Introduction
Sepsis remains an important challenge when treating
patients in Intensive Care Units (ICUs). The pathology of sepsis is
based on a systemic inflammatory response that is characterized by
the upregulation of inflammatory cytokines, particularly in
response to gram-negative bacteria (1,2).
Lipopolysaccharides (LPS) are a major component of the outer
membrane of gram-negative bacteria, which triggers inflammation and
gram-negative sepsis. Patients that develop severe sepsis or septic
shock often have a low survival rate, which may be associated with
the host response to refractory hyperglycemic stress. However, the
molecular mechanism underlying this remains to be elucidated. It is
possible that LPS may directly limit the capacity of pancreatic
β-cells to induce oxidative stress and apoptosis. It may be one of
the mechanisms behind hyperglycemic stress in patients with
sepsis.
The imbalance between the oxidation system and the
antioxidant defense frequently results in oxidative stress. This
imbalance may induce an inflammatory cascade and limit the function
of pancreatic β-cells by the direct effect of free radicals
(3). High levels of reactive
oxygen species (ROS) in the pancreas and low levels of antioxidant
defense mechanisms may lead to increased oxidative damage and
reduced insulin secretion (4). Our
previous study determined that LPS stress led to increased ROS
production and reduced insulin secretion in Ins-1 pancreatic
β-cells (5). Toll-like receptors
(TLRs) are a family of pattern-recognition receptors that serve
important functions in the innate immune system, by activating
pro-inflammatory signaling pathways in response to the detection of
microbial products (6). TLR4 is
expressed in pancreatic β-cells and insulin-sensitive tissues,
including adipocytes and muscle cells (7–9).
TLR4 has been directly associated with proinflammatory signaling in
β-cell dysfunction (10,11). The effect of TLR4 signaling in
insulin resistance is a well-established concept; however, the
specific function of TLR4 during LPS-induced oxidative stress on
pancreatic β-cells remains to be elucidated. The present study
blocked TLR4 using a TLR4 antibody and TLR4-short hairpin (shRNA)
to observe its effect on LPS-induced dysfunction of pancreatic
β-cells. It is possible that TLR4 activation may mediate oxidative
stress within pancreatic β-cells, resulting in their apoptosis and
dysfunction of insulin secretion. The current study aimed to reveal
the possible mechanisms of infection-induced stress
hyperglycemia.
Materials and methods
Animals
A total of six Male Sprague-Dawley rats were
provided by SLAC Laboratory Animal Co., Ltd. (Shanghai, China). The
rats were 6–8 weeks of age and weighed 250–300 g. They were
maintained in a sterile environment prior to the start of the
study. The rats were provided with standard chow and water, and
were exposed to a 12 h light/dark cycles at 26°C in 50% humidity.
The protocol was approved by the Ethics Committee of Shanghai
Jiaotong University School of Medicine (approval no.
XHEC-F-2012-003). The experiments were performed in accordance with
the guidance for the Care and Use of Laboratory Animals of the
Nationals Institutes of Health (Bethesda, MD, USA).
Reagents
Lipopolysacharide, collagenase V and Ficoll 400 were
purchased from Sigma-Aldrich; Merck Millipore (Darmstadt, Germany).
The β-actin antibody (cat. no. sc-81178) and mouse anti-TLR4
antibody (cat. no. sc-12511) and TLR4-short hairpin (sh)RNA were
obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Fetal calf serum (FCS) and RPMI-1640 medium were obtained from
Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA, USA). The
2′,7′-dichlorofluorescin diacetate (DCFH-DA), annexin V-fluorescein
isothiocyanate (FITC) kit and bicinchoninic acid protein assay kit
were purchased from Beyotime Institute of Biotechnology (Haimen,
China). The TRIzol reagent, PrimeScript RT reagent kit and SYBR
Premix Ex Taq kit were obtained from Takara Biotechnology Co., Ltd.
(Shiga, Japan).
Islet isolation and culture
Sprague-Dawley rats were fasted for 12 h and were
subsequently sacrificed using an intraperitoneal injection of 2.5%
sodium pentobarbital (Sinopharm Chemical Reagent Co., Ltd.,
Shanghai, China) (60 mg/kg body weight). The pancreatic and common
bile ducts were exposed and 3 ml precooled Hank's Balanced Salt
Solution (HBSS), combined with 6 ml of precooled collagenase V
(Sigma-Aldrich; Merck Millipore) (1 mg/ml), were immediately
injected through the pancreatic duct. The pancreas was subsequently
removed and digested for 10 min in a water bath at 37°C. The
tissues were then passed through a 600 µm mesh stainless steel
filter. Precooled HBSS containing 10% FCS was used to terminate the
digestion. The islets were purified using Ficoll 400 and density
gradient centrifugation with 600 × g at 4°C. The isolated
islets were cultured in RPMI-1640 media supplemented with 10% FCS
at 37°C and an atmosphere of 5% CO2.
Knockdown of TLR4 by shRNA
The isolated islets were seeded into 24-well plates
at a density ~0.5×103/ml and cultured overnight.
TLR4-shRNA and negative control shRNA were transfected into cells
using TLR4 shRNA (r) Lentiviral Particles (cat. no. sc-156001) and
Control shRNA Lentiviral Particles (cat. no. sc-108080) (Santa Cruz
Biotechnology, Inc.) according to the manufacturer's protocol.
Subsequently, the cells were cultured by puromycin dihydrochloride
(Sigma-Aldrich; Merck Millipore) for 3–4 days. Transfection
efficiency was determined by western blot analysis.
ROS production determined using
DCFH-DA probe
The islets were grouped into the following treatment
groups: i) Control (CON) group treated with phosphate-buffered
saline (PBS); ii) LPS group treated with 10 µg/ml LPS; iii)
anti-TLR4 antibody intervention group pretreated with anti-TLR4
antibody (20 µg/ml) for 1 h, then treated with LPS (10 µg/ml); and
iv) TLR4-shRNA intervention group pretreated with TLR4-shRNA (10
µmol/l) for 1 h, then treated with LPS (10 µg/ml). The islets were
cultured for 24 h and were subsequently incubated in the presence
of 10 µmol/l DCFH-DA for 1 h at 37°C in the dark, and then washed
three times with PBS. The ROS production of islets was assessed by
fluorescent microscopy (excitation 488 nm and emission 525 nm).
Apoptosis of rat islets detected by
flow cytometry analysis
Following the incubation (the isolated islets were
seeded into 24-well plates), the islets were harvested and
centrifuged at 100 × g for 2 min at 4°C and then digested
into single cells with trypsin and DNAse. The cells were
subsequently centrifuged at 1,000 × g for 5 min at 4°C and
washed with PBS twice. The cells were suspended at a density of
1.0×106 cells/well with annexin V-FITC composite liquid
(195 µl), and the islets cells were incubated with annexin V-FITC
(5 µl). Finally, the cells were stained with 10 µl propidium iodide
for 15 min at room temperature in the dark. The apoptotic rate was
detected by a flow cytometer (FACSCalibur; BD Biosciences, Franklin
Lakes, NJ, USA) and analyzed by CellQuest software version 7.5.3
(BD Biosciences).
Western blotting
Isolated islets were cultured and treated as
aforementioned, and were subsequently washed twice with PBS, placed
immediately in precooled lysis buffer and centrifuged at 12,000 ×
g for 10 min at 4°C. The protein concentration of the
extracts was determined using Enhanced BCA Protein assay kit
(Beyotime Institute of Biotechnology). The samples (30–50 µg) were
separated by electrophoresis on 8 or 12% sodium dodecyl
sulphate-polyacrylamide gels and transferred onto nitrocellulose
membranes. Non-specific binding sites of the samples were blocked
using Tris-buffered saline and Tween-20 with 5% non-fat milk for 1
h at 4°C. Following blocking, the membraness were incubated
overnight at 4°C with the anti-mouse TLR4 primary antibody (1:200),
followed by incubation with the secondary goat anti-mouse
horseradish peroxidase-conjugated antibody (1:2,000; Beyotime
Institute of Biotechnology; cat. no. A0216) for 1 h at room
temperature. β-actin (1:1,000) expression was used as an internal
control. The blots were visualized using an enhanced
chemiluminescence system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). The density of protein bands was analyzed using Image Lab
2.0.1 (Bio-Rad Laboratories, Inc.) and normalized against the
intensity of β-actin.
mRNA expression levels of TLR4 and
B-cell lymphoma (Bcl)-2/Bcl-2 associated X protein (Bax) detected
by reverse transcription-quantitative PCR (RT-qPCR)
The total RNA was isolated using TRIzol reagent. The
mRNA expression levels of TLR4 and Bax/Bcl-2 were determined using
RT-qPCR. The total RNA concentration was 1,000 ng. The cDNA were
synthesized using PrimeScript RT reagent kit and the samples were
subsequently subjected to PCR amplification with primer sets
(presented in Table I). RT-qPCR
was performed using SYBR Premix Ex Taq kit. The thermocycling
conditions were as follows: 95°C for 30 sec, 40 cycles of 5 sec at
95°C and 1 min at 60°C. To confirm that the specific PCR product
was formed, a dissociation step of 15 sec at 95°C, 15 sec at 60°C
and 15 sec at 95°C was added to confirm the melting temperature.
Gene expression was determined by the 2−ΔΔCq method
(12) and was normalized against
GAPDH.
 | Table I.Primer sequences used in reverse
transcription-polymerase chain reaction. |
Table I.
Primer sequences used in reverse
transcription-polymerase chain reaction.
| Gene | Sequence (5′-3′) |
|---|
| TLR4 |
|
|
Forward |
AGTTGGCTCTGCCAAGTCTCAGAT |
|
Reverse |
TGGCACTCATCAGGATGACACCAT |
| Bcl-2 |
|
|
Forward |
TTGTGGCCTTCTTTGAGTTCGGTG |
|
Reverse |
TCATCCACAGAGCGATGTTGTCCA |
| Bax |
|
|
Forward |
TTTGCAGACGGCAACTTCAACTGG |
|
Reverse |
TGTCCAGCCCATGATGGTTCTGAT |
| GAPDH |
|
|
Forward |
TGATGCTGGTGCTGAGTATGTCGT |
|
Reverse |
AGGTGGAAGAATGGGAGTTGCTGT |
Glucose-stimulated insulin secretion
(GSIS) in islets
Isolated rat islets were seeded into 96-well plates
at concentration about 0.5×103 cells/well. The cells
were subjected to the indicated treatment for 24 h, and
subsequently, the medium was removed and islets were balanced in
Krebs Ringer Buffer [KRB; 115 mM NaCl; 4.7 mM KCl; 2.6 mM
CaCl2; 1.2 mM KH2PO4; 1.2 mM MgSO4; 10 mM
NaHCO3; 10 mM HEPES; 0.1% bovine serum albumen (pH
7.4)], containing 3.3 mM D-glucose, for 30 min. The islets were
subsequently incubated in KRB containing 3.3 mM D-glucose for
another 1 h and KRB containing 27.8 mM D-glucose for 1 h. The
buffer was collected from the 3.3 and 27.8 mM incubations and
stored at −80°C until analysis. Insulin concentration in the medium
was determined using a Rat Insulin Radioimmunoassay kit (Diagnostic
Systems Laboratories, Inc., Webster, TX, USA).
Statistical analysis
The data are presented as the mean ± standard
deviation. The experiments were repeated at least three times for
each group. Analysis was performed using SPSS version 19.0 software
(IBM SPSS, Armonk, NY, USA). Statistical significance of
differences between groups was determined using one-way analysis of
variance. P<0.05 was were considered to indicate a statistically
significant difference.
Results
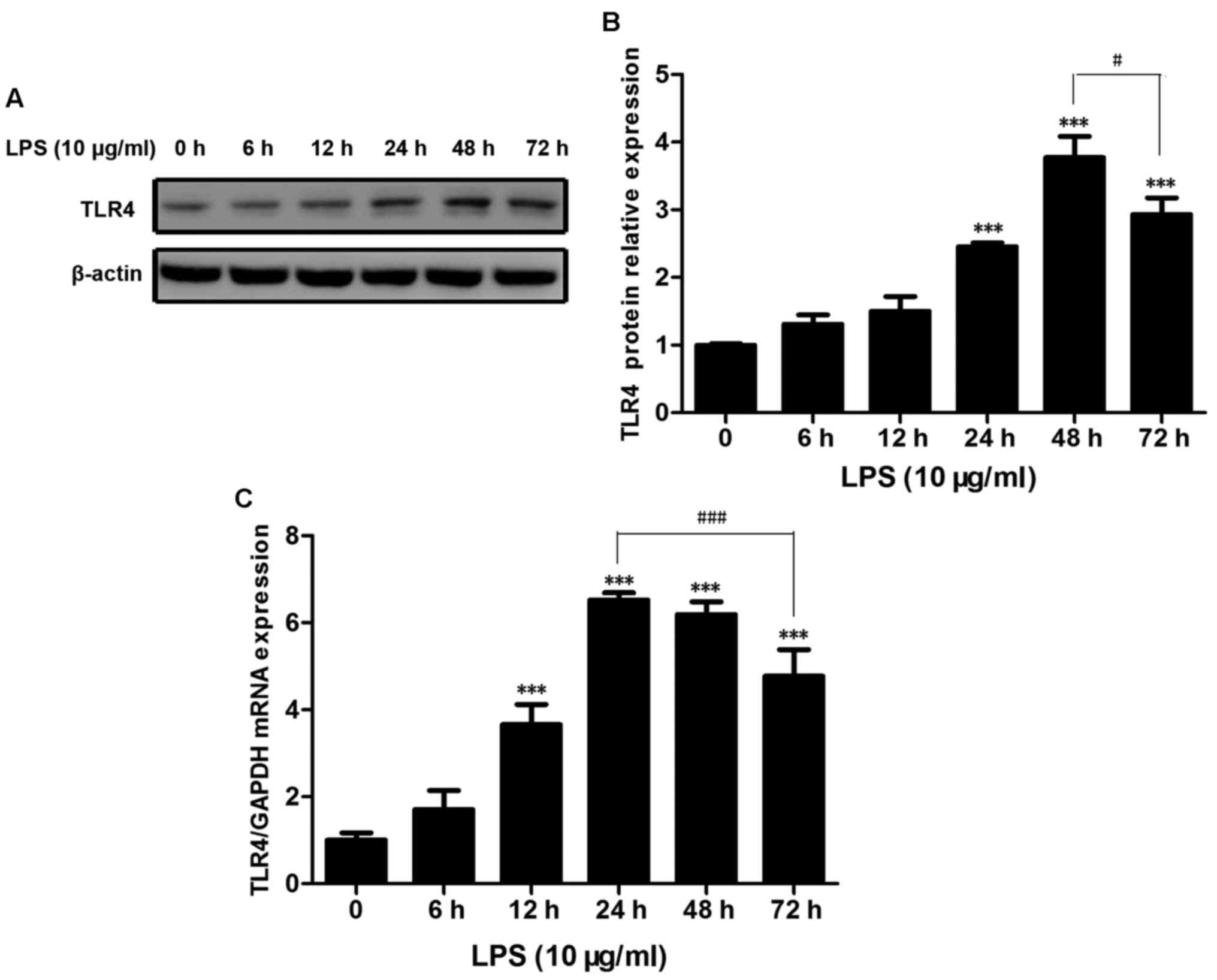
TLR4 expression in rat islets
increases with LPS treatment
The present study detected TLR4 expression levels in
rat islets when stimulated by LPS for various durations. The
protein and mRNA expression levels of TLR4 were upregulated
subsequent to LPS stimulation. A significant increase in TLR4
protein expression at 24 and 48 h, which was 1.5-fold and 4-folds
greater when compared with the control group (P<0.001; Fig. 1A and B). The TLR4 protein
expression levels were significantly reduced when treated for
>48 h (P<0.05; Fig. 1B). The
mRNA expression levels of TLR4 were significantly increased at 12
h, and were 4-fold higher compared with the control group
(P<0.001; Fig. 1C) and
increased by 7-fold at 24 h (P<0.001; Fig. 1C). TLR4 expression levels were
elevated following stimulation with LPS for 48 h (P<0.001;
Fig. 1C) and 72 h (P<0.001;
Fig. 1C). However, the expression
levels fell with prolonged LPS treatment for >24 h (Fig. 1C).
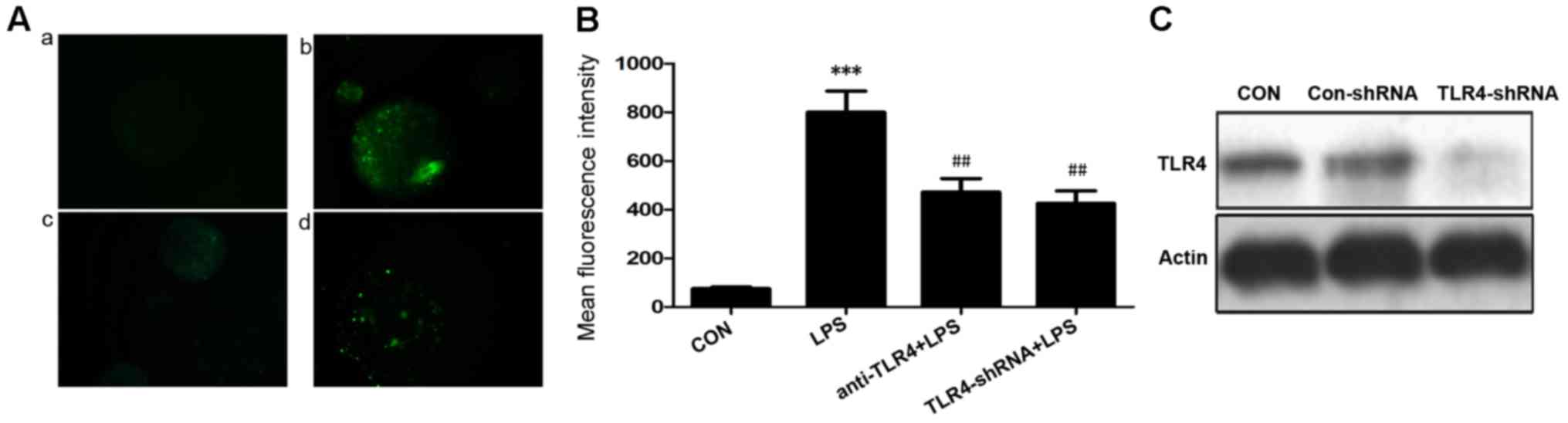
Downregulation of TLR4 in LPS-treated
rat isolated islets reduces ROS production
ROS production was observed as green fluorescence in
the cells. The generation of ROS in rat islets was significantly
increased following exposure to LPS (10 µg/ml) for 24 h when
compared with the control (P<0.001; Fig. 2A and B). The mean fluorescence
intensity of the LPS group increased by 10-fold compared with the
control group (75.3±12.1 vs. 800±150.9; Fig. 2A). In order to investigate the
function of TLR4 during oxidative stress in rat islets exposed to
LPS, TLR4 expression was inhibited by using an anti-TLR4 antibody
and TLR4 shRNA. The ROS production was significantly reduced by
pretreatment of LPS-exposed cells with anti-TLR4 antibody or
TLR4-shRNA when compared with the LPS only group (P<0.01;
Fig. 2B). The efficiency of the
TLR4-shRNA transfection was determined by western blot analysis. It
was revealed that TLR4 expression was reduced by TLR4-shRNA
(Fig. 2C). These findings
suggested that TLR4 may contribute to the process of the oxidative
stress induced by LPS.
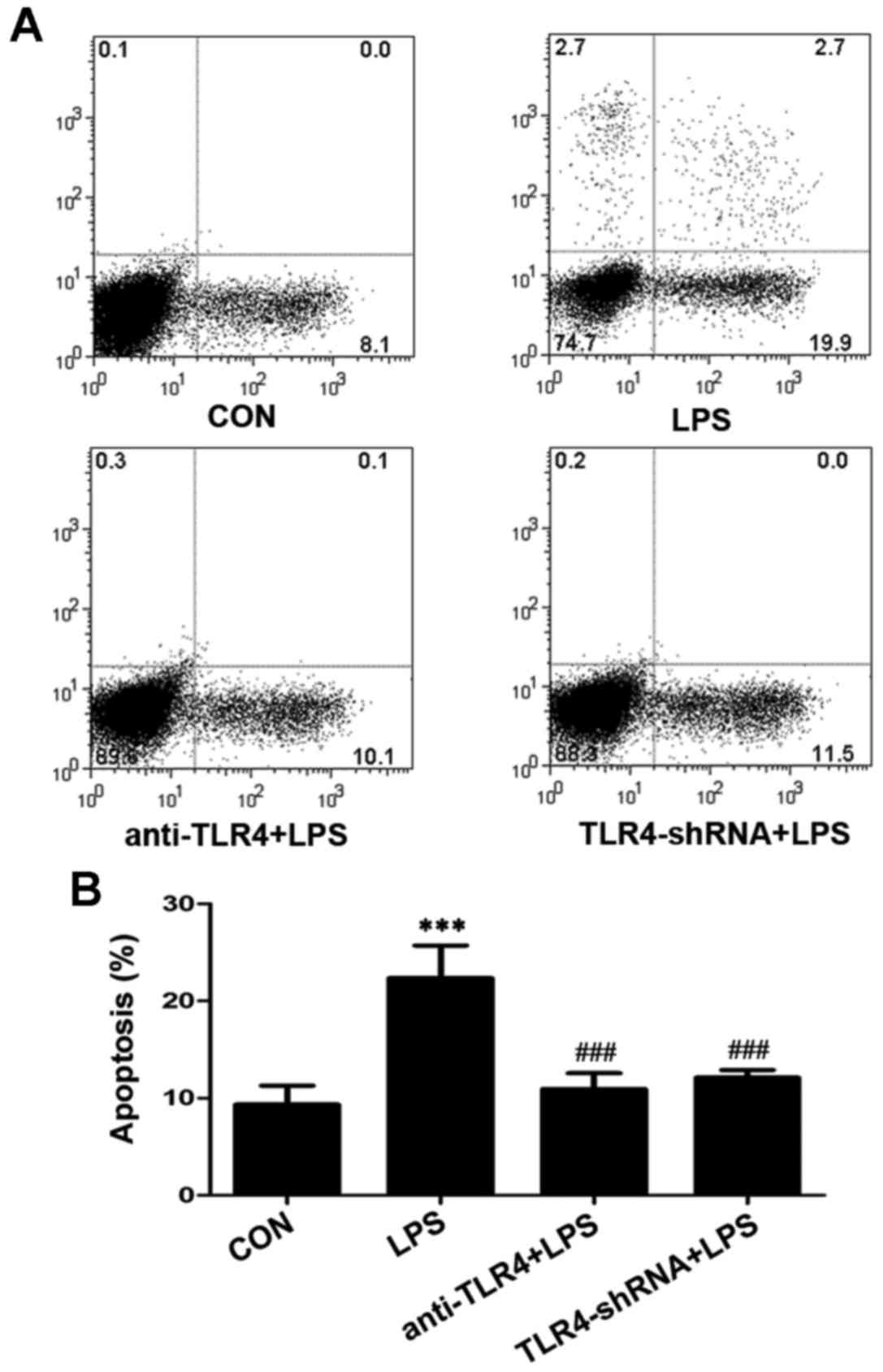
Suppression of TLR4 decreases
LPS-induced apoptosis in islet cells
Apoptosis is an important mechanism that may lead to
the dysfunction of pancreatic β-cells. The present study revealed
that the apoptotic rate of rat islets was higher following LPS
stimulation for 24 h. The apoptotic rate was significantly higher
(22.37±3.35%) in the LPS group compared with the control
(9.33±1.97%) (P<0.001; Fig. 3).
The activity of TLR4 was also suppressed to determine its
involvement in the apoptosis of islet cells. A significant
reduction on apoptotic rate was observed when cells were pretreated
with anti-TLR4 antibody (P<0.001; Fig. 3) and TLR4-shRNA (P<0.001;
Fig. 3) when compared with the
LPS-treated group. The anti-TLR4 antibody and TLR4-shRNA decreased
the LPS-induced apoptosis ratio to 10.60±1.04% and 12.10±0.79%,
respectively. This revealed that the inhibition of TLR4 may protect
rat islet cells from LPS-induced apoptosis.
TLR4 downregulation reduces the
expression levels of apoptosis-associated genes and proteins
The dysregulation of apoptosis-associated genes and
proteins may lead to apoptotic cell death. The present study
examined the effects of TLR4 on the expression levels of caspase-3,
poly ADP-ribose polymerase (PARP) and the ratio of Bax/Bcl-2.
Treatment with 10 µg/ml LPS for 24 h led to significantly
upregulated expression levels of caspase-3 and PARP increased by
2-fold compared with the control group (P<0.001; Fig. 4A and B), whereas the expression
ratio of Bax/Bcl-2 was 1.5-fold higher when the LPS-treated group
was compared with the control (P<0.05; Fig. 4A). Cells pretreated with anti-TLR4
antibody in order to inhibit TLR4 expression significantly reduced
the LPS-induced upregulation of caspase-3 (P<0.05; Fig. 4A), and PARP (P<0.01; Fig. 4A). However, no significant
difference was identified between cells pretreated with TLR4
antibodies and the LPS treatment group. Cells pretreated with
TLR4-shRNA exhibited significantly reduced expression levels of
caspase-3 (P<0.001; Fig. 4B),
PARP (P<0.01; Fig. 4B) and
Bac/Bcl-2 ratio (P<0.01; Fig.
4B) when compared with the LPS treatment group. The mRNA
expression level ratio of Bax/Bcl-2 was significantly increased by
2.2-fold in the LPS treatment group compared with the control group
(P<0.001; Fig. 4C).
Pretreatment with TLR4 antibody and TLR4-shRNA resulted in
significantly reduced expression ratio levels, when compared with
the LPS treatment group (P<0.01 and P<0.001, respectively;
Fig. 4C). Therefore, it is
possible that TLR4 is involved in LPS-induced apoptosis in rat
islet cells.
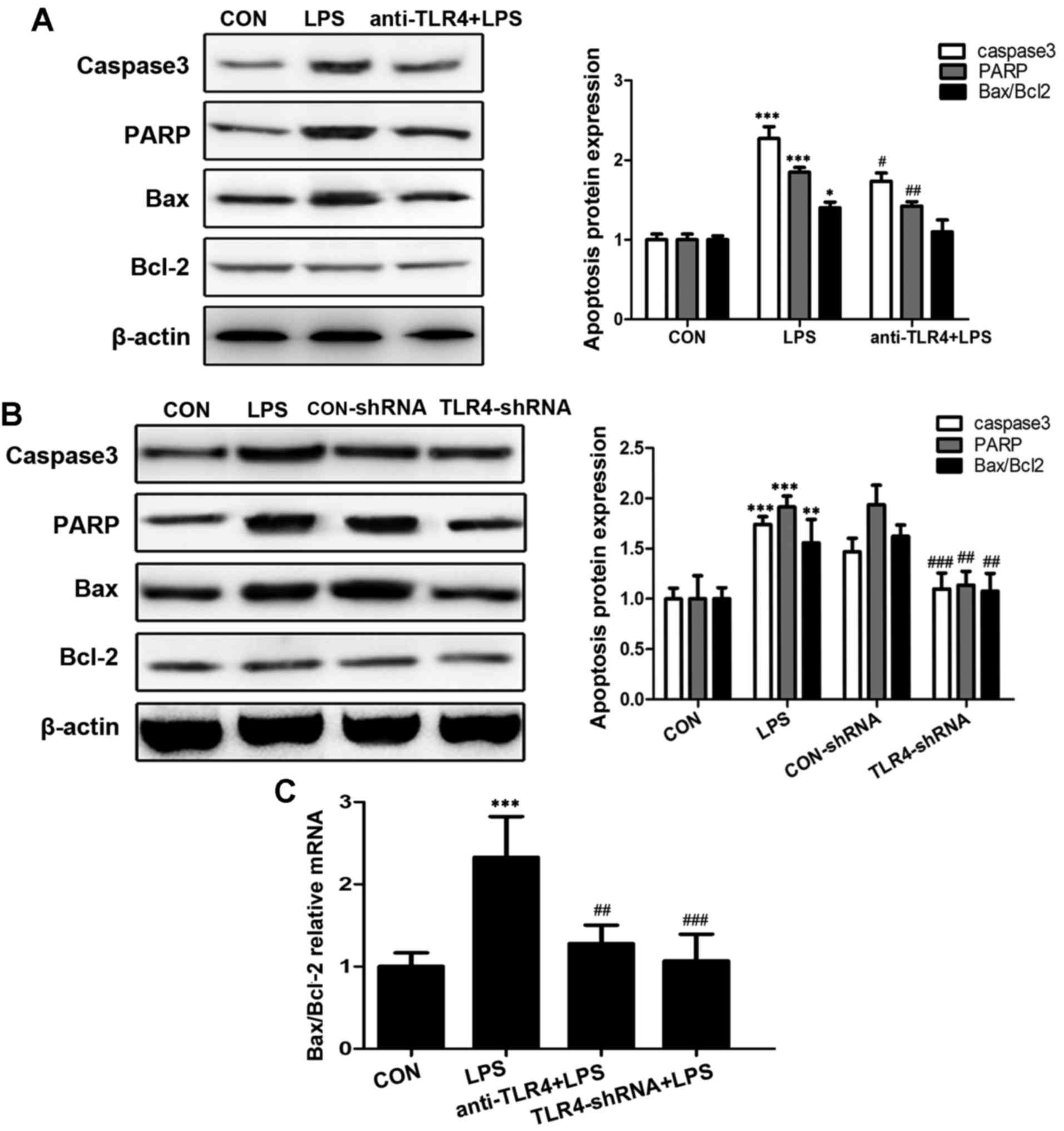 | Figure 4.Expression levels of
apoptosis-associated genes and proteins. (A) The protein expression
levels of caspase-3, PARP, Bax and Bcl-2 were assessed following
pre-treatment with anti-TLR4 antibody by western blotting. The
expression levels were quantified relative to β-actin. (B) The
protein expression levels of the apoptotic proteins, caspase-3,
PARP and Bax/Bcl-2, were assessed following knockdown of TLR4 by
shRNA. The protein expression levels were calculated relative to
β-actin. (C) The Bax/Bcl-2 ratio was determined by reverse
transcription-quantitative polymerase chain reaction. The data are
presented as the mean ± standard deviation of three independent
experiments (***P<0.001, **P<0.01, *P<0.05 vs. control;
###P<0.001, ##P<0.01,
#P<0.05 vs. LPS). CON, control; LPS,
lipopolysaccharides; TLR4, toll-like receptor 4; shRNA, small
hairpin RNA; PARP, poly ADP-ribose polymerase; Bcl-2, B-cell
CLL/lymphoma 2; Bax, BCL2 associated X. |
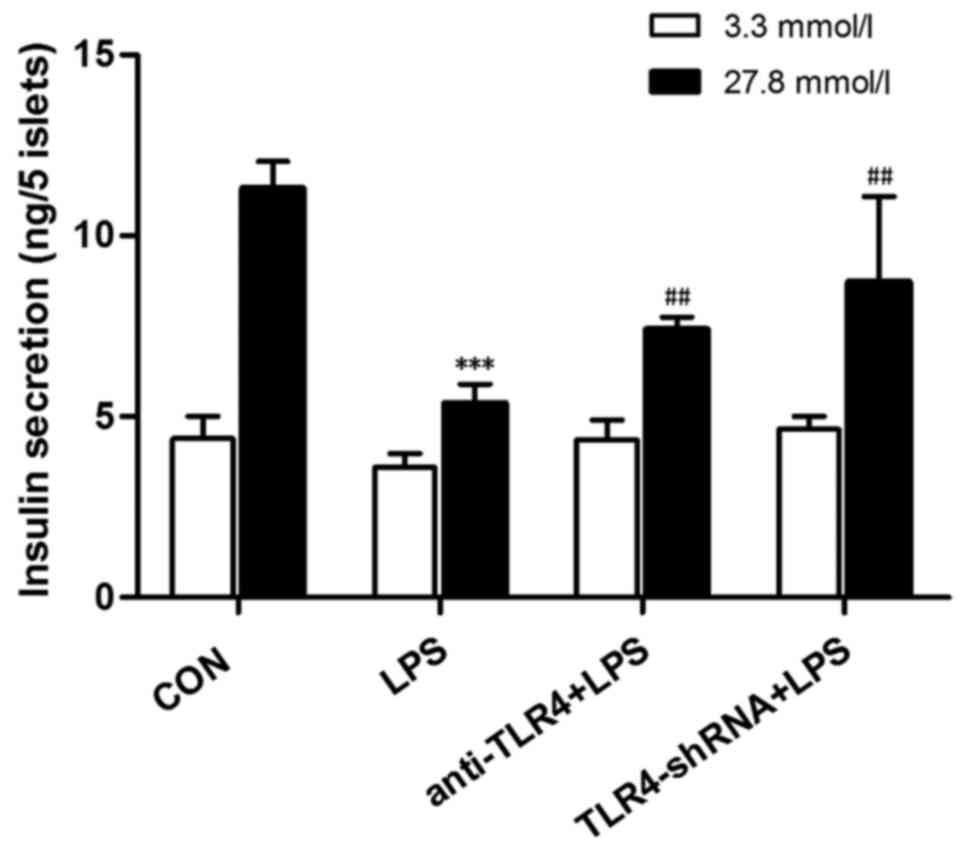
Downregulation of TLR4 expression
levels in LPS-treated increases insulin secretion in islet
cells
In order to determine the importance of TLR4 in
LPS-induced rat pancreatic β-cell dysfunction GSIS assays were
performed. Following the aforementioned treatments, the isolated
islet cells were stimulated by 3.3 and 27.8 mmol/l glucose, and
then insulin secretion was quantified. Isolated islet cells
incubated with 10 µg/ml LPS for 24 h exhibited reduced insulin
secretion. The insulin value was significantly reduced by 2.1-fold
in the LPS-treated group compared with control group when
stimulated with 27.8 mmol/l glucose (P<0.001; Fig. 5). The insulin secretion in isolated
rat islets was significantly increased following pretreatment with
anti-TLR4 antibody and TLR4-shRNA (P<0.01; Fig. 5). This suggested that inhibition of
TLR4 may protect islet cells from LPS-induced damage.
Discussion
Stress hyperglycemia is common in ICU patients,
particularly in patients with severe sepsis and septic shock. It
directly increases the mortality of critically ill patients,
despite the absence of pre-existing conditions, including diabetes
mellitus. The inflammatory response has been regarded as a
mechanism associated with insulin signal transduction, which may
alter the normal structure of β-cells, induce insulin resistance
and decrease insulin secretion (10,13).
Acute severe hyperglycemia during a critical illness may
excessively increase the existing systemic inflammatory response
(14). However, no benefit of
strict glucose control was observed in patients with severe sepsis
in a previous study (15) and the
glucose levels remained difficult to control when developed
anti-inflammatory therapy was administered. Oxidative stress is a
major contributing factor to the high mortality rates associated
with several inflammatory diseases, including severe sepsis
(16). ROS production is
associated with the occurrence and deterioration of organ
dysfunction. Islet β-cell damage is the direct cause of abnormal
blood glucose. Pancreatic islet β-cells are sensitive to ROS levels
due to their low expression levels of antioxidant enzymes. A
previous study determined that acute hyperglycemia enhanced
oxidative stress and the generation of ROS led to reduced glucose
control (17). The effect of
oxidative stress on patients with severe infection and stress
hyperglycemia remains to be elucidated; however, it may be used as
a novel strategy to treat refractory hyperglycemia in the
future.
The generation of ROS is usually balanced by
antioxidants. Oxidative stress results from the imbalance between
the formation and sequestration of free radicals. Various
pathological processes disrupt this balance by increasing ROS or
decreasing the level of available antioxidants. Oxidative stress
contributes to loss of islet mass and function and may trigger cell
death via apoptosis and necrosis (18,19).
Armann et al (20)
determined that ROS production in islets was associated with the
percentage of cells undergoing apoptosis and islet functional
potency in vivo. Duprez et al (21) also demonstrated that oxidative
stress resulted in the apoptosis of rat islet β-cells and
dysfunction of insulin secretion. Our previous study revealed that
LPS-induced oxidative stress and apoptosis results in the loss of
GSIS in Ins-1 pancreatic β-cells (5). It is possible that oxidative stress
may be the key mechanism contributing to the dysfunction of
pancreatic β-cells. In order to investigate the importance of
oxidative stress induced by LPS treatment in pancreatic β-cells,
the present study was performed on isolated rat islet cells. ROS
production in islet cells was significantly enhanced following LPS
treatment for 24 h. This confirmed the increase in LPS-induced ROS
production. Additionally, it was revealed that this process may
partially depend on the TLR4 signaling pathway.
TLR4 is the signal-transducing molecule of the LPS
receptor complex. The LPS-TLR4 signaling pathway has been
identified as a critical upstream event in the pathogenesis of
gram-negative sepsis (22).
However, the function of TLR4 in pancreatic β-cells of patients
with stress hyperglycemia remains to be elucidated. The protein and
mRNA expression levels of TLR4 have previously been observed in
human, mouse and rat pancreatic islets (9,23).
Previous studies established that the dysfunction of pancreatic
islet β-cells was triggered by LPS stimulation via the TLR4
signaling pathway (11,24). The TLR4 signaling pathway was
involved in the early stage of pro-inflammatory and pro-oxidant of
islet cells in vitro (3).
TLR4 stimulation reduced the function of islet β-cells in mice by
limiting GSIS (10). Shi et
al (8) observed that the
absence of TLR4 improved obesity-induced insulin resistance. It is
possible that in the present study, TLR4 was involved in the
process of LPS-induced oxidative stress, apoptosis and dysfunction
in rat islet cells.
The present study revealed that the mRNA and protein
expression levels of TLR4 significantly increased when isolated rat
islet cells were cultured in the presence of LPS (10 µg/ml). LPS
induced the upregulation of TLR4 expression, which may have led to
increased ROS production. TLR4-shRNA and anti-TLR4 antibody
pretreatments were used to suppress TLR4 activity, and the results
demonstrated that inhibition of TLR4 reversed LPS-induced oxidative
stress in rat isolated islets. It was confirmed that LPS-induced
oxidative stress was associated with the expression levels of TLR4.
Knockdown of TLR4 by shRNA demonstrated the involvement of TLR4 in
LPS-induced subsequent effect.
A previous study determined that the permeability of
the mitochondrial membrane was impaired in patients with sepsis and
severe oxidative stress (25).
Various apoptosis genes have been identified to be involved in
mitochondrial apoptotic pathways associated with oxidative stress,
including the caspase family and Bax/Bcl-2. Caspase-3 is crucial to
the process of apoptosis. PARP, one of the substrates of caspase
protein family, is associated with DNA repair and genetic integrity
monitoring. The ratio of Bax/Bcl-2 has been established as an
important regulator of apoptosis. A previous study reported that
defective mitochondrial oxidative phosphorylation contributed to
the dysfunction of pancreatic β-cells (26). The present study determined that
LPS promoted apoptosis in rat islet cells by upregulation of the
pro-apoptosis protein and gene expression levels. It was revealed
that the expression levels of caspase-3, PARP and the ratio of
Bax/Bcl-2 were increased following LPS stimulation. This effect was
reversed by the inhibition of TLR4 expression. It is possible that
LPS altered the permeability of the mitochondrial membrane
resulting in an increased apoptotic rate.
The increase in ROS production and upregulation of
apoptotic genes in pancreatic β-cells also resulted in dysfunction
of insulin secretion. The present study clearly demonstrated
dysfunction of insulin secretion in rat islet cells due to exposure
to LPS. The reduction of insulin secretion was suggested to be
associated with upregulation of TLR4 expression levels in
pancreatic β-cells. Inhibition of TLR4 by anti-TLR4 antibody and
TLR4-shRNA may increase the insulin secretion levels in LPS-induced
islet cells.
It is of note that the observations of the present
study are different to the results presented by Vivot et al
(27). In this previous study,
inhibition of TLR4 expression levels led to an increase in ROS
production. They also determined that the generation of ROS and
various inflammatory mediators, including cytochrome c
oxidase subunit 2, interleukin-6 and C-C motif chemokine ligand 2
were two distinct pathways that depend on TLR4 expression (27). Additionally it is possible that the
upstream regulator of TLR4 may be different under different
conditions, such as apoptosis, oxidative stress or inflammatory
reactions; however, to fully understand this, further investigation
is required. This may be the molecular mechanism behind the poor
clinical outcomes in patients with stress hyperglycemia. The
observations of the present study highlighted that the control of
ROS release must be taken into consideration, in order to improve
the preservation of pancreatic islet cells in vitro.
The present study determined that the structure of
islet cells sourced from Sprague-Dawley rats began to develop an
irregular shape and exhibit reduced insulin secretion following
treatment with LPS for 48 h, which led to adverse effects in
follow-up experiments; therefore, islets were treated with LPS for
24 h instead of 48 h.
The results of the present study indicated that LPS
may act directly on pancreatic islets via the TLR4 signaling
pathway and induce oxidative stress and apoptosis, which may lead
to dysfunction of pancreatic β-cells. Increase of ROS production
may trigger an inflammatory cascade and promote severe
hyperglycemia. Reducing ROS production via TLR4 inhibition may
inhibit the positive-feedback of oxidative stress-induced damage to
the pancreas in critically ill patients with stress hyperglycemia.
This suggested that antioxidant therapy combined with
anti-inflammatory therapy may be beneficial to patients with severe
infections.
Acknowledgements
The present study was supported by the National
Nature Science Foundation of China (no. 81000354) and the Shanghai
Nature Science Foundation (no. 14ZR1427000).
References
|
1
|
Beutler B and Rietschel ET: Innate immune
sensing and its roots: The story of endotoxin. Nat Rev Immunol.
3:169–176. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zahar JR, Timsit JF, Garrouste-Orgeas M,
Français A, Vesin A, Descorps-Declere A, Dubois Y, Souweine B,
Haouache H, Goldgran-Toledano D, et al: Outcomes in severe sepsis
and patients with septic shock: Pathogen species and infection
sites are not associated with mortality. Crit Care Med.
39:1886–1895. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Giribabu N, Kumar KE, Rekha SS, Muniandy S
and Salleh N: Chlorophytum borivilianum root extract maintains near
normal blood glucose, insulin and lipid profile levels and prevents
oxidative stress in the pancreas of streptozotocin-induced adult
male diabetic rats. Int J Med Sci. 11:1172–1184. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Coskun O, Kanter M, Korkmaz A and Oter S:
Quercetin, a flavonoid antioxidant, prevents and protects
streptozotocin-induced oxidative stress and beta-cell damage in rat
pancreas. Pharmacol Res. 51:117–123. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Du SC, Ge QM, Lin N, Dong Y and Su Q:
ROS-mediated lipopolysaccharide-induced apoptosis in INS-1 cells by
modulation of Bcl-2 and Bax. Cell Mol Biol (Noisy-le-grand). 58
Suppl:OL1654–OL1659. 2012.PubMed/NCBI
|
|
6
|
Medzhitov R: Toll-like receptors and
innate immunity. Nat Rev Immunol. 1:135–145. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reyna SM, Ghosh S, Tantiwong P, Meka CS,
Eagan P, Jenkinson CP, Cersosimo E, Defronzo RA, Coletta DK,
Sriwijitkamol A and Musi N: Elevated toll-like receptor 4
expression and signaling in muscle from insulin-resistant subjects.
Diabetes. 57:2595–2602. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shi H, Kokoeva MV, Inouye K, Tzameli I,
Yin H and Flier JS: TLR4 links innate immunity and fatty
acid-induced insulin resistance. J Clin Invest. 116:3015–3025.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li M, Song L, Gao X, Chang W and Qin X:
Toll-like receptor 4 on islet beta cells senses expression changes
in high-mobility group box 1 and contributes to the initiation of
type 1 diabetes. Exp Mol Med. 44:260–267. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cucak H, Mayer C, Tonnesen M, Thomsen LH,
Grunnet LG and Rosendahl A: Macrophage contact dependent and
independent TLR4 mechanisms induce β-cell dysfunction and apoptosis
in a mouse model of type 2 diabetes. PLoS One. 9:e906852014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y: Attenuation of berberine on
lipopolysaccharide-induced inflammatory and apoptosis responses in
β-cells via TLR4-independent JNK/NF-κB pathway. Pharm Biol. Nov
5–2013.(Epub ahead of print).
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hotamisligil GS: Inflammation and
metabolic disorders. Nature. 444:860–867. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoneyama S, Terashima H, Yamaguchi R,
Tadano S and Ohkohchi N: The manner of the inflammation-boosting
effect caused by acute hyperglycemia secondary to overfeeding and
the effects of insulin therapy in a rat model of sepsis. J Surg
Res. 185:380–387. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Viana MV, Moraes RB, Fabbrin AR, Santos MF
and Gerchman F: Assessment and treatment of hyperglycemia in
critically ill patients. Rev Bras Ter Intensiva. 26:71–76. 2014.(In
Portuguese). View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Muhl D, Woth G, Drenkovics L, Varga A,
Ghosh S, Csontos C, Bogár L, Wéber G and Lantos J: Comparison of
oxidative stress & leukocyte activation in patients with severe
sepsis & burn injury. Indian J Med Res. 134:69–78.
2011.PubMed/NCBI
|
|
17
|
Ling PR, Smith RJ and Bistrian BR: Acute
effects of hyperglycemia and hyperinsulinemia on hepatic oxidative
stress and the systemic inflammatory response in rats. Crit Care
Med. 35:555–560. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Henriksnas J, Lau J, Zang G, Berggren PO,
Kohler M and Carlsson PO: Markedly decreased blood perfusion of
pancreatic islets transplanted intraportally into the liver:
Disruption of islet integrity necessary for islet
revascularization. Diabetes. 61:665–673. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu HQ, Gao Y, Guo HR, Kong QZ, Ma Y, Wang
JZ, Pan SH, Jiang HC and Dai WJ: Pretreatment with bilirubin
protects islet against oxidative injury during isolation and
purification. Transplant Proc. 43:1810–1814. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Armann B, Hanson MS, Hatch E, Steffen A
and Fernandez LA: Quantification of basal and stimulated ROS levels
as predictors of islet potency and function. Am J Transplant.
7:38–47. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Duprez J, Roma LP, Close AF and Jonas JC:
Protective antioxidant and antiapoptotic effects of ZnCl2 in rat
pancreatic islets cultured in low and high glucose concentrations.
PLoS One. 7:e468312012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roger T, Froidevaux C, Le Roy D, Reymond
MK, Chanson AL, Mauri D, Burns K, Riederer BM, Akira S and Calandra
T: Protection from lethal gram-negative bacterial sepsis by
targeting Toll-like receptor 4. Proc Natl Acad Sci USA.
106:2348–2352. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Y, Zhou ZG, Xia QJ, Zhang J, Li HG, Cao
GQ, Wang R, Lu YL and Hu TZ: Toll-like receptor 4 detected in
exocrine pancreas and the change of expression in cerulein-induced
pancreatitis. Pancreas. 30:375–381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Amyot J, Semache M, Ferdaoussi M, Fontés G
and Poitout V: Lipopolysaccharides impair insulin gene expression
in isolated islets of langerhans via toll-like receptor-4 and NF-κB
signalling. PLoS One. 7:e362002012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Apostolova N, Garcia-Bou R,
Hernandez-Mijares A, Herance R, Rocha M and Victor VM:
Mitochondrial antioxidants alleviate oxidative and nitrosative
stress in a cellular model of sepsis. Pharm Res. 28:2910–2919.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lu H, Koshkin V, Allister EM,
Gyulkhandanyan AV and Wheeler MB: Molecular and metabolic evidence
for mitochondrial defects associated with beta-cell dysfunction in
a mouse model of type 2 diabetes. Diabetes. 59:448–459. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vivot K, Langlois A, Bietiger W, Dal S,
Seyfritz E, Pinget M, Jeandidier N, Maillard E, Gies JP and Sigrist
S: Pro-inflammatory and pro-oxidant status of pancreatic islet in
vitro is controlled by TLR-4 and HO-1 pathways. PLoS One.
9:e1076562014. View Article : Google Scholar : PubMed/NCBI
|