Introduction
Parkinson's disease (PD) is a movement disorder
characterized by motor and behavioral disturbances, caused by the
gradually progressive and selective degeneration of dopaminergic
neurons in the substantia nigra pars compacta (SNpc) (1).
The pathogenesis of PD remains to be fully
elucidated, however, multiple studies have linked oxidative stress
to dopaminergic neuron degeneration in PD. Increased oxidative
stress contributes to DNA damage, leading to dopaminergic neuron
degeneration and the pathogenesis of PD (2). Postmortem samples of PD have shown
increased DNA oxidative damage selectively in dopaminergic neurons
of the SNpc, indicating the link between DNA oxidation and the loss
of dopaminergic neurons in PD (2).
The classical widely-used pharmacological and toxic agent to model
PD is 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or its
toxic metabolite 1-methyl-4-phenylpyridinium (MPP+),
which cause the production of reactive oxygen species by inhibiting
mitochondrial complex I, leading to DNA oxidative damage and
subsequent neuronal death (3).
These previous reports support the hypothesis that DNA
damage-induced cell death is a mechanism involved in the
pathogenesis of PD. Proliferating cell nuclear antigen (PCNA) is a
well-known protein, which is involved in DNA repair in a wide range
of pathological conditions, including oxidative stress-mediated
damage of DNA by interacting with a number of enzymes and
regulatory proteins (4–6). The PCNA-dependent repair of damaged
DNA is crucial in preserving its integrity under oxidative
conditions (7,8). Currently, the importance of this
neuroprotective strategy to prevent or reverse the degeneration of
dopaminergic neurons has been emphasized in the treatment of PD,
which relies on the effective inhibition of the pathogenesis in
neurodegenerative process. α-lipoic acid (ALA) is a naturally
occurring dithiol compound, which is synthesized enzymatically in
the mitochondria from octanoic acid and cysteine. Its protective
activities have been reported in vivo and in vitro
against a range of pathophysiological insults (9), including MPP+-induced
toxicity in neuronal cells (10).
ALA has been in common clinical use for several diseases associated
with increased oxidative stress, and its administration in moderate
doses has produced no evidence of serious side-effects (10–16).
Several studies have shown that ALA exerts protective effects in
in vivo and in vitro models of neurodegenerative
diseases, including Alzheimer's disease (AD), macular degeneration
and PD (17–19).
The present study was designed to investigate the
effects of ALA on an MPP+-induced PD model and to
examine the mechanisms underlying these actions. The results
demonstrated that ALA effectively prevented MPP+-induced
PC12 cell apoptosis, suggesting the neuroprotective role of ALA in
the neurodegenerative condition. The protein expression of PCNA was
significantly decreased by MPP+ treatment, supporting
the hypothesis that PCNA-dependent apoptotic pathway is one
potential molecular mechanism involved in the neuronal death in PD.
Of note, ALA markedly reversed the decreased expression of PCNA in
the MPP+-induced PD model. The effects of ALA on the
PCNA upstream regulator, p53, were also examined. P53 interacts
with the PCNA promoter to regulate the production of this protein,
and a higher concentration of wild-type p53 inhibits the PCNA
promoter, which results in a decrease in the production of PCNA
(20,21). ALA treatment markedly reduced the
expression levels of p53, however, the expression of PCNA was
upregulated. Together, these results confirmed that ALA upregulated
the protective expression of protective PCNA and provided
neuroprotection against the MPP+-induced neurotoxicity
via the p53 pathway.
Materials and methods
Drugs and chemicals
All reagents and chemicals were purchased from
Sigma-Aldrich; Merck Millipore (Darmstadt, Germany) unless stated
otherwise.
Cell culture
The PC12 cells, obtained from the Cell Bank of the
Chinese Academy of Sciences (Shanghai, China) were grown in high
glucose Dulbecco's modifid Eagle's medium supplemented with 10%
heat-inactivated fetal bovine serum, 4.00 mM L-glutamine, 100 U/ml
penicillin and 100 µg/ml streptomycin (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The cells, seeded at a density
of 30,000 cells/cm2, were plated onto 75-cm tissue
culture flasks and incubated in a humidified 5% CO2
atmosphere at 37°C. The cell monolayers were passaged on reaching
80–90% confluence, and passages 10–20 were used in the subsequent
experiments.
Modified
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT)
assay
Cell viability was measured using an MTT
colorimetric assay. MTT is readily taken up into cells, and is then
reduced, predominantly by mitochondrial enzymes, to form blue
formazan crystals, which are impermeable to cell membranes and are
trapped in living cells. The PC12 cells were plated at the density
of 30,000 cells/cm2 in 96-well plates and incubated for
24 h at 37°C in a humidified 5% CO2 incubator. To assess
the toxicity of MPP+ towards the PC12 cells, the cells
were exposed to different doses (0.5, 1 and 2 mM) of
MPP+ and incubated for 48 h at 37°C. To assess the
neuroprotective effects of ALA on MPP+-induced toxicity
in the PC12 cells, the cells were pre-treated with 0.01 µM ALA for
1 h, and then exposed to 1 mM MPP+ for 48 h at 37°C,
which were previously reported to be optimal conditions leading to
significant protective effects (22). Following treatments, MTT solution
(5 mg/ml) was added to each well, and the formed formazan crystals
were dissolved in dimethyl sulfoxide. The absorbance of the colored
solution was measured at 570 nm using a microplate reader (BioTek,
Epoch, USA). The results are expressed as the percentage of the
absorbance measured in control culture wells. The experiment was
repeated three times.
Lactate dehydrogenase (LDH)
cytotoxicity assay
Cell injury was further confirmed by measuring the
activity of LDH, which is expressed in all mammalian cells and is
released from damaged cells into the culture medium. The activity
of the released LDH in the culture supernatant was measured using
LDH-Cytotoxicity Assay kits (BioVision Research Products, Mountain
View, CA, USA) according to the manufacturer's protocol. Briefly,
the PC12 cells were plated at a concentration of 30,000
cells/cm2 for 24 h at 37°C in a humidified 5%
CO2 incubator, followed by treatment with 0.01 µM ALA
prior to the addition of 1 mM MPP+ for 1 h. Following
treatment, the cells were centrifuged at 5,000 × g for 10 min at
4°C, and 50 µl of the resulting supernatant was transferred into a
separate 96-well plate. A 100 µl volume of the LDH reaction mixture
was added to each well. Following incubation for 30 min at room
temperature, the LDH activity was quantified as absorbance values
at 490 nm using a multiwell spectrometer (BD Biosciences, San
Diego, CA, USA). The data are expressed as a percentage of the
fluorescence values in the untreated control.
Morphological observation of nuclear
change
Apoptosis is a major type of cell death,
characterized by a series of nuclear morphological changes,
including reduced nuclear size, chromatin condensation, intense
fluorescence and nuclear fragmentation. These changes can be
detected by Hoechst 33258 staining, which is used for the
quantification of apoptotic cells. Briefly, following treatment,
the cells were washed with phosphate-buffered saline three times,
and the cells were stained with 10 µg/ml Hoechst 33258 for 10 min
at room temperature in the dark. Subsequently, the numbers of
apoptotic cells were randomly counted under a fluorescence
microscope (IX71; Olympus, Tokyo, Japan). The number of apoptotic
cells is expressed as a percentage of the total cells counted.
Western blot analysis
Following treatment, the PC12 cells were collected
and lysed with cell lysis solution containing 4% sodium dodecyl
sulfate, 2 mM EDTA and 50 mM Tris-HCl (pH 6.8). Protein
concentration was determined using the Bradford method (GE
Healthcare Life Sciences, Chalfont, UK). Equal quantities of
protein (40 µg) were separated by 12% polyacrylamide gel
electrophoresis and transferred onto PVDF membranes (Amersham
Biosciences, Upsalla, Sweden). The membranes were then incubated in
Tris-buffered saline/Tween buffer supplemented with 5% fat-free
milk for 1 h to block nonspecific binding. Western blot analysis
was performed using rabbit anti-PCNA (1:1,000; cat. no. 610664; BD
Biosciences) and anti-P53 (1:1,000; cat. no. 554157; BD
Biosciences) overnight at 4°C, and horseradish
peroxidase-conjugated anti-rabbit antibodies (1:1,000; cat. no.
R-21455; Thermo Fisher Scientific, Inc.) were used as the secondary
antibodies and incubated at room temperature for 2 h. The blots
were analyzed using an enhanced chemiluminescence system (GE
Healthcare Life Sciences).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Statistical analysis was performed using one-way analysis
of variance, followed by Dunnett's multiple-comparisons test.
Analyses were performed using SPSS version 15.0 (SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference between mean values were.
Results
Neuroprotective ALA reduces
MPP+-induced toxicity in neuronal cells
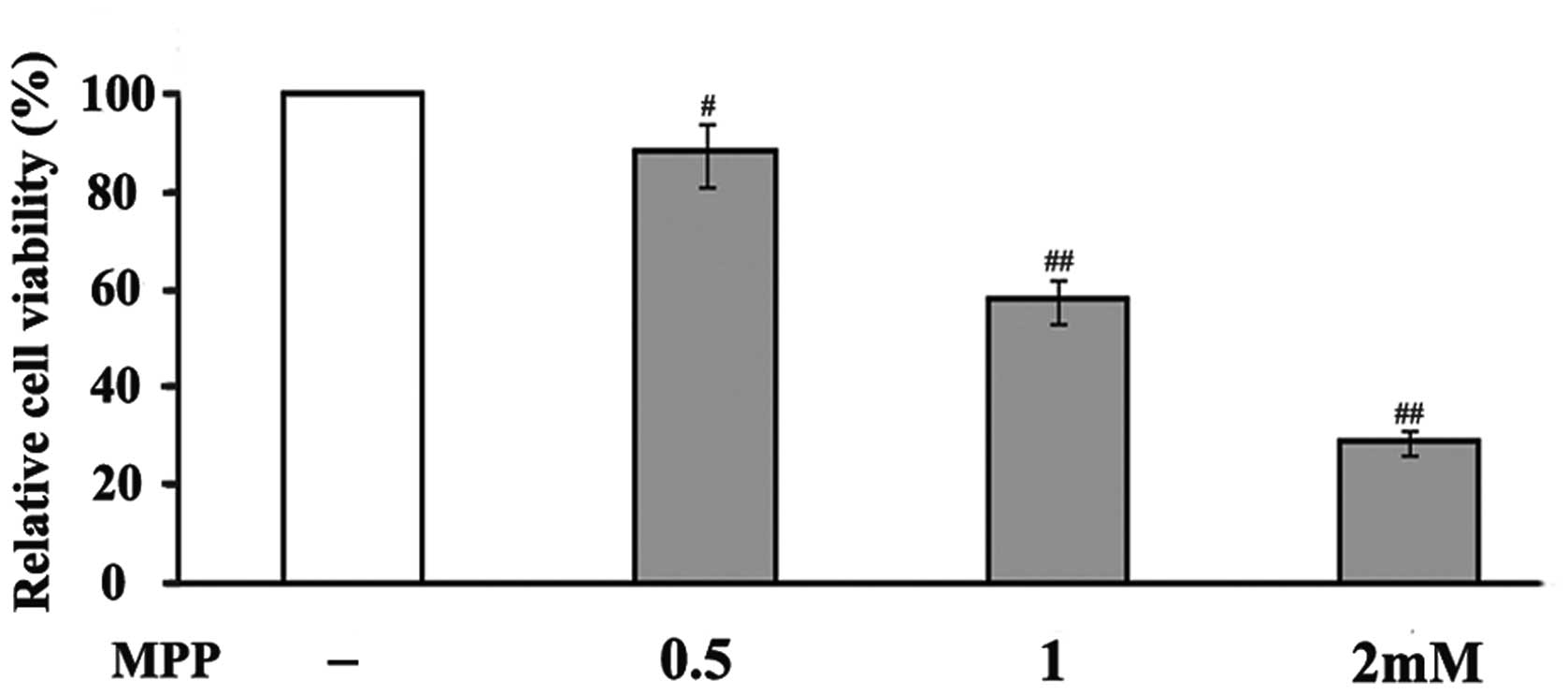
In an attempt to investigate the toxicity of
MPP+ in PC12 cells, the cells were exposed to different
doses (0.5, 1 and 2 mM) of MPP+. MPP+
treatment significantly reduced the viability of the PC12 cells in
a concentration-dependent manner (Fig.
1). After 48 h treatment, 0.5 mM MPP+ reduced cell
viability to 88% compared to that of untreated cells, whereas 1 and
2 mM MPP+ decreased cell viability to 58 and 29%,
respectively. Based on these results, a concentration of 1 mM
MPP+ was used in the following experiments to examine
the neuroprotective effects of ALA on MPP+-induced
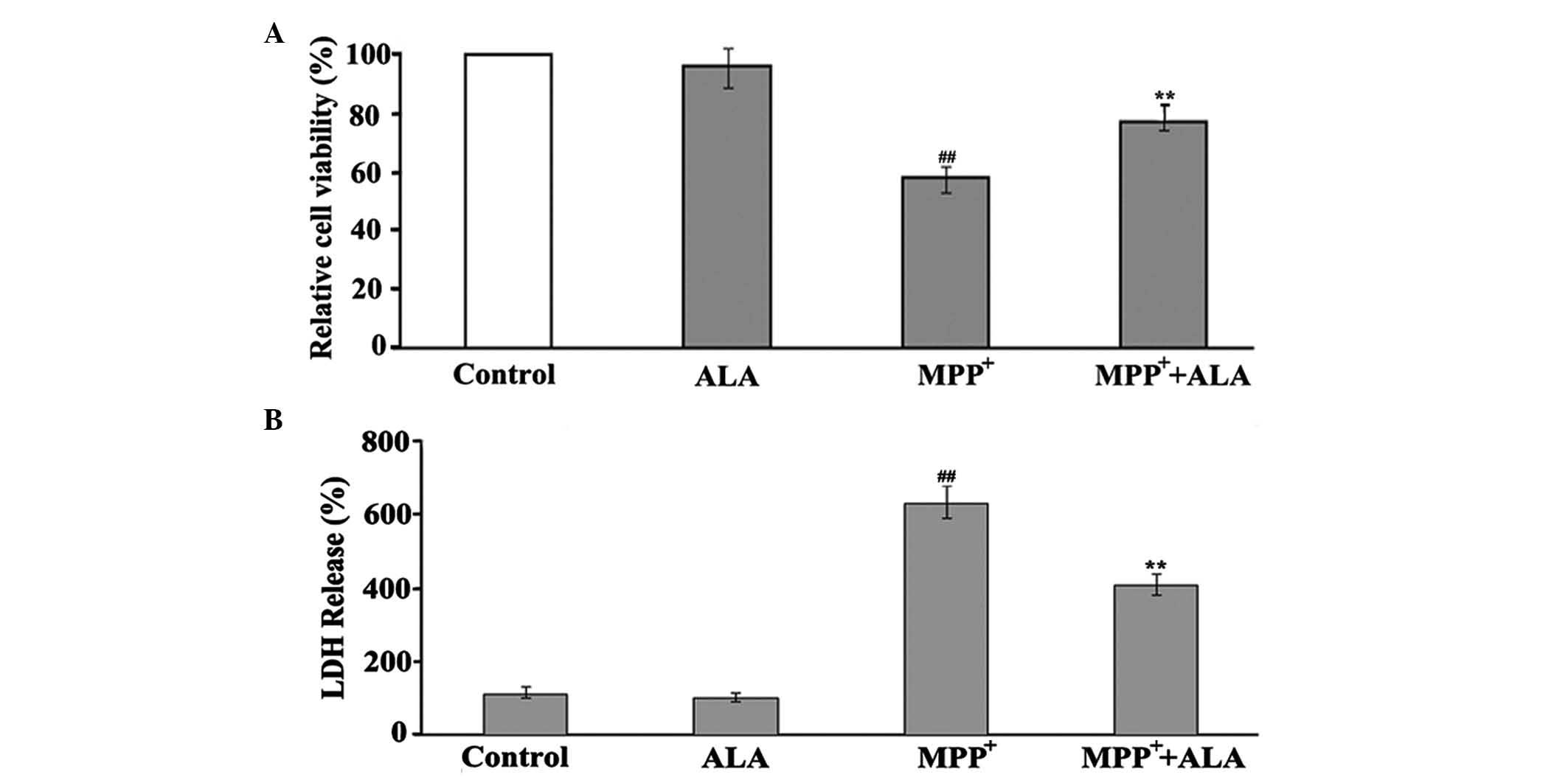
toxicity in PC12 cells. The measurements revealed that the addition
of 0.01 µM ALA to the cells significantly improved the viability of
the PC12 cells to 77% (Fig. 2A),
showing the protective action of ALA in MPP+-induced
neuron damage. The neuroprotective role of ALA was further
confirmed using an LDH assay, which showed that, in the same
conditions, ALA significantly reduced the activity of LDH induced
by MPP+ treatment in the PC12 cells. When the PC12 cells
were treated with 1 mM MPP+ for 48 h, the activity of
LDH was significantly increased, however 0.01 µM ALA significantly
reduced MPP+-induced LDH activity. ALA did not appear to
affect the basal activity of LDH (Fig.
2B).
ALA reduces MPP+-induced
apoptosis in PC12 cells
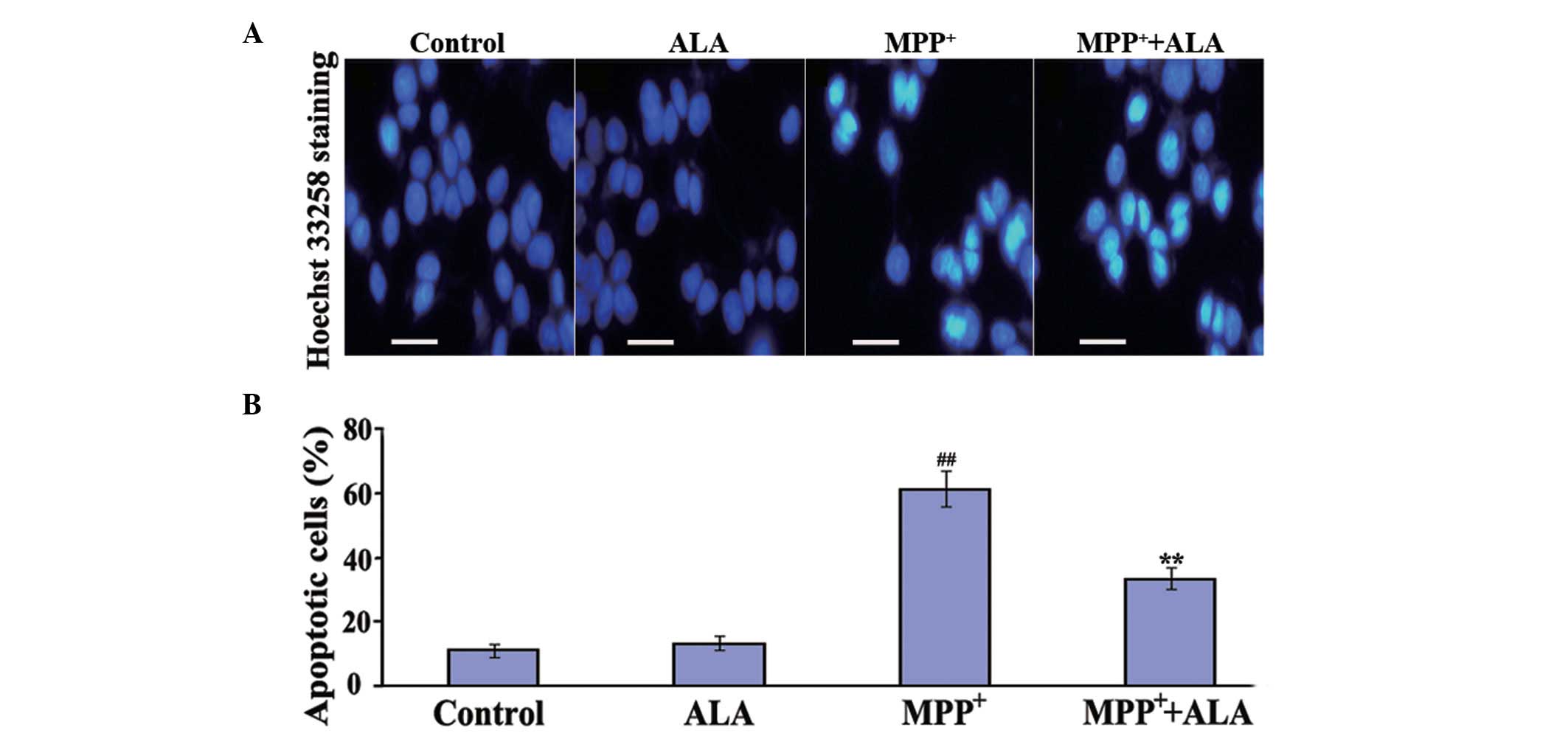
To determine whether ALA prevents
MPP+-induced apoptosis in PC12 cells, Hoechst 33258
staining assays were performed. Apoptosis characterized by a series
of distinct nuclear morphological changes can be detected using
Hoechst 33342 staining, a compound that binds nucleic acids. The
administration of ALA alone did not induce changes in the number of
apoptotic cells. The administration of MPP+
significantly increased the number of apoptotic cells to 61%,
compared with the cells in the control group, whereas in the cells
pre-treated with 0.01 µM ALA, the proportion of apoptotic cells
induced by MPP+ treatment significantly reduced to 33%
(Fig. 3), confirming the
anti-apoptotic activities of ALA against the neurotoxicity of
MPP+ in the neuronal cells.
Neuroprotective ALA increases
production of PCNA in MPP+-treated PC12 cells
To investigate the mechanism underlying the
neuroprotective activity of ALA, the present study investigated
whether ALA had an effect on the expression of PCNA in the
MPP+-induced PD model. Following treatment, the cells
extracts were prepared, and western blot analyses were performed on
the homogenates to examine the effect of ALA on the expression of
PCNA. Consistent with our previous studies (data not shown),
MPP+ treatment significantly reduced the expression of
PCNA in the PC12 cells, confirming the involvement of this protein
in dopaminergic neuron loss in neurodegenerative conditions
(6). Notably, ALA markedly
increased the expression of PCNA altered by MPP+
treatment in the PC12 cells. When the PC12 cells were treated with
1 mM MPP+ for 48 h, the protein expression of PCNA was
markedly decreased, whereas 0.01 µM ALA significantly increased the
expression levels of PCNA in the MPP+-induced PC12
cells. ALA did not affect the basal expression of PCNA (Fig. 4). These results suggested that the
PCNA protein was involved in dopaminergic neuron degeneration, and
ALA exerted its neuroprotive action against the MPP+
neurotoxicity in the dopaminergic neurons, at least in part, via
modulating the production of PCNA.
ALA represses the expression of p53
induced by MPP+ in PC12 cells
In order to investigate the mechanism by which ALA
repressed the expression of PCNA in the MPP+-treated
PC12 cells, the present study examined the effects of ALA on the
PCNA upstream regulator, p53. P53 interacts with the PCNA promoter
to regulate the production of this protein, and a higher
concentration of wild-type p53 inhibits the PCNA promoter,
resulting in a decrease in the production of PCNA (20,21).
The role of the p53 protein in the pathogenesis of several
neurodegenerative disorders, including PD, has been well documented
(22,23). The results of the present study
revealed that the administration of MPP+ caused a
significant upregulation of the expression of p53 in PC12 cells.
This expression pattern was in contrast to that of PCNA, as ALA
treatment markedly reduced the expression levels of p53 induced by
MPP+, but upregulated the expression of PCNA (Fig. 5). These results indicated that the
mechanism by which ALA increased the expression of PCNA in the
MPP+-treated neuronal cells was associated with
repression of the induction of p53.
Discussion
The present study demonstrated for the fist time, to
the best of our knowledge, that ALA exerts its neuroprotective
action mediated by upregulating the protein expression of PCNA via
the p53 pathway in a cellular model of PD.
PD is a movement disorder, which is characterized by
the gradually progressive and selective degeneration of
dopaminergic neurons in the SNpc (24). The pathogenesis of PD remains to be
fully elucidated, however, multiple studies have linked oxidative
stress to dopaminergic neuron degeneration. Cell survival is
dependent on DNA integrity. Under physical and pathological
conditions, DNA is frequently subjected to damage by endogenous and
environmental toxic agents, particularly in the SNpc, which results
from oxidative stress due to its high levels of lipids, iron and
dopamine metabolism (25).
Increased oxidative stress causes oxidative DNA damage, which
subsequently leads to dopaminergic neuron degeneration and the
pathogenesis of PD. Postmortem samples of PD have shown increased
DNA oxidative damage selectively in dopaminergic neurons of the
SNpc, indicating the link between DNA oxidation and the loss of
dopaminergic neurons (2). The
association between DNA damage-induced cell death and the
neurodegenerative process of PD is also supported by the presence
of oxidized DNA in the brain tissues of mice treated with MPTP and
other neuronal toxins, inducing a PD-like pathology (3). To counteract damage, repair
mechanisms for DNA are required to preserve its integrity,
particularly for dopaminergic neurons, which are more prone to
oxidative damage (26–30). PCNA is a well-known protein, which
is involved in DNA repair in a wide range of pathological
conditions by interacting with a number of enzymes and regulatory
proteins (4,5). The PCNA-dependent repair of DNA
damage is crucial in preserving the integrity of DNA under
oxidative conditions (7,8). Our previous in vitro
investigation of the mechanism underlying the degeneration of
dopaminergic neurons in MPP+-induced PC12 cells
indicated that PCNA was involved in DNA damage-induced cell death
in oxidative conditions (data not shown) (6). In the present study, MPP+
treatment significantly reduced the expression of PCNA in the PC12
neuronal cells, and increased the number of apoptotic cells,
indicating that a PCNA-dependent apoptotic pathway is one potential
molecular mechanism involved in neuronal death in the pathogenesis
of PD. Thus, effects of PCNA in reversing degeneration may be
beneficial in neurodegenerative conditions.
ALA is a naturally occurring dithiol compound,
synthesized enzymatically in mitochondria from octanoic acid and
cysteine. In addition to its function as an essential cofactor for
mitochondrial bioenergetic enzymes in the production of energy, ALA
is involved in a set of biochemical activities with potential
pharmacotherapeutic value against a range of pathophysiological
insults (9,31). Several studies have shown that
exogenous ALA can readily cross the blood-brain barrier (32,33).
Notably, the neuroprotective actions of ALA have been reported in
in vivo and in vitro models of neurodegenerative
diseases, including AD, macular degeneration and PD (17–19).
The present study showed that the addition of ALA markedly
increased the expression levels of PCNA in the
MPP+-induced PC12 cells and reduced cell apoptosis,
suggesting that ALA upregulated the expression of the protective
PCNA protein, in addition to providing neuroprotection against the
MPP+-induced neurotoxicity.
The mechanisms underlying the effect of ALA on the
expression of PCNA remain to be fully elucidated. It may be
associated with its ability to regulate the p53 protein, as P53 is
the most well-characterized mechanism for modulating the production
of PCNA through the binding of its promoter (20,21).
P53 was originally identified as a tumor suppressor gene, and has
been considered to be a key contributor in neuronal cell death and
dopaminergic neuron degeneration (34,35).
The pharmacologic inhibition of p53 has been shown to preserve
dopamine neurons against the neurotoxic effects of MPTP and other
neuronal toxins that induce PD-like pathology in in vivo and
in vitro models of PD (23,36–39).
The classical trigger for p53 activation is oxidative stress, and
p53-dependent apoptosis in neuronal cells is predominantly mediated
by DNA damage (34,35). P53 is an upstream inducer of PCNA,
and a higher concentration of wild-type p53 inhibits the PCNA
promoter and reduces the production of PCNA (20,21).
The results of the present study showed that MPP+
significantly increased the expression of P53 in the dopaminergic
neuronal cells and reduced the cell viability, indicating that P53
is a contributor in the pathogenesis of PD. The decrease in the
expression of PCNA was also observed in the MPP+-induced
PD model, and this expression pattern was in contrast to that of
the expression of P53, suggesting a correlation between P53 and the
expression of PCNA in oxidative conditions. Furthermore, LA
efficiently reduced the expression of p53 induced by
MPP+ in the PC12 cells, and upregulated the expression
of PCNA. These results suggested that ALA protected the
dopaminergic neurons against MPP+-induced neurotoxicity
through its ability to upregulate the DNA repair protein, PCNA, via
the P53 pathway.
The present study provided the first evidence, to
the best of our knowledge, that neuroprotective ALA exerts
anti-apoptotic effects on neuronal cells by upregulating the
expression of PCNA via repression of p53 in an
MPP+-induced cellular model of PD. Previous studies have
shown that ALA has anti-inflmmatory and anti-oxidative properties
in a range of cell types and tissues (40–43),
which may be beneficial in neurodegenerative conditions.
Preclinical and clinical data have indicated that ALA is
bioavailable and safe in moderate doses (8). Further investigations are required to
fully elucidate the mechanisms responsible for the protective
effects of ALA in neurodegenerative conditions, which may provide a
potential effective neuroprotection strategy for the treatment of
PD by targeting DNA damage-mediated neuronal degeneration.
Glossary
Abbreviations
Abbreviations:
|
PD
|
Parkinson's disease
|
|
SNpc
|
substantia nigra pars compacta
|
|
MPP+
|
1-methyl-4-phenylpyridinium
|
|
MPTP
|
1-methyl-4-phenyl-1,2,3,6-tetrahydropy-ridine
|
|
PCNA
|
proliferating cell nuclear antigen
|
|
ALA
|
α-lipoic acid
|
References
|
1
|
de Lau LM and Breteler MM: Epidemiology of
Parkinson's disease. Lancet Neurol. 5:525–535. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alam ZI, Jenner A, Daniel SE, Lees AJ,
Cairns N, Marsden CD, Jenner P and Halliwell B: Oxidative DNA
damage in the parkinsonian brain: An apparent selective increase in
8-hydroxyguanine levels in substantia nigra. J Neurochem.
69:1196–1203. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mandir AS, Przedborski S, Jackson-Lewis V,
Wang ZQ, Simbulan-Rosenthal CM, Smulson ME, Hoffman BE, Guastella
DB, Dawson VL and Dawson TM: Poly (ADP-ribose) polymerase
activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine
(MPTP)-induced parkinsonism. Proc Natl Acad Sci USA. 96:5774–5779.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moldovan GL, Pfander B and Jentsch S:
PCNA, the maestro of the replication fork. Cell. 129:665–679. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mailand N, Gibbs-Seymour I and
Bekker-Jensen S: Regulation of PCNA-protein interactions for genome
stability. Nat Rev Mol Cell Biol. 14:269–282. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li DW, Li GR, Zhang BL, Feng JJ and Zhao
H: Damage to dopaminergic neurons is mediated by proliferating cell
nuclear antigen through the p53 pathway under conditions of
oxidative stress in a cell model of Parkinson's disease. Int J Mol
Med. 37:429–435. 2016.PubMed/NCBI
|
|
7
|
Burkovics P, Hajdú I, Szukacsov V, Unk I
and Haracska L: Role of PCNA-dependent stimulation of
3′-phosphodiesterase and 3′-5′ exonuclease activities of human Ape2
in repair of oxidative DNA damage. Nucleic Acids Res. 37:4247–4255.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Amoroso A, Concia L, Maggio C, Raynaud C,
Bergounioux C, Crespan E, Cella R and Maga G: Oxidative DNA damage
bypass in Arabidopsis thaliana requires DNA polymerase λ and
proliferating cell nuclear antigen 2. Plant Cell. 23:806–822. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shay KP, Moreau RF, Smith EJ, Smith AR and
Hagen TM: Alpha-lipoic acid as a dietary supplement: Molecular
mechanisms and therapeutic potential. Biochim Biophys Acta.
1790:1149–1160. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wollin SD and Jones PJ: Alpha-lipoic acid
and cardiovascular disease. J Nutr. 133:3327–3330. 2003.PubMed/NCBI
|
|
11
|
McNeilly AM, Davison GW, Murphy MH, Nadeem
N, Trinick T, Duly E, Novials A and McEneny J: Effect of α-lipoic
acid and exercise training on cardiovascular disease risk in
obesity with impaired glucose tolerance. Lipids Health Dis.
10:2172011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stanković MN, Mladenović D, Ninković M,
Ethuričić I, Sobajić S, Jorgačević B, de Luka S, Vukicevic RJ and
Radosavljević TS: The effects of α-lipoic acid on liver oxidative
stress and free fatty acid composition in methionine-choline
deficient diet-induced NAFLD. J Med Food. 17:254–261. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hatami S, Zavareh S, Salehnia M,
Lashkarbolouki T, Ghorbanian MT and Karimi I: Total oxidative
status of mouse vitrified pre-antral follicles with pre-treatment
of alpha lipoic acid. Iran Biomed J. 18:181–188. 2014.PubMed/NCBI
|
|
14
|
Showkat A, Bastnagel WR and Hudson JQ:
Effect of α-lipoic acid on oxidative stress in end-stage renal
disease patients receiving intravenous iron. ISRN Nephrol.
2014:6345152014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ziegler D, Hanefeld M, Ruhnau KJ, Meissner
HP, Lobisch M, Schütte K and Gries FA: Treatment of symptomatic
diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic
acid. A 3-week multicentre randomized controlled trial (ALADIN
Study). Diabetologia. 38:1425–1433. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ziegler D, Hanefeld M, Ruhnau KJ, Hasche
H, Lobisch M, Schütte K, Kerum G and Malessa R: Treatment of
symptomatic diabetic polyneuropathy with the antioxidant
alpha-lipoic acid: A 7-month multicenter randomized controlled
trial (ALADIN III Study). ALADIN III study group. Alpha-lipoic acid
in diabetic neuropathy. Diabetes Care. 22:1296–1301. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sancheti H, Kanamori K, Patil I, Brinton R
Diaz, Ross BD and Cadenas E: Reversal of metabolic deficits by
lipoic acid in a triple transgenic mouse model of Alzheimer's
disease: A 13C NMR study. J Cereb Blood Flow Metab. 34:288–296.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mansoor S, Gupta N, Luczy-Bachman G, Limb
GA, Kuppermann BD and Kenney MC: Protective effects of lipoic acid
on chrysene-induced toxicity on Müller cells in vitro. Mol Vis.
19:25–38. 2013.PubMed/NCBI
|
|
19
|
Li DW, Li GR, Lu Y, Liu ZQ, Chang M, Yao
M, Cheng W and Hu LS: α-lipoic acid protects dopaminergic neurons
against MPP+-induced apoptosis by attenuating reactive oxygen
species formation. Int J Mol Med. 32:108–114. 2013.PubMed/NCBI
|
|
20
|
Morris GF, Bischoff JR and Mathews MB:
Transcriptional activation of the human proliferating-cell nuclear
antigen promoter by p53. Proc Natl Acad Sci USA. 93:895–899. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shivakumar CV, Brown DR, Deb S and Deb SP:
Wild-type human p53 transactivates the human proliferating cell
nuclear antigen promoter. Mol Cell Biol. 15:6785–6793. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martin LJ: p53 is abnormally elevated and
active in the CNS of patients with amyotrophic lateral sclerosis.
Neurobiol Dis. 7:613–622. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Duan W, Zhu X, Ladenheim B, Yu QS, Guo Z,
Oyler J, Cutler RG, Cadet JL, Greig NH and Mattson MP: p53
inhibitors preserve dopamine neurons and motor function in
experimental parkinsonism. Ann Neurol. 52:597–606. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Forno LS: Neuropathology of Parkinson's
disease. J Neuropathol Exp Neurol. 55:259–272. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dias V, Junn E and Mouradian MM: The role
of oxidative stress in Parkinson's disease. J Parkinsons Dis.
3:461–491. 2013.PubMed/NCBI
|
|
26
|
Montine KS, Quinn JF, Zhang J, Fessel JP,
Roberts LJ II, Morrow JD and Montine TJ: Isoprostanes and related
products of lipid peroxidation in neurodegenerative diseases. Chem
Phys Lipids. 128:117–124. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sadrzadeh SM and Saffari Y: Iron and brain
disorders. Am J Clin Pathol. 121:(Suppl). S64–S70. 2004.PubMed/NCBI
|
|
28
|
Jomova K and Valko M: Advances in
metal-induced oxidative stress and human disease. Toxicology.
283:65–87. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nagatsu T and Sawada M: Molecular
mechanism of the relation of monoamine oxidase B and its inhibitors
to Parkinson's disease: Possible implications of glial cells. J
Neural Transm Suppl. 53–65. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Núñez MT, Urrutia P, Mena N, Aguirre P,
Tapia V and Salazar J: Iron toxicity in neurodegeneration.
Biometals. 25:761–776. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Goraca A, Huk-Kolega H, Piechota A,
Kleniewska P, Ciejka E and Skibska B: Lipoic acid-biological
activity and therapeutic potential. Pharmacol Rep. 63:849–858.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Panigrahi M, Sadguna Y, Shivakumar BR,
Kolluri SV, Roy S, Packer L and Ravindranath V: Alpha-Lipoic acid
protects against reperfusion injury following cerebral ischemia in
rats. Brain Res. 717:184–188. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Arivazhagan P, Shila S, Kumaran S and
Panneerselvam C: Effect of DL-alpha-lipoic acid on the status of
lipid peroxidation and antioxidant enzymes in various brain regions
of aged rats. Exp Gerontol. 37:803–811. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chipuk JE and Green DR: Dissecting
p53-dependent apoptosis. Cell Death Differ. 13:994–1002. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Culmsee C and Mattson MP: p53 in neuronal
apoptosis. Biochem Biophys Res Commun. 331:761–777. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mandir AS, Simbulan-Rosenthal CM, Poitras
MF, Lumpkin JR, Dawson VL, Smulson ME and Dawson TM: A novel in
vivo post-translational modification of p53 by PARP-1 in
MPTP-induced parkinsonism. J Neurochem. 83:186–192. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nair VD: Activation of p53 signaling
initiates apoptotic death in a cellular model of Parkinson's
disease. Apoptosis. 11:955–966. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Biswas SC, Ryu E, Park C, Malagelada C and
Greene LA: Puma and p53 play required roles in death evoked in a
cellular model of Parkinson disease. Neurochem Res. 30:839–845.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nakaso K, Yoshimoto Y, Yano H, Takeshima T
and Nakashima K: p53-mediated mitochondrial dysfunction by
proteasome inhibition in dopaminergic SH-SY5Y cells. Neurosci Lett.
354:213–216. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Busse E, Zimmer G, Schopohl B and
Kornhuber B: Influence of alpha-lipoic acid on intracellular
glutathione in vitro and in vivo. Arzneimittelforschung.
42:829–831. 1992.PubMed/NCBI
|
|
41
|
Talebi A, Zavareh S, Kashani MH,
Lashgarbluki T and Karimi I: The effect of alpha lipoic acid on the
developmental competence of mouse isolated preantral follicles. J
Assist Reprod Genet. 29:175–183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Packer L, Witt EH and Tritschler HJ:
Alpha-Lipoic acid as a biological antioxidant. Free Radic Biol Med.
19:227–250. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Packer L, Tritschler HJ and Wessel K:
Neuroprotection by the metabolic antioxidant alpha-lipoic acid.
Free Radic Biol Med. 22:359–378. 1997. View Article : Google Scholar : PubMed/NCBI
|