Introduction
Selenium was discovered by the Swedish chemist Jöns
Jacob Berzelius in 1817. It was considered a toxin until 1957, when
the trace element was identified as an essential micronutrient for
animals and humans (1,2). Selenium serves a role in numerous
biological processes, including immune function, brain development
and reproduction (3,4). In vivo, selenium is found in
selenoproteins, which co-translationally incorporate
selenium-containing selenocysteine (Sec), the 21st genetically
encoded amino acid (5,6). Selenoproteins are critical for
numerous physiological functions, including antioxidant defense,
thyroid hormone production (4) and
inflammation and immunity (7).
Primary selenoproteins include glutathione peroxidases (GPxs),
thioredoxin reductases (TrxRs) and iodothyronine deiodinases
(8,9). Sec and selenoprotein biosynthesis
requires a multitude of protein factors, including Sep
(O-phosphoserine) transfer (t)RNA:Sec (selenocysteine) synthase
(SepSecS), tRNA Sec 1 associated protein 1 (Trnau1ap), and Sec
insertion sequence binding protein 2 (SBP2) (10).
Trnau1ap, originally named SECp43, is a highly
conserved 43-kDa tRNA[Ser] Sec-binding protein
identified by affinity purification (11). It has been identified to interact
with the selenocysteyl-tRNA[Ser] Sec-Sec-specific
elongation factor (EFsec) complex in vitro, and
co-expression of Trnau1ap facilitates interaction between EFsec and
SBP2 in vivo. In addition, Trnau1ap mediates Sec
incorporation and upregulates selenoprotein mRNA expression levels
(12). Furthermore, when SepSecS
and Trnau1ap are co-expressed in vitro, Trnau1ap induces
SepSecS to localize to the cytoplasm rather than the nucleus, where
Trnau1ap typically resides (13).
A previous study demonstrated that downregulation of Trnau1ap and
SepSecS resulted in an extensive reduction of selenoprotein
expression levels, whereas downregulation of SepSecS alone had no
effect. Additionally, downregulation of Trnau1ap reduced GPx1
expression levels (14). These
findings suggest that Trnau1ap serves a key role in the
biosynthesis of selenoproteins.
Selenium deficiency results in a variety of
cardiovascular diseases, including Keshan disease. Keshan disease,
a cardiomyopathy endemic to China, is characterized by heart
failure and severe cardiomyopathy, typically with arrhythmia and
congestive heart failure (15,16).
Our previous study demonstrated that reactive oxygen species
accumulation and myocardial injury were features of a mouse model
of selenium deficiency (17).
However, the underlying mechanisms of myocardial injury induced by
selenium deficiency remain unclear.
The present study hypothesized that Trnau1ap may
participate in selenium deficiency-induced myocardial injury. The
role of Trnau1ap in myocardial cell proliferation and apoptosis was
investigated using H9c2 cells. Furthermore, the phosphoinositide
3-kinase (PI3K)/protein kinase B (AKT) signaling pathway was
examined to provide further insight into the molecular mechanisms
underlying cardiomyocyte injury.
Materials and methods
Cell culture
The H9c2rat cardiac myoblast cell line (purchased
from the American Type Culture Collection, Manassas, VA, USA), was
maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 5.5 mM
glucose supplemented with 10% fetal bovine serum (Biological
Industries, Kibbutz Beit Haemek, Israel) and 100 U/ml
penicillin/streptomycin (Biological Industries) in an atmosphere of
5% CO2 at 37°C.
Plasmids, small interfering (si)RNA
and transfection
The open reading frame of Trnau1ap was cloned into a
pcDNA3.1(+) vector (pcDNA3.1(+)-Trnau1ap; Thermo Fisher Scientific,
Inc.), and transfection was performed using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol, when cells were at 70–80%
confluence. H9c2 cells transfected with the empty vector
(H9c2-mock) or pcDNA3.1(+)-Trnau1ap (H9c2-Trnau1ap) were analyzed
24–72 h post-transfection. A total of three siRNAs targeting
Trnau1ap were obtained from Guangzhou RiboBio Co., Ltd. (Guangzhou,
China). The sequences were as follows: 5′-GCCGAGAAGTGTTTGCATA-3′
(Trnau1ap siRNA-1), 5′-GGACGAUGGCAUGCUGUAU-3′ (Trnau1ap siRNA-2)
and 5′-GCAGACAUAUGAAGAGGUU-3′ (Trnau1ap siRNA-3). A negative
control siRNA was additionally obtained from Guangzhou RiboBio Co.,
Ltd. Transfection was conducted using Lipofectamine 2000 according
to the manufacturer's protocol, when cells were at 60–70%
confluence. Analysis was conducted 24–72 h post-transfection.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA samples were isolated from cells 24 h
post-transfection with TRIzol® (Thermo Fisher
Scientific, Inc.) and reverse transcribed into cDNA using the
High-Capacity cDNA RT kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. qPCR
was performed using SYBR® Green PCR Master mix and the
7500Real-Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The primers for Trnau1ap were as follows:
Forward, 5′-AGGTTGGGGATGATGCACTG-3′ and reverse,
5′-CGGGGATCTCTGATGACACG-3′. Rat β-actin served as the endogenous
control, with the following primer sequences: Forward,
5′-CCACCATGTACCCAGGCATT-3′ and reverse, 5′-CGGACTCATCGTACTCCTGC-3′.
The PCR was performed with an initial stage of 95°C for 10 min,
followed by 40 cycles of 95°C for 15 sec and elongation at 60°C for
30 sec. A dissociation curve was run for each plate to confirm the
production of a single product. All assays were performed in
triplicate. The relative RNA expression levels were determined
using the 2-∆∆Cq method (18).
Western blotting
Total protein samples were extracted from cells 72 h
post-transfection using radioimmunoprecipitation assay lysis buffer
(EMD Millipore, Billerica, MA, USA). Following incubation at 4°C
for 15 min, the lysates were centrifuged at 6,000 × g for 10
min at 4°C. Protein concentrations were determined using the
Bicinchoninic Acid Assay kit (Pierce; Thermo Fisher Scientific,
Inc.). Samples (50 µg) were mixed with loading buffer, denatured
for 5 min at 95°C and separated by 12% SDS-PAGE. The separated
proteins were transferred onto polyvinylidene difluoride membranes
(EMD Millipore), blocked with 5% non-fat milk in Tris-buffered
saline/Tween-20 for 2 h at 25°C, and incubated with antibodies for
12 h at 4°C. The following primary antibodies were used: rabbit
anti-Trnau1ap (1:1,000; catalog no. 15053-1-AP), rabbit anti-PCNA
(1:1,000; catalog no. 10205-2-AP) and rabbit anti-Bax (1:1,000;
catalog no. 50599-2-Ig) from ProteinTech Group, Inc. (Chicago, IL,
USA); rabbit anti-selenoprotein K (SelK; 1:1,000; catalog no.
ab139949) and rabbit anti-thioredoxin reductase 1 (Txnrd1; 1:1,000;
catalog no. ab16840) from Abcam (Cambridge, MA, USA); rabbit
anti-GPx1 (1:1,000; catalog no. PA5-30593; Thermo Fisher
Scientific, Inc.); rabbit anti-Bcl-2 (1:1,000; catalog no. B0774;
Assay Biotechnology, Sunnyvale, CA, USA); rabbit anti-AKT (1:1,000;
catalog no. 9272) and rabbit anti-phosphorylated (p)-AKT (1:1,000;
catalog no. 4058) from Cell Signaling Technology, Inc. (Danvers,
MA, USA); and mouse anti-β-actin (1:1,000; catalog no. BM0627,
Boster Biological Technology, Wuhan, Hubei, China). The secondary
antibodies used were goat anti-rabbit horseradish peroxidase
(HRP)-conjugated IgG (1:2,000; catalog no. ZB-2301) and goat
anti-mouse HRP IgG (1:2,000; catalog no. ZB-2305) purchased from
Zhongshan Golden Bridge Biotechnology, Co., Ltd. (Beijing, China).
The membranes were incubated with the secondary antibodies for 2 h
at room temperature. The signals were detected by BioSpectrum 810
Imaging system (UVP, LLC, Upland, CA, USA) using enhanced
chemiluminescence system (Pierce ECL Western Blotting substrate;
Thermo Fisher Scientific, Inc.). The relative densities of bands
were analyzed with Quantity One 4.62 software (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Cell proliferation assays
A total of 24 h post-transfection with siRNA or
plasmid, H9c2 cells were seeded into 96-well plates at a density of
2,000 viable cells per well for cell proliferation assays, which
were performed using
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich; Merck Millipore, Darmstadt, Germany), diluted to 5
mg/ml with serum-free DMEM. A total of 48 h after seeding, MTT was
added to each well and incubated for 4 h at 37°C. Formazan crystals
were dissolved in dimethyl sulfoxide, and absorbance at a
wavelength of 490 nm was measured using a SpectraMax®
M3microplate spectrophotometer (Molecular Devices, LLC, Sunnyvale,
CA, USA).
Flow cytometry
A total of 1×106 cells were seeded into a
60-mm dish 24 h prior to the experiment. A total of 48 h
post-transfection, annexin V-fluorescein isothiocyanate (FITC) and
propidium iodide (PI) staining were performed according to the
manufacturer's protocol (Beyotime Institute of Biotechnology,
Haimen, China). The stained cells were acquired using a
FACSCalibur™ Flow Cytometer (BD Biosciences, Franklin Lakes, NJ,
USA). All data were analyzed and visualized using FACSDiva™ 3.0
software (BD Biosciences).
Caspase-3 activity
Caspase-3 activity was determined using the
Caspase-3 Activity assay kit (Beyotime Institute of Biotechnology),
according to the manufacturer's protocol. Assays were conducted in
96-well microplates by incubating 10 µl cell lysate with 80 µl
reaction buffer (1% NP-40, 20 mM Tris-HCl at pH 7.5, 137 mM NaCl
and 10% glycerol) and 10 µl 2 mM caspase-3 substrate (Ac-DEVD-pNA).
The lysates were incubated at 37°C for 4 h. The absorbance of
samples was measured at a wavelength of 405 nm using the
SpectraMaxM3 microplate reader.
Mitochondrial membrane potential
assay
Mitochondrial depolarization in the cells was
measured using a Mitochondrial Membrane Potential assay kit with a
JC-1 probe (Beyotime Institute of Biotechnology). Briefly,
mitochondria were isolated from the cells following treatment,
using the Cell Mitochondria Isolation kit (Beyotime Institute of
Biotechnology), and incubated with 1 ml JC-1 staining solution (5
µg/ml) for 20 min at 37°C, and subsequently with phosphate-buffered
saline. The mitochondrial membrane potentials were measured using
the relative quantities of dual emissions from mitochondrial JC-1
monomers or aggregates using the SpectraMaxM3 microplate
spectrophotometer. The excitation wavelength was set at 485 nm.
Fluorescence intensity was detected at wavelengths of 525 nm for
monomers and 590 nm for aggregates. Mitochondrial depolarization
was indicated by an increase in the 525/590 nm fluorescence
intensity ratio.
Statistical analysis
All experiments were performed at least three times.
Normal distribution data are presented as the mean ± standard
deviation, and differences between two groups were evaluated using
the Student's t-test. One-way ANOVA followed by the
Dunnett's multiple comparison test was used to examine differences
of treated groups with the control group. P<0.05 was considered
to indicate a statistically significant difference. All statistical
analyses were performed using SPSS software version 17.0 (SPSS,
Inc., Chicago, IL, USA).
Results
Modulation of Trnau1ap expression
levels in H9c2 cells
A total of three different siRNAs targeting Trnau1ap
were transfected into H9c2 cells. After a 24-h incubation, the most
effective siRNA (siRNA-1) was determined by RT-qPCR (Fig. 1A; P<0.001for H9c2-siRNA1
compared with H9c2; P<0.001 for H9c2-siRNA2 compared with H9c2;
P<0.001 for H9c2-siRNA3 compared with H9c2) and used for
subsequent experiments. Western blotting analysis was performed 48
h post-transfection to confirm under-and overexpression of Trnau1ap
in H9c2 cells, compared with the control siRNA or H9c2-mock groups,
respectively (Fig. 1B, P<0.01
compared with Control siRNA; Fig.
1C, P<0.05 compared with H9c2-mock). Modulating Trnau1ap
expression levels affected GPx1, Txnrd1 and SelK expression levels,
demonstrating that Trnau1ap affects the expression levels of a
variety of selenoproteins (Fig.
1D, P<0.05 and P<0.01 compared with Control siRNA;
Fig. 1E, P<0.05 and P<0.01
compared with H9c2-mock).
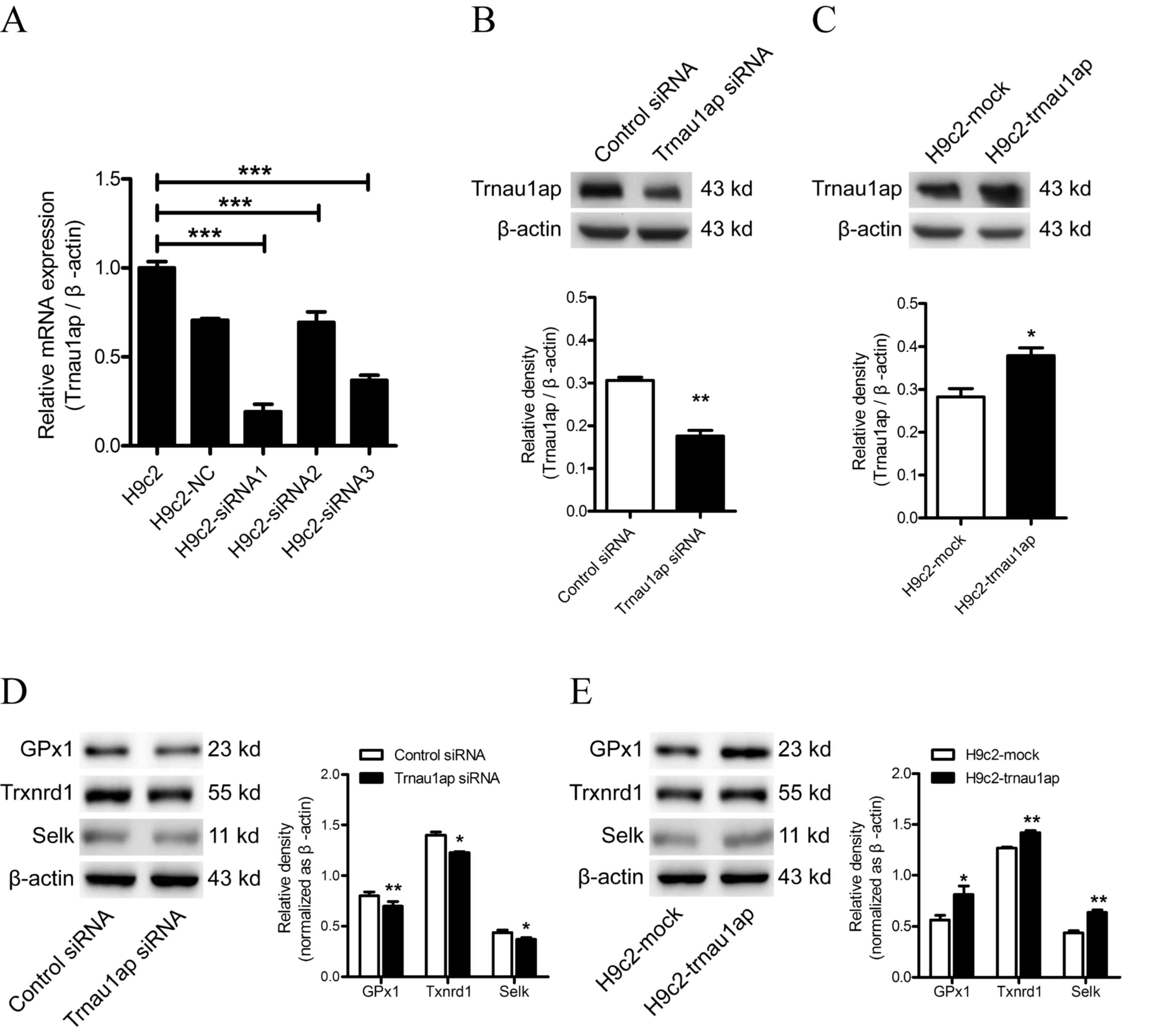 | Figure 1.Expression levels of Trnau1ap in H9c2
cells. (A) Expression levels of Trnau1ap were determined by RT-qPCR
24 h post-transfection with Trnau1ap siRNAs or control siRNA.
Western blot analysis of Trnau1ap protein expression levels in (B)
H9c2 cells transfected with Trnau1ap siRNA or control siRNA for 48
h and (C) H9c2 cells transfected with empty vector or pcDNA
3.1(+)-Trnau1ap, 48 h post-transfection. Western blot analysis of
GPx1, Txnrd1 and SelK levels in (D) Trnau1ap-underexpressing, and
(E) Trnau1ap-overexpressing, cells. β-actin served as an endogenous
control for western blotting and RT-qPCR. All data are presented as
the mean ± standard deviation (n=3). *P<0.05, **P<0.01 vs.
control siRNA or H9c2-mock, ***P<0.001. Trnau1ap, transfer RNA
selenocysteine 1 associated protein 1; siRNA, small interfering
RNA, GPx1, glutathione peroxidase 1; Txnrd1, thiorexin reductase 1;
Selk, selenoprotein K; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction. |
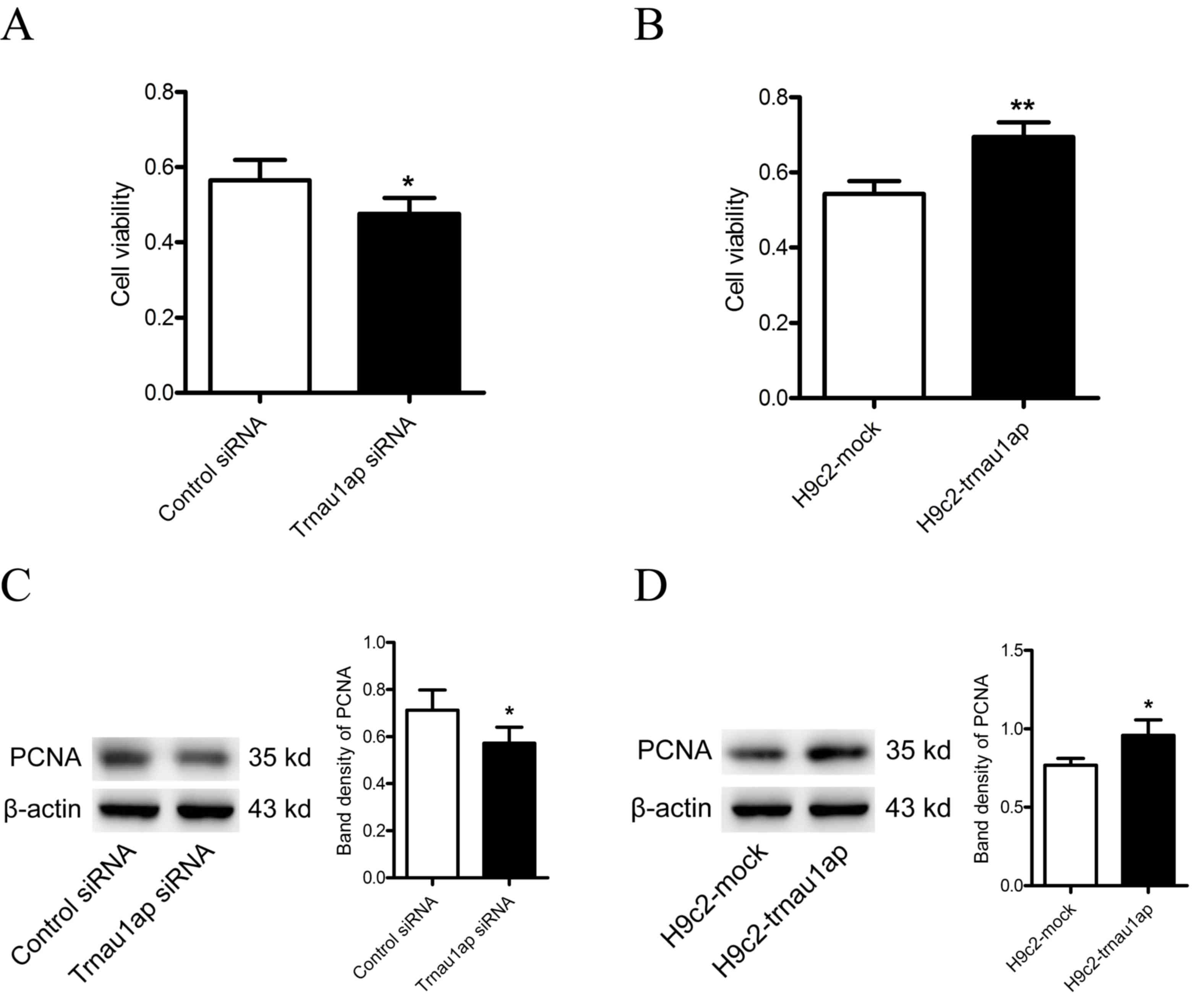
Downregulation of Trnau1ap inhibits
the proliferation of H9c2 cells
To examine whether Trnau1ap affects the
proliferation of H9c2 cells, an MTT assay was performed and
proliferating cell nuclear antigen (PCNA) expression levels were
measured. Compared with the control group, cells transfected with
Trnau1ap siRNA exhibited decreased proliferation (Fig. 2A, P<0.05 compared with
H9c2-siRNA). Conversely, Trnau1ap-overexpressing cells exhibited
increased proliferation, compared with mock-transfected cells
(Fig. 2B, P<0.01 compared with
H9c2-mock). Expression levels of PCNA were reduced in
Trnau1ap-underexpressing cells, compared with control
siRNA-transfected cells (Fig. 2C,
P<0.05 compared with H9c2-siRNA), and increased in
Trnau1ap-overexpressing cells compared with control
mock-transfected cells (Fig. 2D,
P<0.05 compared with H9c2-mock).
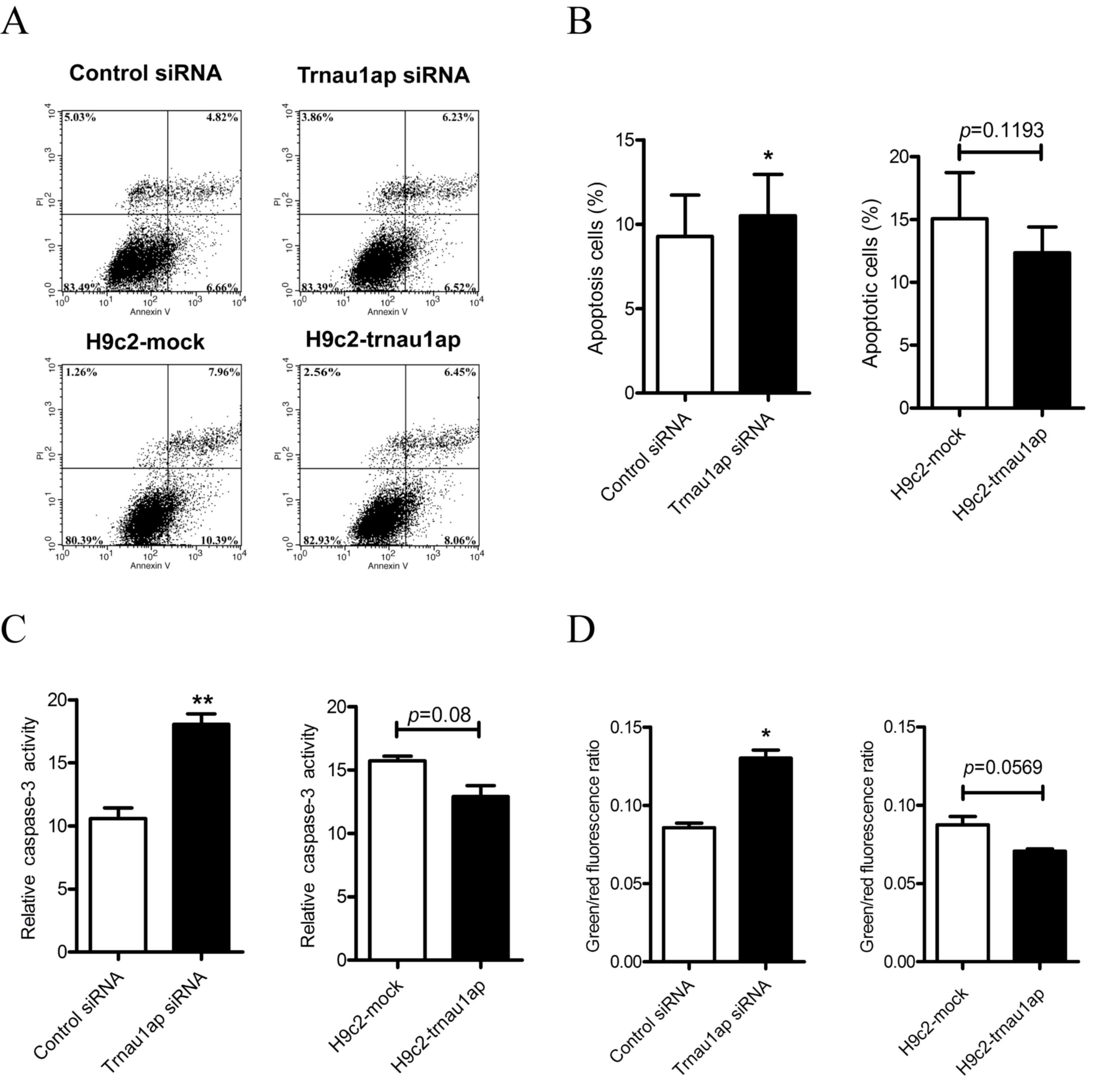
Downregulation of Trnau1ap induces
apoptosis
To examine the role of Trnau1ap in apoptosis in H9c2
cells, annexin V-FITC and PI double staining was performed, and
flow cytometric analysis was performed to measure the apoptotic
rate of cells (Fig. 3A and B,
P<0.05 compared with H9c2-siRNA). Compared with the control
group, the apoptotic rate was increased in the Trnau1ap
siRNA-transfected group. No significant differences were observed
in the apoptotic rates of cells between the Trnau1ap overexpression
and empty vector groups.
Additionally, a Caspase-3 Activity
assay kit was used to evaluate the apoptotic status of the various
groups
In the underexpression group, caspase-3 activity was
markedly increased compared with the control group (Fig. 3C, P<0.01 compared with
H9c2-siRNA). In comparison, there were no significant differences
between the overexpression and mock-transfected groups (Fig. 3C, P=0.08 compared with
H9c2-mock).
A Mitochondrial Membrane Potential
assay was performed using a JC-1 probe
In cells with healthy mitochondria, JC-1 is
aggregated and emits a red fluorescence. When the inner
mitochondrial membrane potential is dissipated, JC-1 is
disaggregated and emits a green fluorescence. Therefore,
depolarization of the mitochondrial membrane potential is detected
as an increase in the green/red fluorescence ratio. The Trnau1ap
underexpression group exhibited a depolarization of the
mitochondrial membrane potential (Fig.
3D, P<0.05 compared with H9c2-siRNA), whereas no significant
differences were observed between the overexpression and
mock-transfected groups (Fig. 3D,
P=0.0569 compared with H9c2-mock).
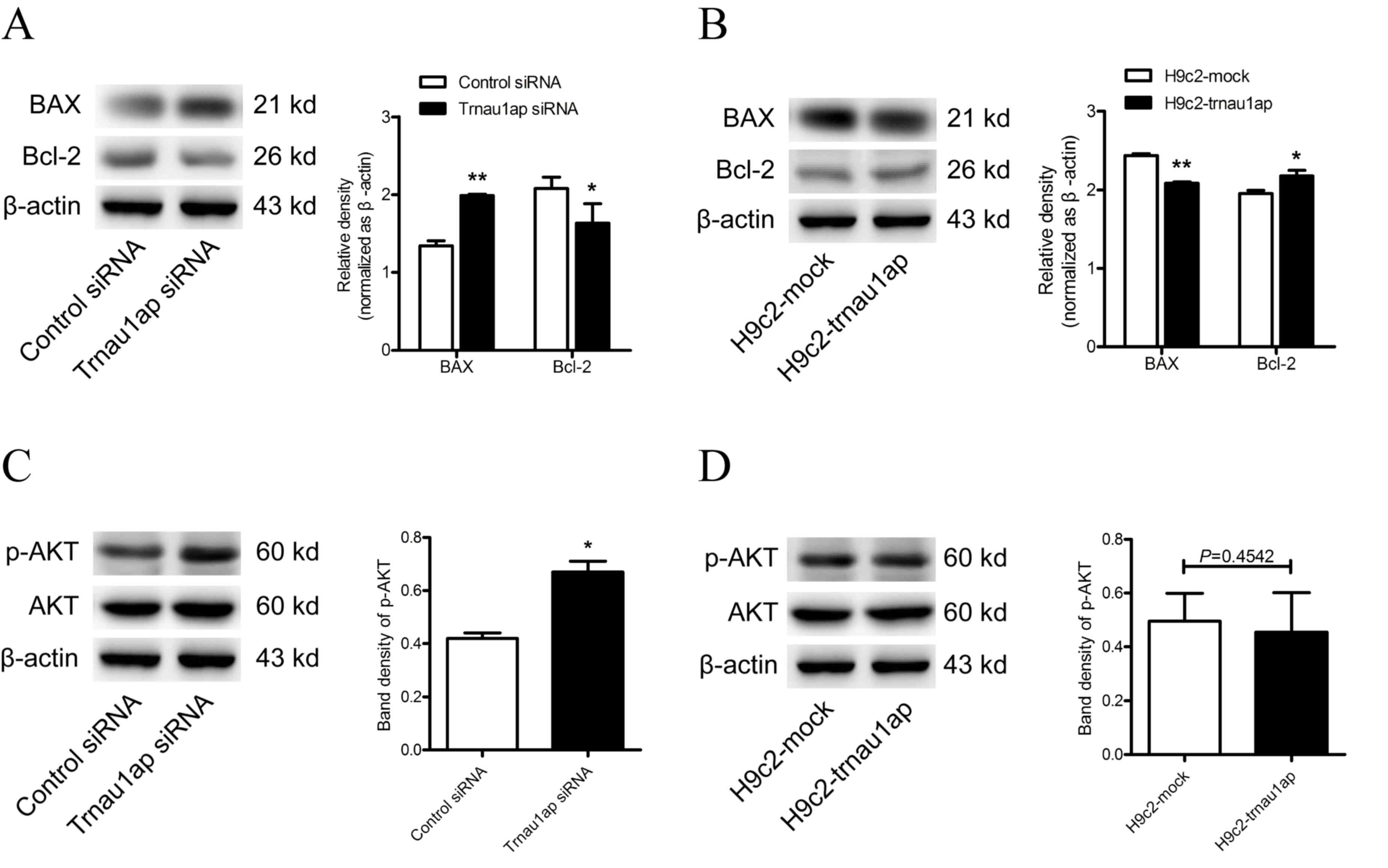
To further examine the role of
Trnau1ap in cell death, two apoptosis-associated proteins, Bcl-2
and Bax, were examined by western blotting
Bax expression levels were upregulated, whereasBcl-2
expression levels were downregulated, in the underexpression group
compared with the control group (Fig.
4A, P<0.05 for Bcl-2, P<0.01 for Bax compared with
H9c2-siRNA). By contrast, Bax expression levels were downregulated,
while Bcl-2 expression levels were upregulated, in the
overexpression group compared with mock-transfected cells (Fig. 4B, P<0.05 for Bcl-2, P<0.01
for Bax compared with H9c2-mock). In addition, the PI3K/AKT
signaling pathway was activated in Trnau1ap-underexpressing cells
compared with control cells after 48 h transfection (Fig. 4C, P<0.05 compared with
H9c2-siRNA), although no significant differences were observed in
Trnaulap-overexpressing compared with mock-transfected cells
(Fig. 4D, P=0.4524 compared with
H9c2-mock).
Discussion
The present study demonstrated for the first time,
to the best of our knowledge, that modulating expression levels of
Trnau1ap affected the proliferation and apoptosis of rat
cardiomyocyte-like H9c2 cells. Knockdown of Trnau1ap by siRNA
inhibited proliferation and increased apoptosis. Mitochondrial
membrane potential depolarization was observed in the Trnau1ap
knockdown cells. In addition, the PI3K/AKT pathway was activated in
cells with reduced Trnau1ap expression levels. In comparison,
Trnau1ap-overexpressing cells exhibited an increased proliferative
capacity.
Trnau1ap, additionally known as SECp43, is a highly
conserved protein with two ribonucleoprotein-binding domains and a
polar/acidic carboxy terminus, and is critical for selenoprotein
synthesis (11). A previous study
reported that a decrease in the expression levels of Trnau1ap
induced a downregulation of GPx1 expression levels, and
co-knockdown of Trnau1ap and SepSecS induced a decrease in
selenoprotein expression levels (14). A recent study identified no
significant alteration in the levels of 75Se-labeled
hepatic proteins, or in levels of selenoproteins as determined by
western blot analysis, in hepatocyte-specific Trnau1ap knockout
mice (19). However, in the
present study, knockdown of Trnau1ap in H9c2 cells reduced GPx1,
Txnrd1 and SelK expression levels, and overexpression of Trnau1ap
increased selenoprotein expression levels, indicating that Trnau1ap
serves an important role in selenoprotein production. This
discrepancy in findings suggests that the functional role of
Trnau1ap may vary according to tissue type and species.
Various selenoproteins are crucial antioxidant
enzymes, including GPxs, TrxRs, SelK and selenoprotein W.
Therefore, a decrease in selenoprotein expression levels, as in the
Trnau1ap knockdown group, may disturb cellular redox homeostasis,
eventually leading to apoptosis.
PCNA, an essential cofactor of DNA polymerases,
encircles synthesizing DNA and is used as a molecular marker of
cell proliferation (20,21). Numerous studies have reported that
the proliferation of cells and tissues is associated with PCNA
levels (22–24). In the present study, PCNA levels
were increased in Trnau1ap-overexpressing cells, suggesting that a
potential association exists between Trnau1ap and cell
proliferation.
Mitochondria serve critical roles in oxygen free
radical detoxification (25).
Furthermore, these organelles are important for apoptosis. When
pro-apoptotic proteins, including Bax and BH3 interacting-domain
death agonist, translocate to the mitochondrial membrane,
mitochondrial permeability transition pores promote cytochrome c
release, triggering apoptosis (26). The present study revealed that
mitochondrial membrane depolarization occurred in cells with
decreased Trnau1ap expression levels, suggesting that a reduction
in Trnau1ap levels promoted apoptosis via the mitochondrial
pathway.
Apoptosis is an important cellular process during
development, and is additionally involved in tissue homeostasis by
eliminating damaged cells. Furthermore, apoptosis is involved in
numerous diseases, including cardiovascular disease (27). Caspase (cysteine aspartase) family
members serve primary roles in programmed cell death. In apoptosis,
activation of caspase-3 is a critical event in the execution phase.
Bcl-2 family proteins are key regulators of the apoptotic signaling
pathway. The pro- and anti-apoptotic Bcl-2 family members present
on the mitochondrial membrane determine apoptosis or survival
(28). Bcl-2 is an anti-apoptotic
protein, while Bax is pro-apoptotic. Numerous studies have revealed
that the Bax/Bcl-2 ratio is an important determinant of whether
cells undergo apoptosis (29–31).
Previous studies have demonstrated that selenium
protects neuroblastoma cells from hypoxia-induced apoptosis
(32), and additionally protects
primary human keratinocytes from apoptosis induced by exposure to
ultraviolet radiation (33). The
present study revealed that reducedTrnau1ap expression levels
inhibited proliferation and induced apoptosis, suggesting that this
protein regulates proliferation and apoptosis in H9c2 cells via its
essential role in selenoprotein synthesis.
The PI3K/AKT signaling pathway serves a critical
role in mediating survival and apoptosis (34,35).
Oxidative stress has been demonstrated to activate the PI3K/AKT
signaling pathway in a variety of cells (36). A previous study revealed that the
PI3K/AKT signaling pathway was activated in the muscles of
selenium-deficient chickens (37).
The present study identified that the PI3K/AKT signaling pathway
was activated in cells with reducedTrnau1ap expression levels. This
finding suggests that the downregulation of selenoprotein synthesis
may induce apoptosis via increased levels of oxidative stress.
In conclusion, to the best of our knowledge, these
results demonstrated for the first time that Trnau1ap regulated
proliferation and the mitochondrial apoptotic pathway in
cardiomyocyte-like cells. However, additional studies using other
cell types and species are required to clarify the functional roles
of Trnau1ap. Nonetheless, these findings provide insight into the
mechanisms underlying myocardial injury induced by selenium
deficiency.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81172616 and
81472929).
References
|
1
|
Lu J and Holmgren A: Selenoproteins. J
Biol Chem. 284:723–727. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Combs GF Jr: Biomarkers of selenium
status. Nutrients. 7:2209–2236. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rayman MP: Selenium and human health.
Lancet. 379:1256–1268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mehdi Y, Hornick JL, Istasse L and
Dufrasne I: Selenium in the environment, metabolism and involvement
in body functions. Molecules. 18:3292–3311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Turanov AA, Xu XM, Carlson BA, Yoo MH,
Gladyshev VN and Hatfield DL: Biosynthesis of selenocysteine, the
21st amino acid in the genetic code, and a novel pathway for
cysteine biosynthesis. Adv Nutr. 2:122–128. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Metanis N and Hilvert D: Natural and
synthetic selenoproteins. Curr Opin Chem Biol. 22:27–34. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang Z, Rose AH and Hoffmann PR: The role
of selenium in inflammation and immunity: From molecular mechanisms
to therapeutic opportunities. Antioxid Redox Signal. 16:705–743.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reeves MA and Hoffmann PR: The human
selenoproteome: Recent insights into functions and regulation. Cell
Mol Life Sci. 66:2457–2478. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Diamond AM: The subcellular location of
selenoproteins and the impact on their function. Nutrients.
7:3938–3948. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Squires JE and Berry MJ: Eukaryotic
selenoprotein synthesis: Mechanistic insight incorporating new
factors and new functions for old factors. IUBMB Life. 60:232–235.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ding F and Grabowski PJ: Identification of
a protein component of a mammalian tRNA(Sec) complex implicated in
the decoding of UGA as selenocysteine. RNA. 5:1561–1569. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Small-Howard A, Morozova N, Stoytcheva Z,
Forry EP, Mansell JB, Harney JW, Carlson BA, Xu XM, Hatfield DL and
Berry MJ: Supramolecular complexes mediate selenocysteine
incorporation in vivo. Mol Cell Biol. 26:2337–2346. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Papp LV, Lu J, Holmgren A and Khanna KK:
From selenium to selenoproteins: Synthesis, identity, and their
role in human health. Antioxid Redox Signal. 9:775–806. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu XM, Mix H, Carlson BA, Grabowski PJ,
Gladyshev VN, Berry MJ and Hatfield DL: Evidence for direct roles
of two additional factors, SECp43 and soluble liver antigen, in the
selenoprotein synthesis machinery. J Biol Chem. 280:41568–41575.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Oropeza-Moe M, Wisløff H and Bernhoft A:
Selenium deficiency associated porcine and human cardiomyopathies.
J Trace Elem Med Biol. 31:148–156. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rose AH and Hoffmann PR: Selenoproteins
and cardiovascular stress. Thromb Haemost. 113:494–504. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cui J, Zhong R, Chu E, Zhang XF, Zhang WG,
Fang CF, Dong Q, Li FL and Li H: Correlation between oxidative
stress and L-type calcium channel expression in the ventricular
myocardia of selenium-deficient mice. J Int Med Res. 40:1677–1687.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Method. 25:402–408. 2001.
View Article : Google Scholar
|
|
19
|
Mahdi Y, Xu XM, Carlson BA, Fradejas N,
Günter P, Braun D, Southon E, Tessarollo L, Hatfield DL and
Schweizer U: Expression of selenoproteins is maintained in mice
carrying mutations in SECp43, the tRNA selenocysteine 1 associated
protein (Trnau1ap). PLoS One. 10:e01273492015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang SC: PCNA: A silent housekeeper or a
potential therapeutic target? Trends Pharmacol Sci. 35:178–186.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moldovan GL, Pfander B and Jentsch S:
PCNA, the maestro of the replication fork. Cell. 129:665–679. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ip C, Thompson HJ and Ganther HE: Selenium
modulation of cell proliferation and cell cycle biomarkers in
normal and premalignant cells of the rat mammary gland. Cancer
Epidemiol Biomarkers Prev. 9:49–54. 2000.PubMed/NCBI
|
|
23
|
Chen JH, Tsou TC, Chiu IM and Chou CC:
Proliferation inhibition, DNA damage, and cell-cycle arrest of
human astrocytoma cells after acrylamide exposure. Chem Res
Toxicol. 23:1449–1458. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeng H: Selenium as an essential
micronutrient: Roles in cell cycle and apoptosis. Molecules.
14:1263–1278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Buettner GR: Superoxide dismutase in redox
biology: The roles of superoxide and hydrogen peroxide. Anticancer
Agents Med Chem. 11:341–346. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Orrenius S, Gogvadze V and Zhivotovsky B:
Calcium and mitochondria in the regulation of cell death. Biochem
Biophys Res Commun. 460:72–81. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hotchkiss RS, Strasser A, McDunn JE and
Swanson PE: Cell death in disease: Mechanisms and emerging
therapeutic concepts. N Engl J Med. 361:1570–1583. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brenner D and Mak TW: Mitochondrial cell
death effectors. Curr Opin Cell Biol. 21:871–877. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang X, Chen Y, Gao B, Luo D, Wen Y and
Ma X: Apoptotic effect of koumine on human breast cancer cells and
the mechanism involved. Cell Biochem Biophys. 72:411–416. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang WG, Chen L, Dong Q, He J, Zhao HD,
Li FL and Li H: Mmu-miR-702 functions as an anti-apoptotic mirtron
by mediating ATF6 inhibition in mice. Gene. 531:235–242. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang W, Yu J, Dong Q, Zhao H, Li F and Li
H: A mutually beneficial relationship between hepatocytes and
cardiomyocytes mitigates doxorubicin-induced toxicity. Toxicol
Lett. 227:157–163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sarada SK, Himadri P, Ruma D, Sharma SK
and Pauline T: Mrinalini: Selenium protects the hypoxia induced
apoptosis in neuroblastoma cells through upregulation of Bcl-2.
Brain Res. 1209:29–39. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rafferty TS, Beckett GJ, Walker C, Bisset
YC and McKenzie RC: Selenium protects primary human keratinocytes
from apoptosis induced by exposure to ultraviolet radiation. Clin
Exp Dermatol. 28:294–300. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Burgering BM and Coffer PJ: Protein kinase
B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction.
Nature. 376:599–602. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Franke TF, Yang SI, Chan TO, Datta K,
Kazlauskas A, Morrison DK, Kaplan DR and Tsichlis PN: The protein
kinase encoded by the Akt proto-oncogene is a target of the
PDGF-activated phosphatidylinositol 3-kinase. Cell. 81:727–736.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Martindale JL and Holbrook NJ: Cellular
response to oxidative stress: Signaling for suicide and survival. J
Cell Physiol. 192:1–15. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang JQ, Ren FZ, Jiang YY, Xiao C and Lei
XG: Selenoproteins protect against avian nutritional muscular
dystrophy by metabolizing peroxides and regulating redox/apoptotic
signaling. Free Radic Biol Med. 83:129–138. 2015. View Article : Google Scholar : PubMed/NCBI
|