Introduction
Diabetes mellitus (DM) is traditionally
characterized by dysfunction of pancreatic β-cells and insulin
resistance. Inflammation has been shown to be a key contributing
factor to DM, and the importance of inflammation in insulin
resistance and complications associated with diabetes has been
previously reviewed (1,2). Inflammation may contribute to
pancreatic β-cell apoptosis and pro-inflammatory cytokines,
including interleukin (IL)-β and tumor necrosis factor (TNF)-α, and
activate signaling pathways resulting in pancreatic β-cell death
and dysfunction (2,3). Lipopolysaccharide (LPS) is a cell
membrane endotoxin of Gram-negative bacteria that produces local or
systemic inflammatory reactions in the host following bacterial
infection. In animal models maintained in germ-free environments,
the endotoxin is associated with cardiometabolic abnormalities,
including obesity, insulin resistance and diabetes (4). Both prevalent and incident diabetes
are associated with endotoxemia, suggesting that endotoxemia is a
key player in the pathogenesis of diabetes and that microbes may
have a central role, thereby linking metabolic disorders to
inflammation (5).
High mobility group box 1 (HMGB1) is a nuclear
non-histone chromatin-binding protein that maintains nucleosomal
structure and stability, and regulates gene transcription (6). In addition, intracellular HMGB1
serves a role in a number of fundamental cellular processes,
including transcription, replication, DNA repair and recombination
(7). HMGB1 is commonly found in
mammalian cells, but can be actively secreted into extracellular
spaces in response to external stimuli, including LPS and TNF-α, as
well as being passively released from necrotic cells (8). Extracellular secretion of HMGB1 is
closely associated with increased mortality in animal models of
sepsis and septic patients, and it has been reported that
extracellular HMGB1 may interact with TLR and/or the receptor for
advanced glycation end products (RAGE), which can lead to the
activation of the p38 mitogen-activated protein kinase (MAPK)
signaling pathway (9). Inhibition
of HMGB1 protects animals from lethal doses of LPS and reduces
injurious ventilation-induced lung inflammation (10).
Vitexin is a major bioactive flavonoid compound
derived from the dried leaf of hawthorn (Crataegus
pinnatifida), a widely used conventional folk medicine in China
(11). Vitexin has been shown to
exert a variety of biological and pharmacological activities,
including anticancer, antioxidant, anti-inflammatory and
antimyeloperoxidase functions. Previous findings have indicated
that vitexin has a protective effect against myocardial
ischemia/reperfusion injury in the isolated rat heart model, an
effect that is associated with the release of inflammatory
cytokines and myocardial apoptosis by reducing the gene expression
of Bax, while increasing the gene expression of B-cell lymphoma
(Bcl)2 (12). Vitexin also
inhibits the production of pro-hyperalgesic cytokines and increases
the production of anti-hyperalgesics (13). As DM is associated with
inflammation, the issue of whether vitexin may alleviate
LPS-induced the release of HMGB1 and protect islet cell injury
remains to be examined. Therefore, in the present study, LPS
treated rats and INS-1 cells were used to investigate the effects
of vitexin on HMGB1 release, and to examine possible mechanisms
associated with this effect.
Materials and methods
Reagents
Vitexin, with a purity >99.9%, was obtained from
Xi'an Haoxuan Biotechnology Co. Ltd., (Xi'an, China). The P38 MAPK
inhibitor, SB203580, was purchased from Selleck Chemicals (Houston,
TX, USA). RPMI-1640 medium and fetal bovine serum (FBS) were
purchased from Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). LPS was purchased from Sigma-Aldrich; Merck Millipore
Darmstadt, (Germany). Rabbit polyclonal antibodies against HMGB1
(Rabbit IgG, cat. no. 6893, 1:1,000), Bcl2 (Rabbit IgG, cat. no.
2876, 1:1,000), cleaved caspase-3 (Rabbit IgG, cat. no. 9664,
1:1,000), P38 (Rabbit IgG, cat. no. 14451, 1:1000), phosphorylated
(p-)P38 (Rabbit IgG, cat. no. 4092, 1:1,000) and β-actin (Rabbit
IgG, cat. no. 4970, 1:1,000) were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). The streptavidin-peroxidase
(SP) and 3,3′-diaminobenzidine kits were purchased from Beijing
Zhongshan Golden Bridge Biotechnology; OriGene Technologies, Inc.
(Rockville, MD, USA). The primers (P38, F:GCC TCA CCG CCT CAG TAT ,
R:GCA GTC TTC TCA TTC CCT TG; β-actin, F:TTT TGT GCC TTG ATA GTT
CG, R:GGA GTC CTT CTG ACC CAT AC-3) for P38 (Mapk14) and β-actin
were synthetized by Sangon Biotech Co., Ltd. (Shanghai, China).
HMGB1 and the TNF-α enzyme-linked immunosorbent assay (ELISA) kits
were purchased from R&D Systems (Minneapolis, MN, USA).
Animals and treatments
The present study was approved by the animal care
and use committee of Harbin Medical University (Harbin, China). All
research was performed in accordance with the internationally
accepted principles for laboratory animal use and care.
Sprague-Dawley rats (n=36, 6-weeks-old; weight, 200–220 g; SPF
grade) were obtained from the Animal Center of Harbin Medical
University. All animal protocols were approved by the experimental
animal care and use committee of Harbin Medical University, were
housed under specific pathogen-free conditions. Animals received
humane care according to established standards with free access to
water and food, and they were maintained in an air-conditioned room
at 25°C with a 12-h light/dark cycle. The animals received
intraperitoneal injection of LPS solution (2 mg/kg) on the first,
third and fifth day of experiment. The control rats were fed with
standard diet and tap water. The rats were randomly divided into
five groups (6 rats/group): i) control group with rats fed with
standard diet for 7 days; ii) LPS group with rats receiving
intraperitoneal injection of LPS solution (2 mg/kg) on the first,
third and fifth day of experiment, and fed with standard diet and
tap water for 7 days; iii) vitexin (5 mg/kg) group with rats that
received intraperitoneal injection of LPS solution (2 mg/kg) on the
first, third and fifth day of the experiment, and received
intraperitoneal injection of vitexin solution (5 mg/kg/day), whilst
being fed a standard diet and tap water for 7 days; iv) vitexin (10
mg/kg) group with rats receiving intraperitoneal injection of LPS
solution (2 mg/kg) on the first, third and fifth day of the
experiment, and intraperitoneal injection of vitexin solution (10
mg/kg/day), whilst being fed with standard diet and tap water for 7
days; v) SB203580 group rats receiving intraperitoneal injection of
LPS solution (2 mg/kg) on the first, third and fifth day of the
experiment, and intraperitoneal injection of SB203580 solution (1
µM/kg/day), whilst being fed with standard diet and tap water for 7
days.
ELISA detection
An ELISA was performed to determine serum HMGB1 and
TNF-α levels, according to the manufacturer's protocol (R&D
Systems).
Histology
Samples of the pancreas were fixed in 4%
mediosilicic isotonic formaldehyde for 24 h (4°C), and were
subsequently dehydrated and embedded in paraffin. Tissue sections
(5 µm-thick) were cut from each paraffin embedded tissue sample,
stained with hematoxylin and eosin and observed under a light
microscope to evaluate the degree of pancreatic islets damage.
Immunofluorescence and terminal
deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)
The expression and localization of HMGB1 in the
pancreatic islets were detected by immunofluorescence from the
aforementioned 5 µm-thick tissue sections. To identify the types of
injury, apoptosis-associated analyses were performed using TUNEL.
The TUNEL reaction was performed using the in situ TMR red
cell death detection kit (Roche Diagnostics GmbH, Mannheim,
Germany). Briefly, the slides containing tissue samples were
incubated with the enzyme terminal deoxynucleotidyl transferase at
37°C for 1 h and washed 3 times with PBS. The TUNEL mixture was
added, and the slides were incubated for 30 min at 37°C. Finally,
the positive cells were observed with fluorescent microscopy. For
quantification, the mean number of TUNEL-positive cells was
calculated under a magnification of ×100 in five different
fields.
Cell culture and treatment
The INS-1 cell line was purchased from American Type
Culture Collection (Rockville, MD, USA). The cells were cultured in
RPMI-1640 medium containing 10% fetal bovine serum (FBS) at 5%
CO2, 37°C. INS-1 cells were seeded at a density of 2×105
cells/ml in 6-well plates, then divided into five groups according
to different processing methods: i) Control group, cells were
cultured in RPMI-1640 medium containing 10% FBS at 37°C without
treatment; ii) LPS group, cells were cultured in complete RPMI-1640
medium with LPS (5 µg/ml) for 24 h; iii) Vitexin group, cells were
cultured in complete RPMI-1640 medium with LPS (5 µg/ml) for 24 h,
then cultured in complete RPMI-1640 medium with vitexin (50 µM) for
24 h; iv) P38 MAPK inhibitor (SB203580) group, cells were cultured
in complete RPMI-1640 medium with SB203580 0.5 µM) for 24 h, then
cultured in complete RPMI-1640 medium with LPS (5 µg/ml) for 24 h.
An ELISA was used to determine the HMGB1 levels in cell
supernatants.
Cell viability assay
Cell viability was estimated using a colorimetric
assay based on conversion of a tetrazolium dye (MTT) into a blue
formazan product. Briefly, INS-1 cells were seeded at a density of
1×104 cells/well in 96-well plates. The cells were cultured in
complete RPMI-1640 medium with LPS (5 µg/ml) for 24 h, then vitexin
was added to the wells at different concentrations (20, 30, 40, 50,
100, 200 and 300 µM) and the cells were cultured for 24 h. The
culture medium was subsequently replaced with 20 µl MTT solution.
The MTT solution was removed after 4 h of incubation at 37°C and
the produced formazan was solubilized in 200 µl DMSO. The
absorbance was measured at 490 nm using an automated microplate
reader.
Reverse transcription polymerase chain
reaction (RT-PCR)
The total RNA was isolated from INS-1 cells using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Briefly, cDNA was synthesized from 1 µg RNA in
the presence of ribonuclease inhibitor (Sigma-Aldrich; Merck
Millipore), dNTPs, Oligo (dT) 18 primers, and RevertAid™ M-Mulv
reverse transcriptase (Fermentas; Thermo Fisher Scientific, Inc.)
in a total volume of 25 µl. PCR was performed using a Takara mRNA
Selective PCR kit (Takara Bio, Inc., Otsu, Japan) in a total volume
of 25 µl, under the following cycling conditions: PCR
amplifications were performed in duplicate at 94°C for 2 min,
followed by 35 cycles at 94°C for 5 sec, 56°C for 20 sec and 72°C
for 60 sec, and a final extension step at 72°C for 10 min. The
primers used were as follows: P38 (Mapk14), sense:
5′-GCCTCACCGCCTCAGTAT−3′ and antisense: 5′-GCAGTCTTCTCATTCCCTTG-3′
(252 bp); internal control β-actin, sense:
5′-TTTTGTGCCTTGATAGTTCG-3′ and antisense 5′-GGAGTCCTTCTGACCCATAC-3
(265 bp). The PCR products were separated by 1.5% agarose gel
electrophoresis, followed by ethidium bromide staining. Target
bands were analyzed by densitometry, using a GS-800 calibrated
densitometer (Bio-Rad Laboratories, Hercules, CA, USA) and Gel-Pro
Analyzer 4.0 gel analyzing software (Media Cybernetics, Rockville,
MD, USA). The results were calculated as the ratio of the optical
density value relative to that of β-actin.
Western blotting
INS-1 cells were collected by scraping and washed
with PBS. The cells were lysed in RIPA buffer containing
phosphatase inhibitor cocktail I (Sigma-Aldrich; Merck Millipore)
and protease inhibitor cocktail mini-tablet (Roche Diagnostics,
Indianapolis, IN, USA). The total cellular protein was extracted
and separated using 10 or 12% SDS-PAGE. The proteins were
transferred onto nitrocellulose membranes (Merck Millipore).
Non-specific protein binding was inhibited by incubating the
membranes in blocking buffer (5% milk diluted in PBS). Following
blocking, the membranes were incubated with specific primary
antibodies at 4°C overnight. After washing, the membranes were
incubated with horseradish peroxidase-conjugated anti-IgG
(Anti-rabbit IgG; cat. no. 14708S; 1:3,000; Cell Signaling
Technology, Inc.) at room temperature for 2 h. Signal detection was
carried out with an enhanced chemiluminescence system (Merck
Millipore). Protein bands were quantified using Gel Pro Analyzer
software 4.0 (Media Cybernetics) and the intensity of the bands
were normalized against that of β-actin.
Statistical analysis
The data are presented as the mean ± standard
deviation. The differences among the groups were analyzed using
one-way analysis of variance with SPSS 19.0 statistical software
(IBS SPSS, Chicago, IL, USA). P<0.05 was considered to indicate
a statistically significant difference.
Results
Vitexin inhibits LPS-induced release
of HMGB1 in islet tissue
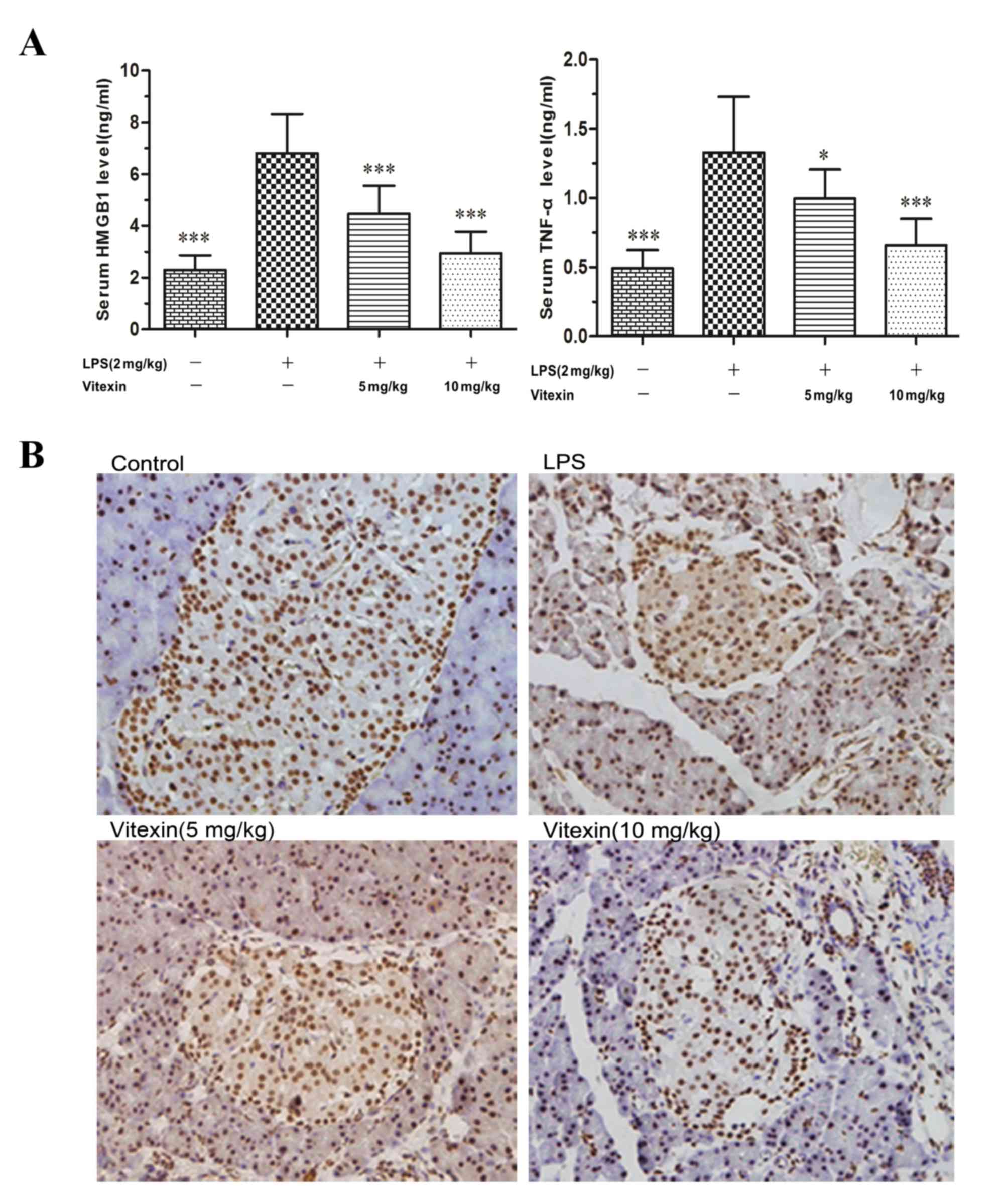
Under basal conditions, HMGB1 is predominantly
located in the nucleus of macrophages, while LPS stimulation can
result in the secretion of this intranuclear HMGB1 into
extracellular spaces (14). As
shown in the in vivo rat model, LPS injections induced the
release of HMGB1 in the islet tissue. Following pretreatment with
vitexin, the expression and distribution of HMGB1 in islet tissue
decreased and attenuated LPS-induced the levels of HMGB1. Similar
results were observed for TNF-α in the serum (Fig. 1). These results provided evidence
to suggest that vitexin alleviates LPS-induced HMGB1 release.
Protective effects of vitexin on
LPS-induced islet cell injury and apoptosis
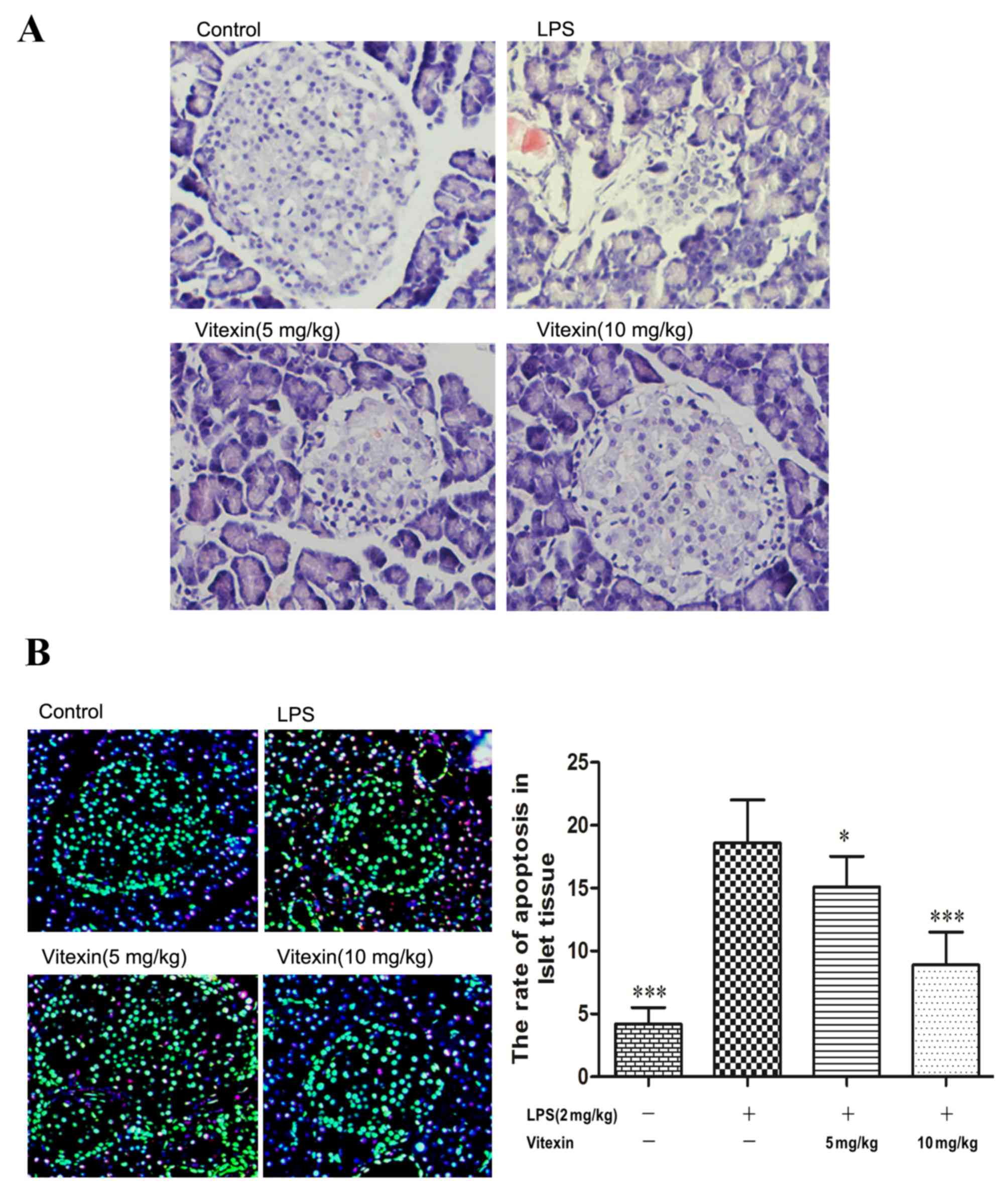
The present study also observed that vitexin
protected against LPS-induced islet tissue injury (Fig. 2A). Apoptosis is regarded as one
mechanism contributing to the inflammatory response of β-cells in
DM. As shown in Fig. 2B, treatment
with LPS increased the levels of extracellular HMGB1 and apoptosis
in islet tissue. However, a significant decrease was observed in
these parameters following pre-treatment with vitexin (5 and 10
mg/kg). These results suggested that vitexin protects against
LPS-induced islet cell injury and apoptosis.
Vitexin enhances INS-1 cell
survival
An effect of vitexin on LPS-induced INS-1 cell
survival was also observed. As determined by MTT assays, the
survival ratios of INS-1 cells were reduced in response to 5 mg/kg
LPS, while INS-1 cell survival ratios increased significantly in a
dose-dependent manner following treatment with vitexin, compared
with LPS alone (Fig. 3A). These
results suggested that vitexin protects INS-1 cells from
LPS-induced death.
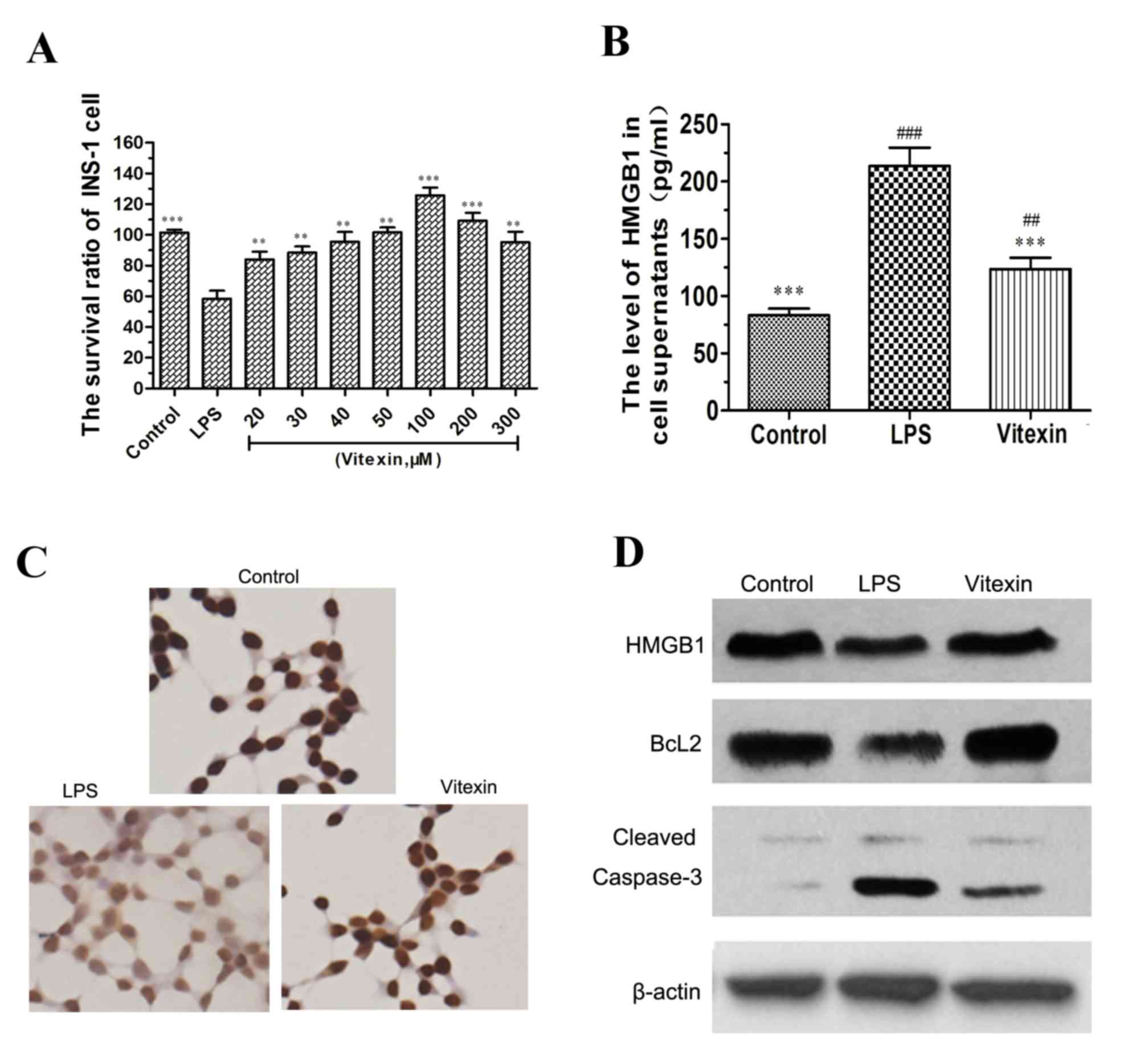 | Figure 3.Vitexin suppresses LPS-induced release
of HMGB1 and caspase-3 activation in INS-1 cells. The effect of
vitexin on INS-1 cell survival rate was assessed. The INS-1 cells
were treated with vitexin at a concentration of 20, 30, 40, 50,
100, 200 and 300 µM in the presence of 5 mg/l LPS. (A) An MMT assay
was performed in INS-1 cells. The data are presented as the mean ±
standard deviation (*P<0.05, ***P<0.001 compared with the LPS
group). (B) The increase of HMGB1 levels stimulated by LPS was
reversed by vitexin pretreatment (50 µmol/l). **P<0.01,
***P<0.001 compared with the LPS group; ##P<0.01
compared with vitexin. (C) Immunocytochemistry was used to observe
the distribution of HMGB1 in INS-1 cells, magnification, ×400. (D)
Western blotting was used to observe the influence of vitexin on
the protein expression levels of HMGB1, Bcl2 and cleaved caspase-3
in LPS-treated INS-1 cells. HMGB1, high mobility group box 1; LPS,
lipopolysaccharide; Bcl2, B-cell lymphoma 2. |
Vitexin suppresses LPS-induced release
of HMGB1 and caspase-3 activation in INS-1 cells
Complementing the results from the present in
vivo experiments, it was also observed that LPS induced the
release of HMGB1 in INS-1 cells, as shown in Fig. 3B. When vitexin was added to these
LPS-treated preparations, the level of HMGB1 in cell supernatants
was reduced. The distribution of HMGB1 in INS-1 cells, as observed
by immunocytochemistry, is presented in Fig. 3C. The dyeing intensity of HMGB1 in
the nucleus faded when INS-1 cells were treated with LPS, however,
vitexin attenuated the change. In addition to suppressing
LPS-induced release of HMGB1, the protein expression of cleaved
caspase-3 in INS-1 cells was also decreased by vitexin, compared
with LPS-alone (Fig. 3D). The
expression of the key antiapoptosis factor, Bcl-2, which
contributes to the caspase pathway of apoptosis, was significantly
increased, while expression levels of the capase-3 protein was
decreased in response to vitexin + LPS, as compared with the LPS
alone group.
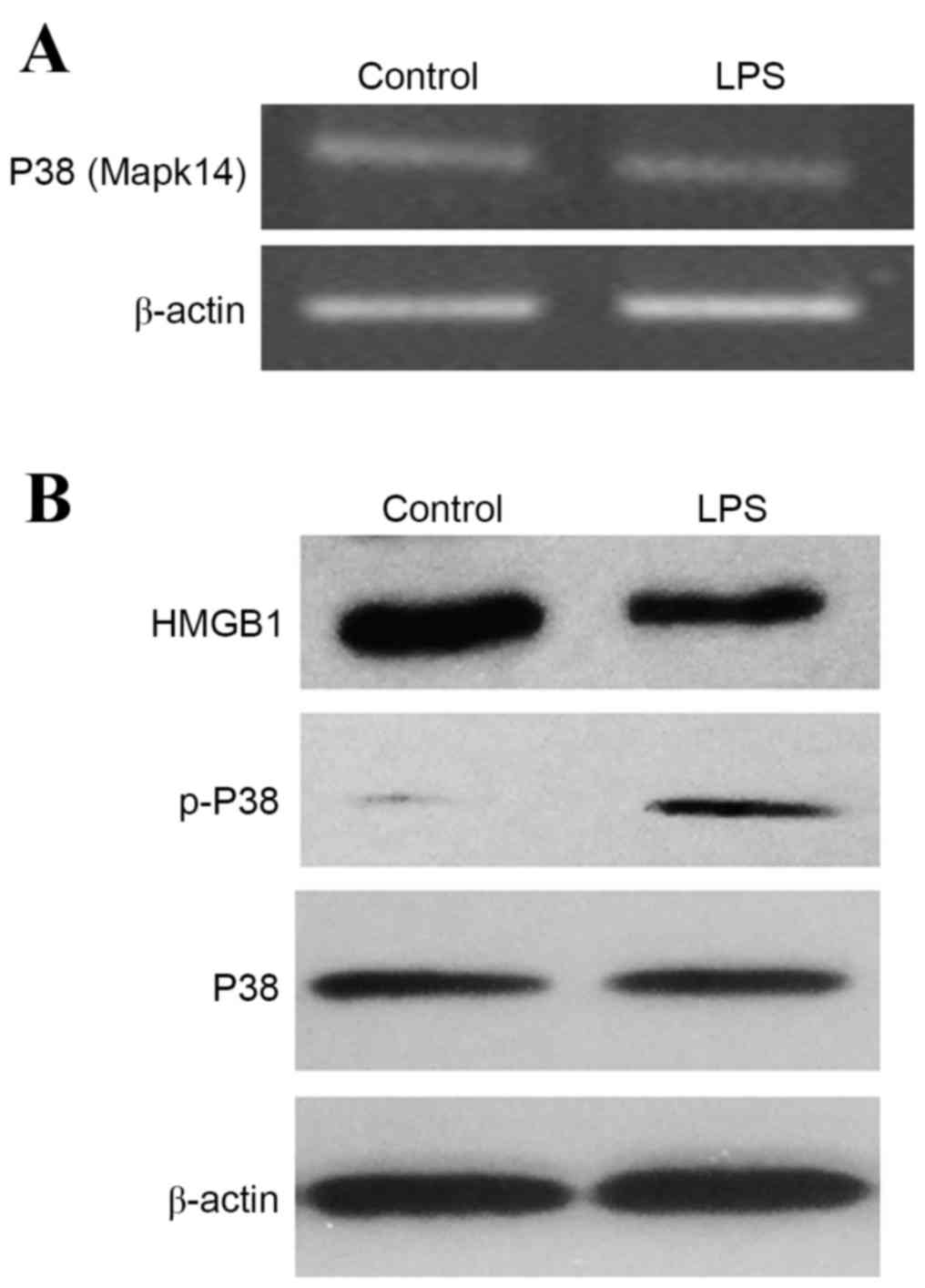
Influence of LPS on the P38 MAPK
pathway in INS-1 cell
The present study observed the release of HMGB1
induced by LPS to further demonstrate the effect of HMGB1 release
on P38 MAPK pathway in pancreatic islet cells. As shown in Fig. 4, compared with the control group,
the mRNA and protein expression levels of P38 (Mapk14) exhibited no
increase following LPS treatment; however, the phosphorylation of
P38 increased following LPS treatment. Therefore, LPS-induced
release of HMGB1 may induce the activation of the P38 signaling
pathways in INS-1 cells.
Vitexin protection of islet cell
injury induced by LPS is dependent on inhibiting HMGB1 release
To further clarify that vitexin inhibits HMGB1
release or the P38 pathway, the present study used a P38 inhibitor
(SB203580). Following stimulation with LPS, the serum level of
HMGB1 and the rate of apoptosis of islet tissue increase
(P<0.001); however, a significant decrease was observed
following treatment with 10 mg/kg vitexin (Fig. 5A and B). Treatment with SB203580
also decreased the serum level of HMGB1 induced by LPS (Fig. 5C) and the apoptosis of islet tissue
in rats (Fig. 5B). Notably, the
level of serum HMGB1 and the rate of apoptosis were markedly higher
compared with the vitexin group (P<0.05). Additionally, the
present study also performed in vitro experiments to confirm
that vitexin inhibits HMGB1 release or the P38 pathway. The level
of HMGB1 in the INS-1 cell supernatants increased following
stimulation with LPS, but decreased following treatment with
vitexin (Fig. 5D). SB203580 also
decreased the level of HMGB1 in INS-1 cell supernatants induced by
LPS, but was markedly higher compared with the vitexin group
(P<0.001). In addition, immunocytochemistry demonstrated that
vitexin inhibited LPS-induced HMGB1 translocation from the nucleus
to the cytoplasmic compartment; however, weak inhibition of
LPS-induced HMGB1 translocation from the nucleus to the cytoplasmic
compartment was observed (Fig.
5E). Following the LPS treatment, INS-1 cells were treated with
vitexin or SB203580. This revealed a significant reduction in the
protein expression levels of p-P38 and cleaved caspase-3, but the
levels of Bcl2 increased (Fig.
5F). A more significant effect was observed in the vitexin
group compared with the SB203580 group. The present results
suggested that vitexin protects cells from LPS-induced islet cell
injury and apoptosis.
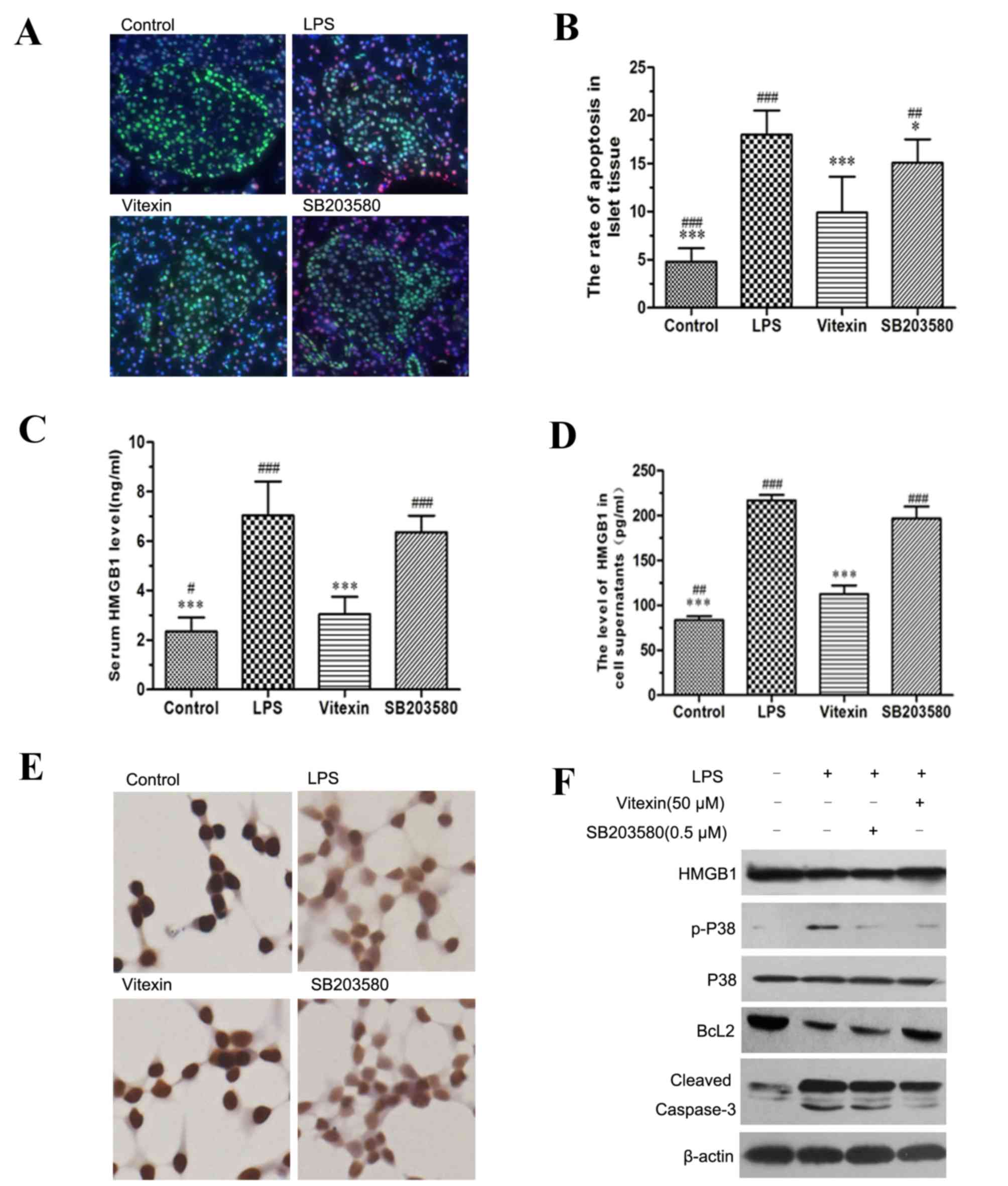 | Figure 5.Vitexin protection of islet cell
injury induced by LPS is dependent on the inhibition of HMGB1
release. (A) The effect of vitexin on LPS-induced islet cell
apoptosis and the distribution of HMGB1 was assessed by
immunostaining. HMGB1 was stained in green and apoptosis was
highlighted in red. (B) The rate of apoptosis in islet tissue was
assessed. The data are presented as the mean ± standard deviation
(**P<0.01, ***P<0.001 compared with LPS,
##P<0.01, ###P<0.001 compared with
vitexin). (C) The rats were treated with LPS, vitexin or SB203580,
and the levels of serum HMGB1 were measured by ELISA. The data are
presented as the mean ± standard deviation (***P<0.001 compared
with LPS, ##P<0.01, ###P<0.001 compared
with vitexin). (D) INS-1 cells were treated with LPS, vitexin or
SB203580, and the levels of HMGB1 in the cell supernatants were
measured by ELISA, The data are presented as the mean ± standard
deviation (***P<0.001 compared with LPS, #P<0.05,
##P<0.01, ###P<0.001 compared with
vitexin). (E) Immunocytochemistry was used to demonstrate HMGB1
translocation from the nucleus to the cytoplasmic compartment
magnification, ×400) Dyeing intensity of HMGB1 in the nucleus
faded. (F) INS-1 cells were treated with LPS, vitexin or SB203580,
and the protein expression levels of the indicated proteins were
measured by western blot analysis. β-actin was used as a loading
control. HMGB1, high mobility group box 1; LPS, lipopolysaccharide;
p-, phosphorylated; Bcl2, B-cell lymphoma 2. |
Discussion
HMGB1 can be passively released from damaged cells
or necrotized tissues (15,16).
The present results demonstrated that LPS can induce the release of
HMGB1 from islet tissue. These findings are in accord with previous
reports indicating that HMGB1 is associated with islet destruction
(17). This LPS-induced release of
HMGB1 was significantly decreased following the addition of
vitexin, suggesting that vitexin protected islet cells from injury
and enhanced their survival. LPS can also induce the activation of
the P38 MAPK pathway, which represents a crucial synergistic
component that contributes to pancreatic islet cells destruction.
Based upon the present results with the P38 inhibitor (SB203580),
it is clear that the protective effect of vitexin upon LPS-induced
islet cell injury and apoptosis resides primarily in its capacity
to inhibit HMGB1 release.
The inflammatory response has been regarded as one
of the mechanisms associated with impaired insulin signal
transduction, as the secretion of inflammatory cytokines can
contribute to alterations in both insulin signaling and insulin
sensitivity. HMGB1 is a vastly abundant and conserved protein that
exerts a number of significant intra and extracellular biological
activities, and is linked with inflammation (18). It is commonly considered a nuclear
molecule, however, when stimulated by LPS, it can be translocated
to the cytoplasm. HMGB1 is also actively secreted by inflammatory
cells and binds with high affinity to several receptors, including
RAGE and the Toll-like receptor (TLR)-2, TLR-4 and TLR-9 (19). The release of HMGB1 into the
extracellular environment, where it can function as an endogenous
danger signal or ‘alarmin’ to promote inflammation, has been
implicated in several diseases, including sepsis, acute lung injury
and type 1 diabetes (20). Type 1
and 2 diabetes are associated with inflammatory cytokines, which
can serve important roles in islet dysfunction and death in DM.
Growing evidence suggests that inflammation, lipids and insulin
sensitivity are tightly interconnected (21). Metabolic disorders, which
contribute to β-cell dysfunction and insulin resistance in patients
with DM, have also been shown to induce an increase in inflammatory
cytokines, including C-reactive protein and IL-6 (22). A growing body of evidence supports
the hypothesis that inflammatory responses may alter the normal
structure of β-cells and induce insulin resistance, as well as
decrease insulin secretion (23).
Therefore, any processes that can attenuate the inflammatory
response will be beneficial for the improvement of β-cell function.
Numerous cytokines have been identified to contribute to the
occurrence and development of β-cell dysfunction. The c-Jun
N-terminal kinases (JNK)/nuclear factor (NF)-κB signaling pathway,
which has been shown to be closely associated with the activation
of TLR4, contributes to the release of inflammatory mediators,
including TNF-a and IL-6 (24).
NF-κB can be activated by MAPKs, including JNK, P38 and
extracellular-regulated kinase. P38 kinases are members of MAPK and
are well documented as being involved a wide range of signaling
pathways and biological processes. The prototypic P38 MAPK, P38α
MAPK, was originally identified as a tyrosine phosphorylated
protein detected in LPS-stimulated macrophages, and is essential
for inflammatory cytokine production (25). In the present study, the levels of
HMGB1 and TNF-α were increased in response to LPS stimulation,
which is consistent with other reports indicating that HMGB1 is
involved in the pathogenesis of DM and involvement of
HMGB1-mediated activation of inflammatory signaling in
Sprague-Dawley rats (19). The
present study revealed that vitexin treatment significantly
inhibited TNF-α and HMGB1 production in response to
LPS-stimulation, as demonstrated both in vitro and in
vivo.
Vitexin, a naturally occurring flavone glycoside in
plants, has been reported to exert a variety of pharmacological
activities, including anti-inflammatory, anticancer,
antinociceptive, antispasmodic, antioxidant and antimyeloperoxidase
effects, as well as a protectant against ischemia/reperfusion
injury and an α-glucosidase inhibitor. Accordingly, a wide range of
potential applications exist for vitexin in the treatment of
cardiovascular diseases, diabetes and cancer. The present study
examined the effects of vitexin upon LPS-induced islet cell injury,
and revealed that pretreatment with vitexin attenuated HMGB1
translocation from the nucleus to the cytoplasm in LPS-stimulated
islet tissue and INS-1 cells. This effect may serve as a
significant factor that decreases LPS-induced islet cell injury. A
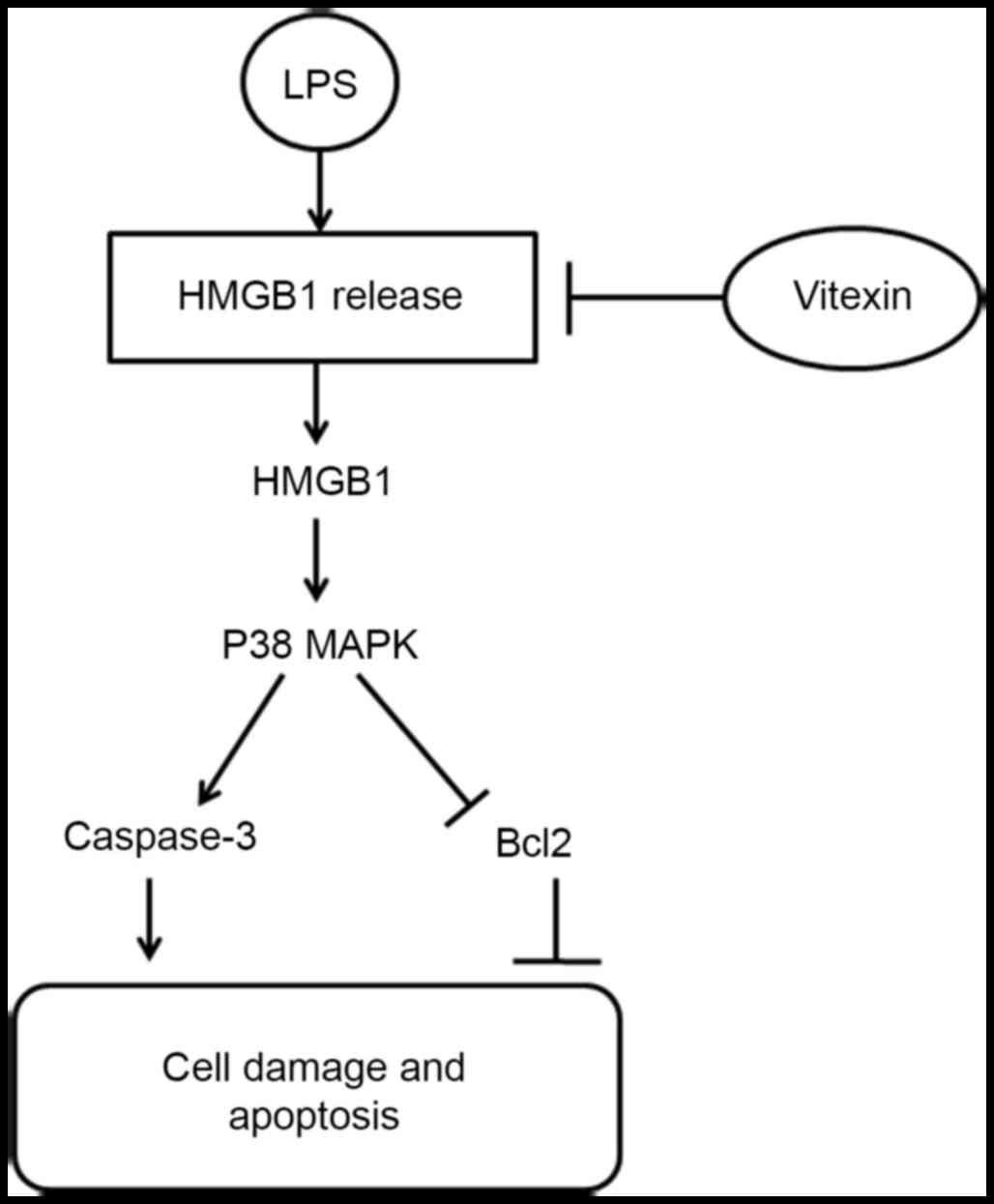
summary of the protective effects of vitexin is presented in
Fig. 6.
In conclusion, the present study evaluated the
therapeutic effects of vitexin in LPS-induced inflammation and
examined its potential mechanism. Vitexin alleviates LPS-induced
islet injury and apoptosis by reducing the release of HMGB1. The
present study shows that vitexin may serve as a promising
therapeutic agent for the treatment of DM.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant. no. 81370929 and
U1404805), The Research Fund for the Clinical Medicine of Chinese
Medical Association (grant. no. 13040670452) and The Science
Foundation of the Education Department of Heilongjiang Province
(grant. no. 12531316).
Glossary
Abbreviations
Abbreviations:
|
DM
|
diabetes mellitus
|
|
LPS
|
lipopolysaccharide
|
|
HMGB1
|
high mobility group box 1
|
|
IR
|
insulin resistance
|
|
RAGE
|
receptor for advanced glycation end
products
|
|
MAPKs
|
mitogen-activated protein kinase
|
References
|
1
|
Mahmoud F and Al-Ozairi E: Inflammatory
cytokines and the risk of cardiovascular complications in type 2
diabetes. Dis Markers. 35:235–241. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Donath MY and Shoelson SE: Type 2 diabetes
as an inflammatory disease. Nat Rev Immunol. 11:98–107. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eizirik DL, Sammeth M, Bouckenooghe T,
Bottu G, Sisino G, Igoillo-Esteve M, Ortis F, Santin I, Colli ML,
Barthson J, et al: The human pancreatic islet transcriptome:
Expression of candidate genes for type 1 diabetes and the impact of
pro-inflammatory cytokines. PLoS Genet. 8:e10025522012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini
V, Mardis ER and Gordon JI: An obesity-associated gut microbiome
with increased capacity for energy harvest. Nature. 444:1027–1031.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pussinen PJ, Havulinna AS, Lehto M,
Sundvall J and Salomaa V: Endotoxemia is associated with an
increased risk of incident diabetes. Diabetes care. 34:392–397.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Landsman D and Bustin M: A signature for
the HMG-1 box DNA-binding proteins. Bioessays. 15:539–546. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lotze MT and Tracey KJ: High-mobility
group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal.
Nat Rev Immunol. 5:331–342. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim JM, Han HJ, Hur YH, Quan H, Kwak SH,
Choi JI and Bae HB: Stearoyl lysophosphatidylcholine prevents
lipopolysaccharide-induced extracellular release of high mobility
group box-1 through AMP-activated protein kinase activation. Int
Immunopharmacol. 28:540–545. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ghavami S, Rashedi I, Dattilo BM, Eshraghi
M, Chazin WJ, Hashemi M, Wesselborg S, Kerkhoff C and Los M:
S100A8/A9 at low concentration promotes tumor cell growth via RAGE
ligation and MAP kinase-dependent pathway. J Leukoc Biol.
83:1484–1492. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ogawa EN, Ishizaka A, Tasaka S, Koh H,
Ueno H, Amaya F, Ebina M, Yamada S, Funakoshi Y, Soejima J, et al:
Contribution of high-mobility group box-1 to the development of
ventilator-induced lung injury. Am J Respir Crit Care Med.
174:400–407. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y, Zhen Y, Wu X, Jiang Q, Li X, Chen
Z, Zhang G and Dong L: Vitexin protects brain against
ischemia/reperfusion injury via modulating mitogen-activated
protein kinase and apoptosis signaling in mice. Phytomedicine.
22:379–384. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dong L, Fan Y, Shao X and Chen Z: Vitexin
protects against myocardial ischemia/reperfusion injury in
Langendorff-perfused rat hearts by attenuating inflammatory
response and apoptosis. Food Chem Toxicol. 49:3211–3213. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Borghi SM, Carvalho TT, Staurengo-Ferrari
L, Hohmann MS, Pinge-Filho P, Casagrande R and Verri WA Jr: Vitexin
inhibits inflammatory pain in mice by targeting TRPV1, oxidative
stress, and cytokines. J Nat Prod. 76:1141–1149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Musumeci D, Roviello GN and Montesarchio
D: An overview on HMGB1 inhibitors as potential therapeutic gents
in HMGB1-related pathologies. Pharmacol Ther. 141:347–357. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Abdulahad DA, Westra J, Limburg PC,
Kallenberg CG and Bijl M: HMGB1 in systemic lupus Erythematosus:
Its role in cutaneous lesions development. Autoimmun Rev.
9:661–665. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin Q, Fang J, Fang D, Li B, Zhou H and Su
SB: Production of recombinant human HMGB1 and anti-HMGB1 rabbit
serum. Int Immunopharmacol. 11:646–651. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jo EH, Hwang YH and Lee DY: Encapsulation
of pancreatic islet with HMGB1 fragment for attenuating
inflammation. Biomater Res. 19:212015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Andersson U, Wang H, Palmblad K, Aveberger
AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M,
Yang H and Tracey KJ: High mobility group 1 protein (HMG-1)
stimulates proinflammatory cytokine synthesis in human monocytes. J
Exp Med. 192:565–570. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bauernfeind FG, Horvath G, Stutz A,
Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks
BG, Fitzgerald KA, et al: Cutting edge: NF-kappaB activating
pattern recognition and cytokine receptors license NLRP3
inflammasome activation by regulating NLRP3 expression. J Immunol.
183:787–791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li M, Song L, Gao X, Chang W and Qin X:
Toll-like receptor 4 on islet β cells senses expression changes in
high-mobility group box 1 and contributes to the initiation of type
1 diabetes. Exp Mol Med. 44:260–267. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shu CJ, Benoist C and Mathis D: The immune
system's involvement in obesity-driven Type 2 diabetes. Semin
Immunol. 24:436–442. 2013. View Article : Google Scholar :
|
|
22
|
Akash MS, Rehman K and Chen S: Role of
inflammatory mechanisms in pathogenesis of type 2 diabetes
mellitus. J Cell Biochem. 114:525–531. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cai K, Qi D, Hou X, Wang O, Chen J, Deng
B, Qian L, Liu X and Le Y: MCP-1 upregulates amylin expression in
murine pancreatic β cells through ERK/JNK-AP1 and NF-kB related
signaling pathways independent of CCR2. PLoS One. 6:e195592011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cui G, Qin X, Zhang Y, Gong Z, Ge B and
Zang YQ: Berberine differentially modulates the activities of ERK,
p38 MAPK, and JNK to suppress Th17 and Th1 T cell differentiation
in type 1 diabetic mice. J Biol Chem. 284:28420–28429. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aggeli IK, Beis I and Gaitanaki C:
Oxidative stress and calpain inhibition induce alpha B-crystallin
phosphorylation via p38-MAPK and calcium signalling pathways in
H9c2 cells. Cell Signal. 20:1292–1302. 2008. View Article : Google Scholar : PubMed/NCBI
|