4.0 | 6.0 | 3.2 | 0.6 |
| Intermittent | 2.2 | 1.8 | 0.8 | 0.8 |
Analysis of MDA and SOD activity
To further identify the association between
oxidative stress and MAPK activation, changes in MDA, a lipid
metabolic product in the oxidative reaction, and SOD, an important
anti-oxidant, in the experimental hypoxia groups. The results
demonstrated that the MDA content increased in the two hypoxia
groups and was significantly elevated compared to that in the
control (P<0.05). The MDA content reached a peak level at the
6th and the 8th week, respectively, in the continued and the
intermittent hypoxia groups. Of note, the MDA content in the
intermittent hypoxia group was significantly higher than that in
the continued hypoxia group at each time-point (P<0.05)
(Table VIII). The SOD activity
was decreased and reached its lowest level at the 6th week in the
two hypoxia groups and this low level of SOD persisted until the
8th week of the experiments. In addition, the SOD activity in the
intermittent hypoxia group was consistently lower than that in the
continued hypoxia group (Table
IX).
| Table VIIIQuantitative analysis of the
malondialdehyde content in the hippocampal CA1 region of rats in
response to hypoxia. |
Table VIII
Quantitative analysis of the
malondialdehyde content in the hippocampal CA1 region of rats in
response to hypoxia.
| Group | Malondialdehyde
content (nM/mg total tissue protein)
|
|---|
| Week 2 | Week 4 | Week 6 | Week 8 |
|---|
| Control | 7.60±1.47 | 7.58±1.48 | 7.58±1.46 | 7.62±1.50 |
| Continued | 9.70±1.83a | 12.70±4.86a | 27.94±7.54a | 22.42±8.94a |
| Intermittent | 14.38±3.36a,b | 18.82±5.58a,b | 34.96±9.32a,b | 39.92±11.28a,b |
| Table IXQuantitative analysis of superoxide
dismutase activity in the hippocampal CA1 region of rats in
response to hypoxia. |
Table IX
Quantitative analysis of superoxide
dismutase activity in the hippocampal CA1 region of rats in
response to hypoxia.
| Groups | Superoxide
dismutase activity (U/mg total tissue protein)
|
|---|
| Week 2 | Week 4 | Week 6 | Week 8 |
|---|
| Control | 87.68±2.32 | 87.70±2.38 | 87.66±2.30 | 87.70±2.36 |
| Continued | 76.68±2.68a | 68.50±2.62a | 60.94±2.54a | 67.42±2.94a |
| Intermittent | 68.62±2.66a,b | 60.82±2.58a,b | 54.96±2.32a,b | 55.92±3.08a,b |
Discussion
OSAS is widely recognized as an independent risk
factor for cerebrovascular diseases, particularly cerebral stroke.
It increases the incidence of cerebral stroke by two times even
after adjustment for potential confounding factors (15). In addition, the majority of
patients with OSAS exhibit symptoms of nervous system damage,
manifested as a dysfunction in learning, memory and decision-making
ability (17). Mitchell et
al (18) reported that
cognitive function in patients with OSAS was only partially
improved, even following surgical treatment. Furthermore, it is
associated with changes in brain structure, including a reduction
in the gray matter in the hippocampus, frontal cortex, anterior
cingulate cortex and other sections as well as volume atrophy
(19–20). A previous study by our group
demonstrated that various degrees of intermittent hypoxia induced
injury to the neural cell ultrastructure as well as nerve cell
loss, which were irreversible (21). In the present study, it was
illustrated that intermittent hypoxia elicited a severe level of
neuronal degeneration and neuronal death in comparison to continued
hypoxia in a rat model, suggesting that intermittent hypoxia in
OSAS may induce marked damage to nerve cells.
The MAPK pathway is activated in the brains of
animal models of cerebral ischemia; furthermore, it has been
demonstrated that the MAPK signaling pathway is important in
cerebral ischemia/reperfusion, the closed head injury model and
perinatal hypoxia-ischemia (22–24).
Further studies have also assessed the timing and location of the
MAPK activation in various brain tissues under different stress
conditions. Guo et al (25)
proposed that ERK1/2 activity, instead of JNK1/2 activity, was
increased 30 min after cerebral ischemia in a model of permanent
forebrain ischemia in rats induced using the four artery ligation
method. In a cerebral ischemia-re-perfusion model, the ERK1/2
activity began to increase at 15 min and peaked at 6 h, while
JNK1/2 activity increased after 1 h and gradually increased to peak
at 21 h; both activated ERKl/2 and JNKl/2 were detected at 24 h
after injury. It is noteworthy that activated ERKl/2 instead of
JNKl/2 was also induced by another re-perfusion (26). In a gerbil model of brain
ischemia-re-perfusion induced by bilateral common carotid artery
blocking, indications of MAPK activation, including ERKl/2
phosphorylation, mainly existed in the CA3/dentate gyrus sub-region
at a short time after ischemia and was lowly expressed in the CA1
sub-region. With the extension of ischemia, JNKl/2 and P38MAPK
phosphorylation levels were increased in the CA1 and CA3
sub-regions (27). In the present
study, western blot analysis indicated that in the continued
hypoxia model, phospho-ERKl/2 was markedly increased at various
time-points and reached a peak level at four weeks, after which it
declined. With increasing time following hypoxia, active P38MAPK
and JNKl/2 were detected. In the intermittent hypoxia group,
phospho-ERKl/2, P38MAPK and JNKl/2 increased markedly from two
weeks after hypoxia, and ERK1/2 activity maintained a stable
increase to the top level from the 2nd to the 8th week in the
experimental period. Activated P38MAPK and JNKl/2 were gradually
increased, reaching their highest levels at the 8th week also. The
results of the present study suggested that the MAPK signaling
pathways were selectively activated under various hypoxic
conditions. It is possible that intermittent hypoxia induced by
OSAS activated MAPK signaling pathways in a continually excessive
manner. It is known that P38MAPK and JNK are similar in nature, and
the activation of either has a negative regulatory role, leading to
cell injury and death (11,12,28).
By contrast, ERKl/2 has a dual effect, which may promote cell
survival and proliferation with appropriate activation in the short
term, but may also cause cell death with excessive activation in
the long term (29,30). Alessandrini et al (31) demonstrated that ERK1/2
phosphorylation and cytochrome C were increased following cerebral
injury, and co-localized within the same nerve cell. The activation
of ERK1/2 may upregulate the protective anti-oxidant system and
promote neuronal survival (32).
The Ras/ERK1/2 cascade has been hypothesized to be a tolerance core
of neurons in cerebral ischemic stress (33). In fact, MAPK activation in
mammalian cells has coordinating effects and the same simulation is
able to activate various MAPK pathways. For example, stress
reactions are able to activate the ERKl/2, P38MAPK and JNK1/2
pathways (34), while epidermal
growth factor may stimulate ERKl/2 and JNKl/2 signals (35). In addition, activated MAPKs may
phosphorylate or activate the same transcription factor, such as
ERK-1, and thus integrate extracellular stimuli, ultimately leading
to biological reactions to induce cell survival or death (36).
In the present study, certain correlations between
activated ERKl/2 and the expression of Bcl-2 family members were
identified. The levels of Bcl-2 and ERK1/2 began to increase at the
2nd week and reached a peak at the 4th week or the 6th week, and
then decreased in the continued hypoxia group. Bax expression
gradually increased and reached a peak at the 8th week in
association with a decrease in phospho-ERKl/2 and increases in
activated P38MAPK and JNKl/2 in the continued hypoxia group. In the
intermittent hypoxia group, activated ERKl/2 maintained a steady
increase reaching the top level at the 8th week; however, Bcl-2
expression reached a peak at 4 weeks and then gradually decreased.
It is well established that Bcl-2 and Bax exhibit the opposite
biological functions. Bcl-2 is able to enhance cell survival
following ischemia and hypoxia by maintaining mitochondrial
membrane integrity, whereas Bax promotes ischemia or
hypoxia-induced cell death via the release of cytochrome C. Thus,
the ratio of Bcl-2 to Bax affects the survival of cells (37,38).
Based on changes in the ratio of Bcl-2 to Bax and MAPK signaling
pathway activation under various hypoxic conditions, the present
study hypothesized that in the continued hypoxia group, ERKl/2
activation may have had a protective effect and enhanced Bcl-2
expression. This may have increased the compensatory function of
nerve cells resistant to low oxygen. However, the intermittent
hypoxia of OSAS may resemble re-perfusion injury, resulting in a
marked stress reaction and continued, excessive activation of the
MAPK signaling pathways. The possible role of ERKl/2 activation at
the early stages may have increased the tolerance to hypoxia for a
short period. However, with prolonged intermittent hypoxia, the
MAPK signaling pathway was excessively activated, leading to
abnormal expression of Bcl-2 and Bax and the death of nerve cells.
This may be an important molecular mechanism for OSAS-associated
injury to the brain.
In the animal model, hypoxia enhanced the production
of MDA and reduced the activity of SOD in a time-dependent manner,
particularly in the intermittent hypoxia group. Hypoxia has been
reported to result in a marked elevation in reactive oxygen species
(ROS) in non-brain tissues (39).
The present study illustrated differences in the type and extent
hypoxia on MAPK activation, including increases of MDA or decreases
of SOD, suggesting an association between changes in the oxidative
stress reaction and MAPK activation in the pathological process of
nerve injury induced by hypoxia. Direct exposure of cells to
exogenous H2O2, to mimic oxidative stress,
leads to the activation of MAPKs (40). The prevention of ROS accumulation
by anti-oxidants blocks MAPK activation in cells exposed to
specific stimuli (41,42). A previous study by our group
reported that oxidative stress initiated the JNK pathway, which
mediated nerve cell injury in a model of severe intermittent
hypoxia (41). ROS may trigger the
ERK1/2 pathway through activation of growth factor receptors
(43) or modification of apoptosis
signal-regulating kinase 1, a member of the MAP3K superfamily,
which targets JNK and P38MAPK. In addition, ROS oxidize the
cysteine residues in MAPK phosphatase, leading to activation of the
JNK and P38 pathways. Under the conditions of intermittent hypoxia
found in OSAS, oxidative reactions due to marked repetitive
re-oxygenation enhance the early damage of nerve cells.
In conclusion, the present animal study indicated
that MAPK signaling pathways were selectively activated in the
hippocampus under intermittent and continued hypoxic conditions. In
addition, excessive activation of MAPK signaling pathways by
intermittent hypoxia elicited abnormal expression of Bcl-2 and Bax
as well as a severe loss of hippocampal nerve cells. This
phenomenon was also closely associated with oxidative stress,
inducing an elevated production of MDA and the downregulation of
SOD. These findings suggested a critical role of MAPK activation in
OSAS-induced brain pathogenesis, and provided therapeutic
strategies for the prevention and treatment of this common
disease.
Acknowledgments
The present study was supported by grants from the
Natural Science Foundation, Department of Education, Hebei Province
(grant no. ZH201120) and the Hebei Province Science and Technology
Project (grant no. 09276103D-11), China.
References
|
1
|
Young T, Palta M, Dempsey J, Skatrud J,
Weber S and Badr S: The occurrence of sleep-disordered breathing
among middle-aged adults. N Engl J Med. 328:1230–1235. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peppard PE, Young T, Palta M and Skatrud
J: Prospective study of the association between sleep-disordered
breathing and hypertension. N Engl J Med. 342:1378–1384. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Durgan DJ and Bryan RM Jr: Cerebrovascular
consequences of obstructive sleep apnea. J Am Heart Assoc.
1:e0000912012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baessler A, Nadeem R, Harvey M, Madbouly
E, Younus A, Sajid H, Naseem J, Asif A and Bawaadam H: Treatment
for sleep apnea by continuous positive airway pressure improves
levels of inflammatory markers-a meta-analysis. J Inflamm (Lond).
10:132013. View Article : Google Scholar
|
|
5
|
Opstad KS, Provencher SW, Bell BA, et al:
Detection of elevated glutathione in meningiomas by quantitative in
vivo1H MRS. Magn Reson Med. 49:632–637. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hagen T, Taylor CT, Lam F and Moncada S:
Redistribution of intracellular oxygen in hypoxia by nitric oxide:
Effect on HIF1alpha. Science. 302:1975–1978. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ohga E, Nagase T, Tomita T, Teramoto S,
Matsuse T, Katayama H and Ouchi Y: Increased levels of circulating
ICAM-1, VCAM-1 and L-selectin in obstructive sleep apnea syndrome.
J Appl Physiol (1985). 87:10–14. 1999.
|
|
8
|
Yuan G, Nanduri J, Bhasker CR, Semenza GL
and Prabhakar NR: Ca2+/calmodulin kinase-dependent activation of
hypoxia-inducible factor 1 transcriptional activity in cells
subjected to intermittent hypoxia. J Biol Chem. 280:4321–4328.
2005. View Article : Google Scholar
|
|
9
|
Broom OJ, Widjaya B, Troelsen J, Olsen J
and Nielsen OH: Mitogen activated protein kinases: A role in
inflammatory bowel disease? Clin Exp Immunol. 158:272–280. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Suganuma T and Workman JL: MAP kinases and
histone modification. J Mol Cell Biol. 4:348–350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kyriakis JM and Avruch J: Mammalian MAPK
signal transduction pathways activated by stress and inflammation:
A 10-year update. Physiol Rev. 92:689–737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lawrence MC, Jivan A, Shao C, Duan L, Goad
D, Zaganjor E, Osborne J, McGlynn K, Stippec S, Earnest S, et al:
The roles of MAPKs in disease. Cell Res. 18:436–442. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kiec-Wilk B, Grzybowska-Galuszka J, Polus
A, Pryjma J, Knapp A and Kristiansen K: The MAPK-dependent
regulation of the Jagged/Notch gene expression by VEGF, bFGF or
PPAR gamma mediated angiogenesis in HUVEC. J Physiol Pharmacol.
61:217–225. 2010.PubMed/NCBI
|
|
15
|
Yoon SY, Lee YJ, Seo JH, Sung HJ, Park KH,
Choi IK, Kim SJ, Oh SC, Choi CW, Kim BS, et al: uPAR expression
under hypoxic conditions depends on iNOS modulated ERK
phosphorylation in the MDA-MB-231 breast carcinoma cell line. Cell
Res. 16:75–81. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao YN, Wang HY, Guo X, et al:
Phospho-JNK expression and significance of hippocampus of rats to
varying degrees intermittent hypoxia. J Jilin Univ. 38:1135–1140.
2012.
|
|
17
|
Sateia MJ: Neuropsychological impairment
and quality of life in obstructive sleep apnea. Clin Chest Med.
24:249–259. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mitchell RB, Kelly J, Call E and Yao N:
Long-term changes in quality of life after surgery for pediatric
obstructive sleep apnea. Arch Otolaryngol Head Neck Surg.
130:409–412. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Alchanatis M, Zias N, Deligiorgis N,
Amfilochiou A, Dionellis G and Orphanidou D: Sleep apnea-related
cognitive deficits and intelligence: An implication of cognitive
reserve theory. J Sleep Res. 14:69–75. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rao BS, Raju TR and Meti BL: Increased
numerical density of synapses in CA3 region of hippocampus and
molecular layer of motor cortex after self-stimulation rewarding
experience. Neuroscience. 91:799–803. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li L, Wang HY and Zhao YN: The effects of
intermittent serious hypoxia on the cognitive function and
hippocampus ultra microstructure in Rats. Xi'an Jiaotong University
(Medical Sciences). 32:687–689. 2011.
|
|
22
|
Hu B, Liu C, Bramlett H, Sick TJ, Alonso
OF, Chen S and Dietrich WD: Changes in TrkB-ERK1/2-CREB/ELK-1
pathways in hippocampal mossy fiber organization after traumatic
brain injury. J Cereb Blood Flow Metab. 24:934–943. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lu K, Cho CL, Liang CL, Chen SD, Liliang
PC, Wang SY and Chen HJ: Inhibition of the MEK/ERK pathway reduces
microglial activation and interleukin-1-beta expression in spinal
cord ischemia/reperfusion injury in rats. J Thorac Cardiovasc Surg.
133:934–941. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee SR and Lo EH: Interactions between p38
mitogen-activated protein kinase and caspase-3 in cerebral
endothelial cell death after hypoxia-reoxygenation. Stroke.
34:2704–2709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guo J, Zhu H and De W: Mitogen-activated
protein kinases ERKs and JNKs differences in brain ischemia
activation and regulation mechanism. Chin J Neurosci. 20:207–221.
2004.
|
|
26
|
Guo J, Meng F, Zhang G and Zhang Q: Free
radical are involved in continuous activation of non receptor
tyrosine protein kinase c-Src after ischemia/reperfusion in rat
hippocampus. Neurosci Lett. 345:101–104. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chandler LJ, Sutton G, Dorairaj NR and
Norwood D: N-Methyl D-aspartate receptor-mediated bidirectional
control of extracellular signal regulated kinase activity in
cortical neuronal cultures. J Biol Chem. 276:2627–2636. 2001.
View Article : Google Scholar
|
|
28
|
Kaminska B, Gozdz A, Zawadzka M,
Ellert-Miklaszewska A and Lipko M: MAPK signal transduction
underlying brain inflammation and gliosis as therapeutic target.
Anat Rec (Hoboken). 292:1902–1913. 2009. View Article : Google Scholar
|
|
29
|
Clausen F, Lundqvist H, Ekmark S, Lewén A,
Ebendal T and Hillered L: Oxygen free radical-dependent activation
of extracellular signal-regulated kinase mediates apoptosis-like
cell death after traumatic brain injury. J Neurotrauma.
21:1168–1182. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Berkeley JL, Decker MJ and Levey AI: The
role of muscarinic acetylcholine receptor-mediated activation of
extracellular signal-regulated kinase 1/2 in pilocarpine-induced
seizures. J Neurochem. 82:192–201. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Alessandrini A, Namura S, Moskowitz MA and
Bonventre JV: MEK1 protein kinase inhibition protects against
damage resulting from focal cerebral ischemia. Proc Natl Acad Sci
USA. 96:12866–12869. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang P, Wang YZ, Kagan E and Bonner JC:
Peroxynitrite targets the epidermal growth factor receptor, Raf-1
and MEK independently to activate MAPK. J Biol Chem.
275:22479–22486. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jin K, Mao XO, Zhu Y and Greenberg DA: MEK
and ERK protect hypoxic cortical neurons via phosphorylation of
Bad. J Neurochem. 80:119–125. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dasari A and Messersmith WA: New
strategies in colorectal cancer: Biomarkers of response to
epidermal growth factor receptor monoclonal antibodies and
potential therapeutic targets in phosphoinositide 3-kinase and
mitogen-activated protein kinase pathways. Clin Cancer Res.
16:3811–3818. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ma FY, Liu J and Nikolic-Paterson DJ: The
role of stress-activated protein kinase signaling in renal
pathophysiology. Braz J Med Biol Res. 42:29–37. 2009. View Article : Google Scholar
|
|
36
|
Ge X, Shi Z, Yu N, Jiao Y, Jin L and Zhang
J: The Role of EGFR/ERK/ELK-1 MAP kinase pathway in the underlying
damage to diabetic rat skin. Indian J Dermatol. 58:101–106. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pirzadeh A, Mammen A, Kubin J, Reade E,
Liu H, Mendoza A, Greeley WJ, Wilson DF and Pastuszko A: Early
regional response of apoptotic activity in newborn piglet brain
following hypoxia and ischemia. Neurochem Res. 36:83–92. 2011.
View Article : Google Scholar :
|
|
38
|
Hagberg H, Mallard C, Rousset CI and Wang
Xiaoyang: Apoptotic mechanisms in the immature brain: Involvement
of mitochondria. J Child Neurol. 24:1141–1146. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Clanton TL: Hypoxia-induced reactive
oxygen species formation in skeletal muscle. J Appl Physiol.
102:2379–2388. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Posen Y, Kalchenko V, Seger R, Brandis A,
Scherz A and Salomon Y: Manipulation of redox signaling in
mammalian cells enabled by controlled photogeneration of reactive
oxygen species. J Cell Sci. 118:1957–1969. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kyaw M, Yoshizumi M, Tsuchiya K, Kirima K
and Tamaki T: Antioxidants inhibit JNK and p38 MAPK activation but
not ERK 1/2 activation by angiotensin II in rat aortic smooth
muscle cells. Hypertens Res. 24:251–261. 2001. View Article : Google Scholar
|
|
42
|
Yeo JE and Kang SK: Selenium effectively
inhibits ROS-mediated apoptotic neural precursor cell death in
vitro and in vivo in traumatic brain injury. Biochim Biophys Acta.
1772:1199–1210. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Akool el-S, Gauer S, Osman B, Doller A,
Schulz S, Geiger H, Pfeilschifter J and Eberhardt W: Cyclosporin A
and tacrolimus induce renal Erk1/2 pathway via ROS-induced and
metalloproteinase-dependent EGF-receptor signaling. Biochem
Pharmacol. 83:286–295. 2012. View Article : Google Scholar
|