Introduction
In their resting state, hepatic stellate cells
(HSCs) are a group of quiescent pericytes that comprise 5–8% of
liver cells (1). They contain a
number of lipid droplets in the cytoplasm primarily involved in
vitamin A storage (2). Upon liver
injury, HSCs become activated, which converts these quiescent cells
into proliferative, fibrogenic and contractile myofibroblast-like
cells (3). Activated HSCs are the
principal collagen-producing cells and are responsible for fibrosis
in chronic liver disease. Aside from their key roles in fibrosis
and extracellular matrix (ECM) remodeling, activated HSCs have
additionally been implicated in the progression of hepatitis and
liver carcinogenesis (4). Due to
their critical function in liver pathogenesis, the majority of
proposed antifibrotic therapeutic approaches focus on modulating
HSC activation.
Bilirubin is a bile pigment resulting from the
degradation of heme, which is one of the breakdown products of red
blood cells (RBCs). It is produced by the liver and is present in
small quantities in the blood; however, excessive quantities cause
toxicity, primarily due to neonatal jaundice and the potential for
mediating irreversible brain damage at high concentrations
(5). The normal range of total
bilirubin concentration in the human body is 2–12 mg/l; elevated
levels are observed in disorders involving liver cell damage,
biliary disease and RBC-processing disorders (6). In addition, a physiological role for
bilirubin as an antioxidant has been reported. Stocker et al
(7) demonstrated that at
physiological concentrations, bilirubin efficiently protected
against oxidation of lipid membranes. The scavenging and
antioxidant activities of bilirubin were revealed to be more potent
than those of vitamins C and E against lipid peroxidation (8,9). As
neonates have impaired antioxidant defenses and are susceptible to
oxygen free radical-mediated diseases, it is hypothesized that
elevated bilirubin levels act as a defense mechanism to counteract
increased exposure to reactive oxygen species (ROS) (10,11).
ROS serve important roles in HSC activation and
hepatic fibrosis. Increased ROS production and the resulting
oxidative stress are commonly observed in alcoholic liver disease,
hepatitis C virus infection and experimental models of liver
fibrogenesis (12). Antioxidant
agents have been demonstrated to mitigate HSC activation and
hepatic fibrosis in various research models (13,14).
However, the effect of bilirubin on HSC activation remains to be
investigated.
In the present study, rat HSCs were cultured in
vitro and the effect of bilirubin treatment on their activation
was assessed, including the effects on ROS production, α-smooth
muscle actin (α-SMA) and ECM remodeling-associated gene expression,
cell proliferation and apoptosis. These results suggested that
within the normal physiological concentration range, bilirubin may
exert beneficial effects.
Materials and methods
Reagents and antibodies
Dulbecco's modified Eagle's medium (DMEM; HyClone;
GE Healthcare Life Sciences, Logan, UT, USA) served as the cell
culture medium. Fetal bovine serum (FBS) was purchased from Gibco;
Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Bilirubin was
obtained from Sigma-Aldrich; Merck Millipore (Darmstadt, Germany).
2′,7′-dichlorofluorescin diacetate (DCFH-DA) was obtained from
eBioscience, Inc. (San Diego, CA, USA). A rabbit anti-rat α-SMA
primary antibody (cat. no. ab5694), a rabbit anti-GAPDH antibody
(cat. no. ab9485) and a fluorescein isothiocyanate (FITC)-labeled
goat anti-rat secondary antibody (cat. no. ab33110) were obtained
from Abcam (Cambridge, UK). Angiotensin converting enzyme
chromogenic reagents were obtained from Wuhan Boster Biological
Technology, Ltd. (Wuhan, China). The Cell Counting Kit-8 (CCK-8)
was purchased from Dojindo Molecular Technologies, Inc. (Kumamoto,
Japan). The Apoptosis assay kit was obtained from Hangzhou
MultiSciences (Lianke) Biotech Co., Ltd. (Hangzhou, China).
Cell culture
HSCs were isolated from 20 six-week-old Male
Sprague-Dawley rats obtained from the Laboratory Animal Center at
Wenzhou Medical College (Wenzhou, China). The care and use of
animals in this study fully complied with relevant governmental and
institutional requirements, guidelines and policies (15). Ethical approval was obtained from
the Human Ethics Committee of Wenzhou Medical University (Wenzhou,
China). The animals were sacrificed by intraperitoneal injection of
3% pentobarbital sodium (60 mg/kg body weight, purchased from
Beijing Propbs Biotechnology Co., Ltd.). The cell isolation method
used was as previously described (16). Isolated HSCs were cultured in DMEM
with L-glutamine and 10% FBS. The medium was replaced every 2–3
days. Following primary culture for ~7 days, the cells underwent
rapid proliferation. Experimental assays were conducted with fifth
generation (passage) activated HSCs.
ROS assay
Cultured HSCs were adjusted to 1×105 cells/ml and
100 µl/well was seeded into 96-well plates. Bilirubin was diluted
in DMEM with 10% FBS and added at concentrations of 0, 1, 10 or 20
mg/l. After 24 h of treatment, the medium was aspirated. DCFH-DA
diluted in DMEM at 10 µmol/l was added to each well and incubated
for 30 min. Cells were subsequently rinsed twice with DMEM and
observed under a fluorescence microscope.
Western blot analysis
Isolated HSCs were treated with 0, 1, 10 or 20 mg/l
bilirubin for 24 h, and subsequently lysed with cell lysis buffer
(cat. no. P0013; Beyotime Institute of Biotechnology, Haimen,
China) for 30 min on ice, and centrifuged at 5,000 × g for 15 min
at 4°C to isolate the proteins in the supernatant. A total of 50 µg
isolated proteins were loaded onto 10% SDS-PAGE gels. Following
SDS-PAGE, the separated proteins were transferred to a
polyvinylidene difluoride membrane. Membranes were subsequently
blocked for 1 h with 5% non-fat milk in TBS with Tween-20 (cat. no.
K873-500ML, Shanghai Haoran Bio Technologies Co., Ltd., Shanghai,
China). The anti-GAPDH antibody diluted at 1:200 and the anti-α-SMA
antibody diluted at 1:100 were added to the membrane and incubated
overnight at 4°C. Subsequently, the membrane was washed three times
with TBS with Tween-20 for 10 min; the secondary antibody was added
at a 1:30,000 dilution and incubated at 37°C for 1 h. Following
this, the membrane was washed again and treated with chromogenic
reagents to visualize the proteins. The western blots were scanned
and analyzed using the Gel-Pro Analyzer system (version, 4.5; Media
Cybernetics, Inc. (Rockville, MD, USA).
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis of mRNA expression
HSCs were treated with various concentrations of
bilirubin for 24 h and collected for RNA isolation (TRIzol kit,
Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), cDNA
synthesis (Beijing Dingguo Changsheng Biotechnology Co., Ltd.,
Beijing, China) and PCR detection (PCR kit cat. no. P3012;
Guangdong Dongsheng Biotech Co., Ltd., Guangzhou, China) of mRNA
expression of matrix metalloproteinase (MMP-2) and tissue inhibitor
of matrix metalloproteinase-1 (TIMP-1). GAPDH served as an internal
control. The primers used in PCR reactions are presented in
Table I. PCR conditions were as
follows: Initial denaturation at 94°C for 5 min, followed by 40
cycles of denaturation for 30 sec at 94°C, annealing for 30 sec at
52.1°C and extension for 45 sec at 72°C. The software used for
densitometry was ImageJ (version, 1.48u; National Institutes of
Health, Bethesda, MD, USA).
 | Table I.Primer sequences for polymerase chain
reaction detection of mRNA expression. |
Table I.
Primer sequences for polymerase chain
reaction detection of mRNA expression.
| Gene | Direction | Sequence (5′-3′) |
|---|
| TIMP-1 | Forward |
CCTCTGGCATCCTCTTGTTG |
|
| Reverse |
CGCTGGTATAAGGTGGTCTC |
| MMP-2 | Forward |
CCCCTATCTACACCTACACCAA |
|
| Reverse |
CACCACGGATCTGAGCAAT |
| GAPDH | Forward |
GAGGACCAGGTTGTCTCCTG |
|
| Reverse |
GGATGGAATTGTGAGGGAGA |
Cell proliferation assay
HSCs were seeded at 1×104 cells/well in 96-well
plates and cultured in serum-free DMEM for 24 h prior to treatment
with bilirubin. The final concentrations of bilirubin were 0, 1, 10
and 20 mg/l, with five replicates per concentration. After
treatment for 24 h, 80 µl CCK-8 solution was added to each well and
incubated for 2 h. The absorbance was read at a wavelength of 450
nm using a plate reader, and cell proliferation rates were
calculated as previously described (16).
Apoptosis assay
Following bilirubin treatment, ~2×105 HSCs per
sample were harvested by trypsinization, centrifuged at 1000 × g
for 5 min at room temperature, washed twice with PBS and
resuspended in 200 µl binding buffer. Following this, 5 µl Annexin
V-FITC and 5 µl propidium iodide were added to each sample. The
samples were incubated at room temperature for 20 min in the dark
prior to flow cytometric analysis, with an excitation wavelength of
488 nm and an emission wavelength of 530 nm. The software used for
flow cytometric analysis was CellQuest Pro (version, 5.1; BD
Biosciences, Franklin Lakes, NJ, USA).
Statistical analysis
All data are expressed as the mean ± standard
deviation. Differences between groups were analyzed by one-way
analysis of variance using SPSS software version 16.0 (SPSS, Inc.,
Chicago, IL, USA). No post hoc analysis was conducted. P<0.05
was considered to indicate a statistically significant
difference.
Results
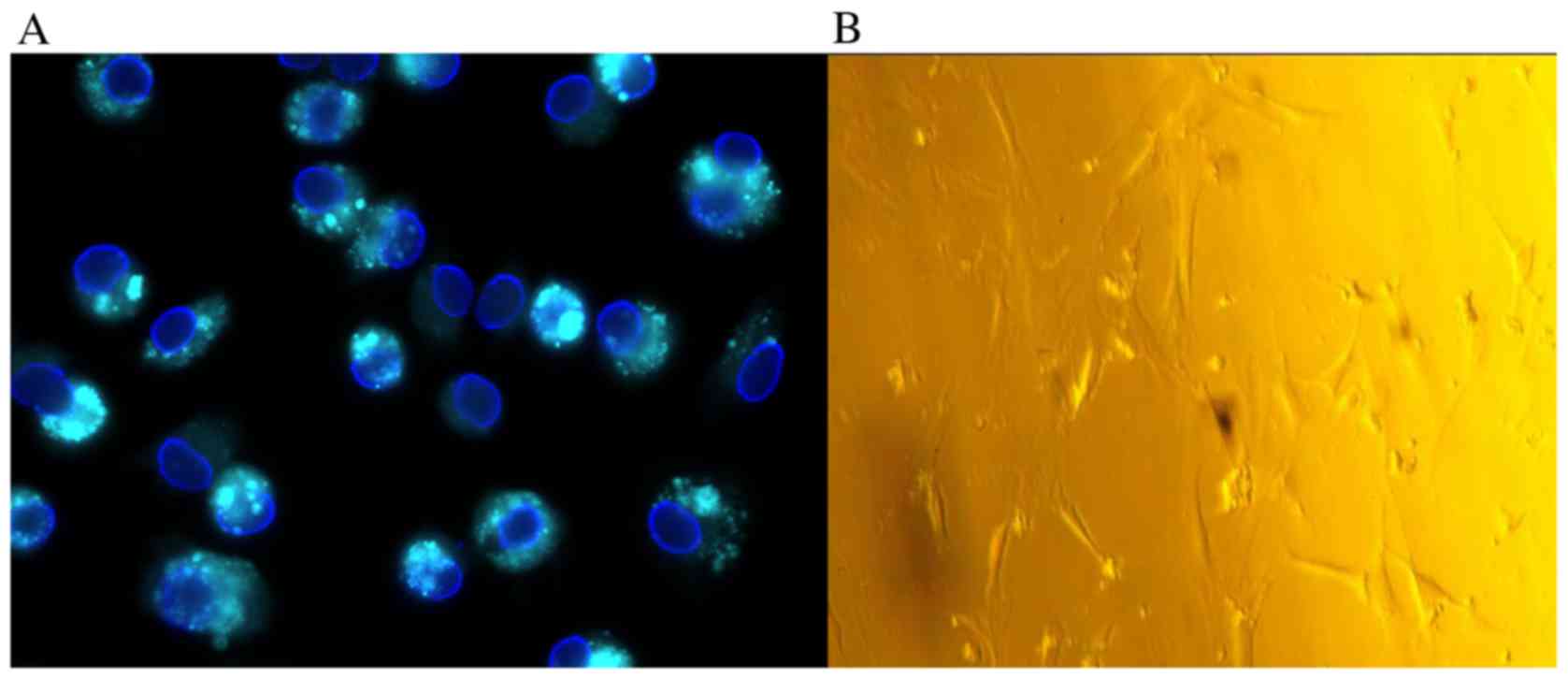
Morphological alterations in HSCs
during primary culture
HSCs, isolated with a yield of 1–2×107 cells/rat,
demonstrated >95% viability, as revealed by trypan blue
staining. Freshly isolated HSCs were rounded in shape and contained
multiple cytoplasmic lipid droplets. On excitation with light at a
wavelength of 328 nm, the droplets produced a green-blue
fluorescence (Fig. 1A). Following
primary culture for 2–3 days, cells became adherent. At days 5–7,
cells appeared to lose their capability for lipid storage, acquired
a star-like shape and demonstrated rapid proliferation (Fig. 1B). Gradually, the isolated
quiescent HSCs in the primary culture transitioned to activated
HSCs. These activated HSCs appeared to exhibit features of an
effective model for the study of HSC-associated liver diseases.
These cells transformed into myofibroblast-like cells with active
contractility, and appeared to induce fibrogenesis, matrix
degradation and proliferation. Activated HSC cultures at passage 5
were used for all subsequent experiments unless otherwise
stated.
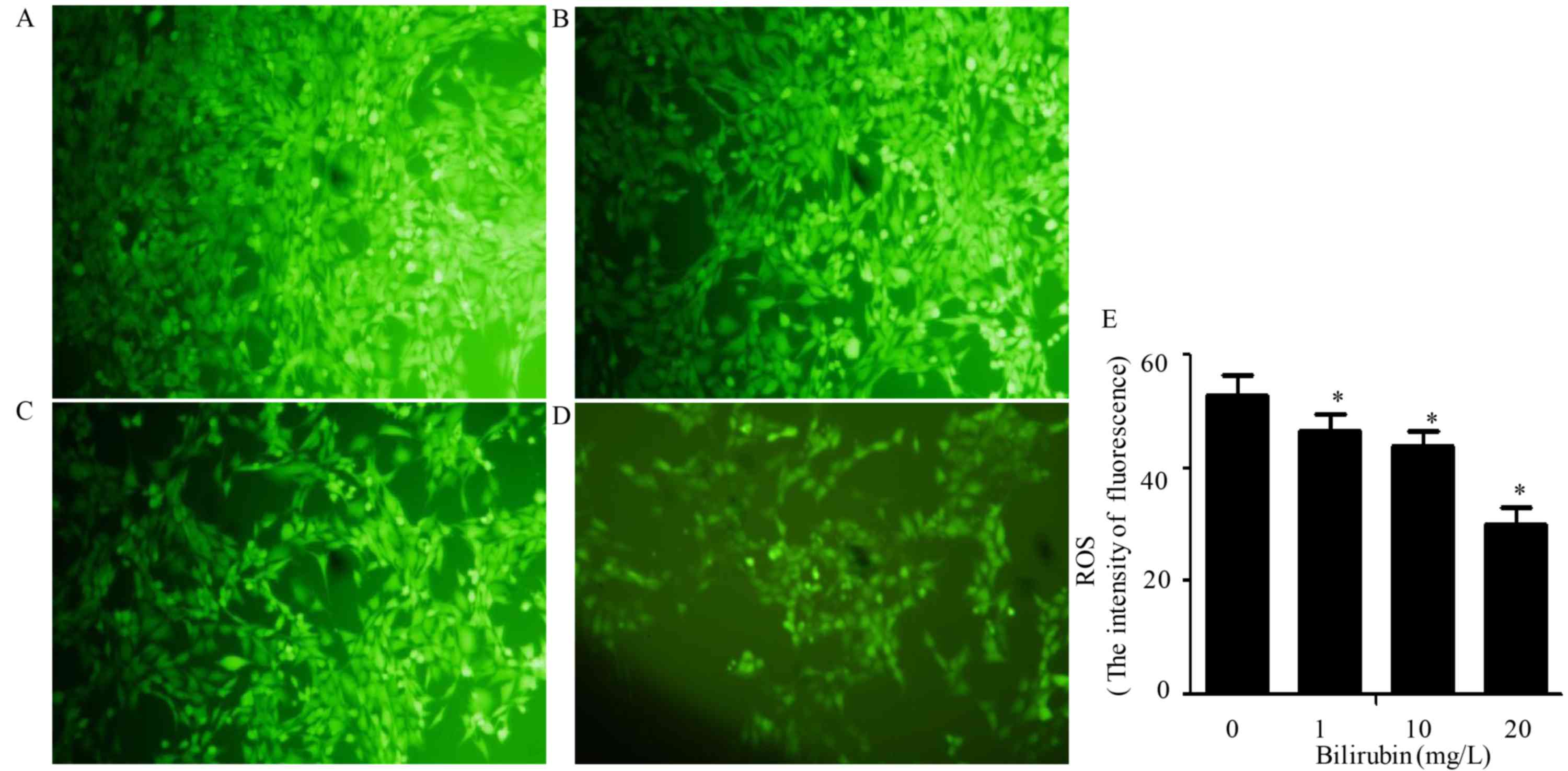
Effect of bilirubin on ROS
production
ROS production is an important feature of HSC
activation, and generated ROS may in turn stimulate the activation
of HSCs. Bilirubin was added to the HSC culture medium at
concentrations of 0, 1, 10 or 20 mg/l for 24 h. Following this,
DCFH-DA was used to assess ROS production. As bilirubin
concentration increased, the cellular fluorescence intensity
decreased (Fig. 2). The ROS
production was measured using an arbitrary fluorescence unit, and
the values were 53.6±6.3, 46.7±5.6, 44.7±5.5 and 31.2±6.1,
respectively (F=187.527, P=0.036). These findings indicated that
bilirubin reduced ROS production in HSCs in a dose-dependent
manner.
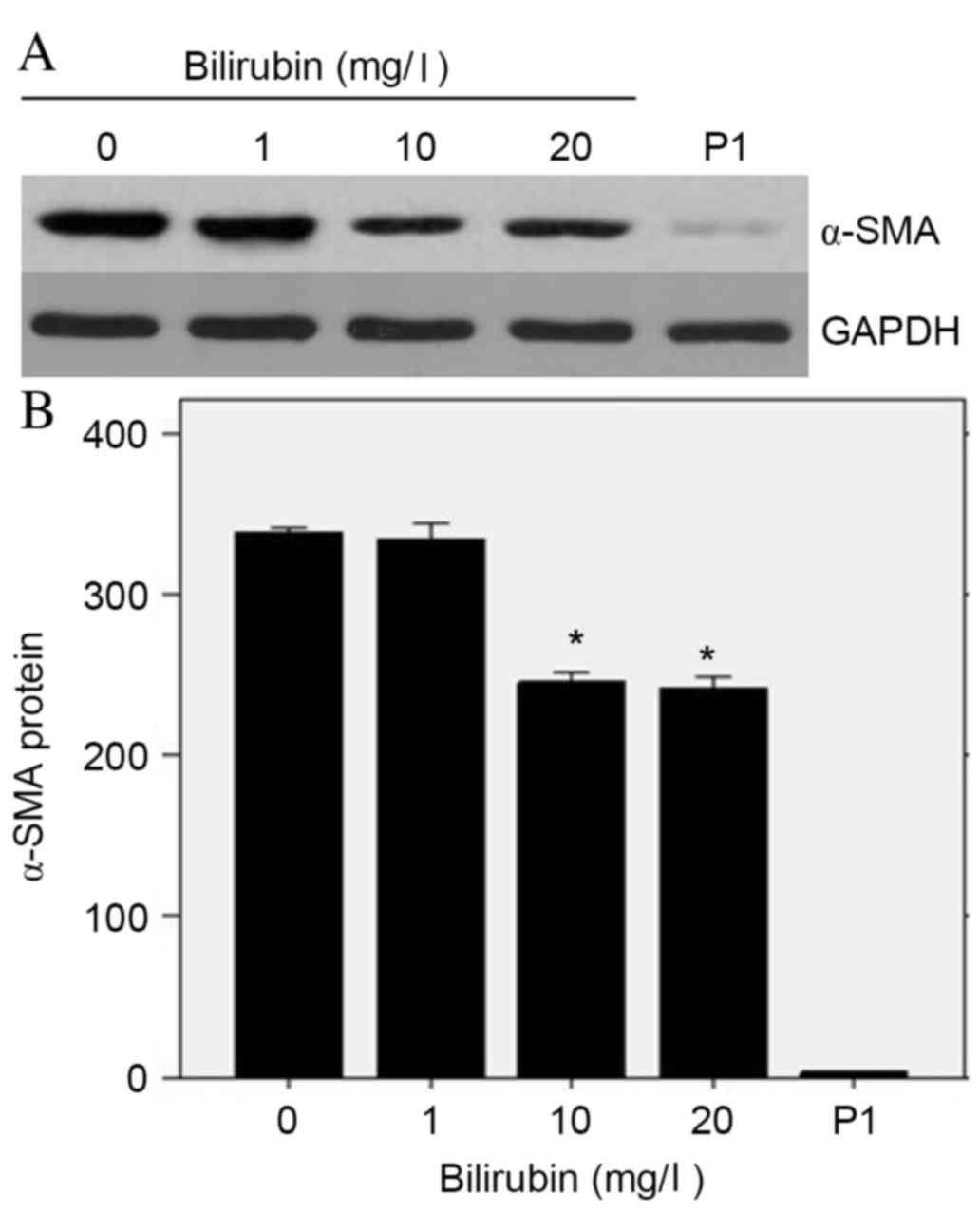
Effect of bilirubin on α-SMA protein
expression levels
The expression levels of α-SMA in HSCs reflect their
transition to myofibroblast-like cells and is therefore a marker of
HSC activation. Western blot analysis demonstrated a dramatic
decrease in α-SMA protein expression levels as bilirubin
concentration increased (Fig. 3A).
At bilirubin concentrations of 0, 1, 10 and 20 mg/l, and passage 1
HSCs, the relative α-SMA protein expression levels were 339±2,
336±10, 246±7, 242±5 and 3.7±0.3, respectively (Fig. 3B). A one-way ANOVA determined the F
ratio of these groups to be 191.107 (P=0.042), thus indicating
significantly reduced expression levels of α-SMA in activated HSCs
treated with bilirubin.
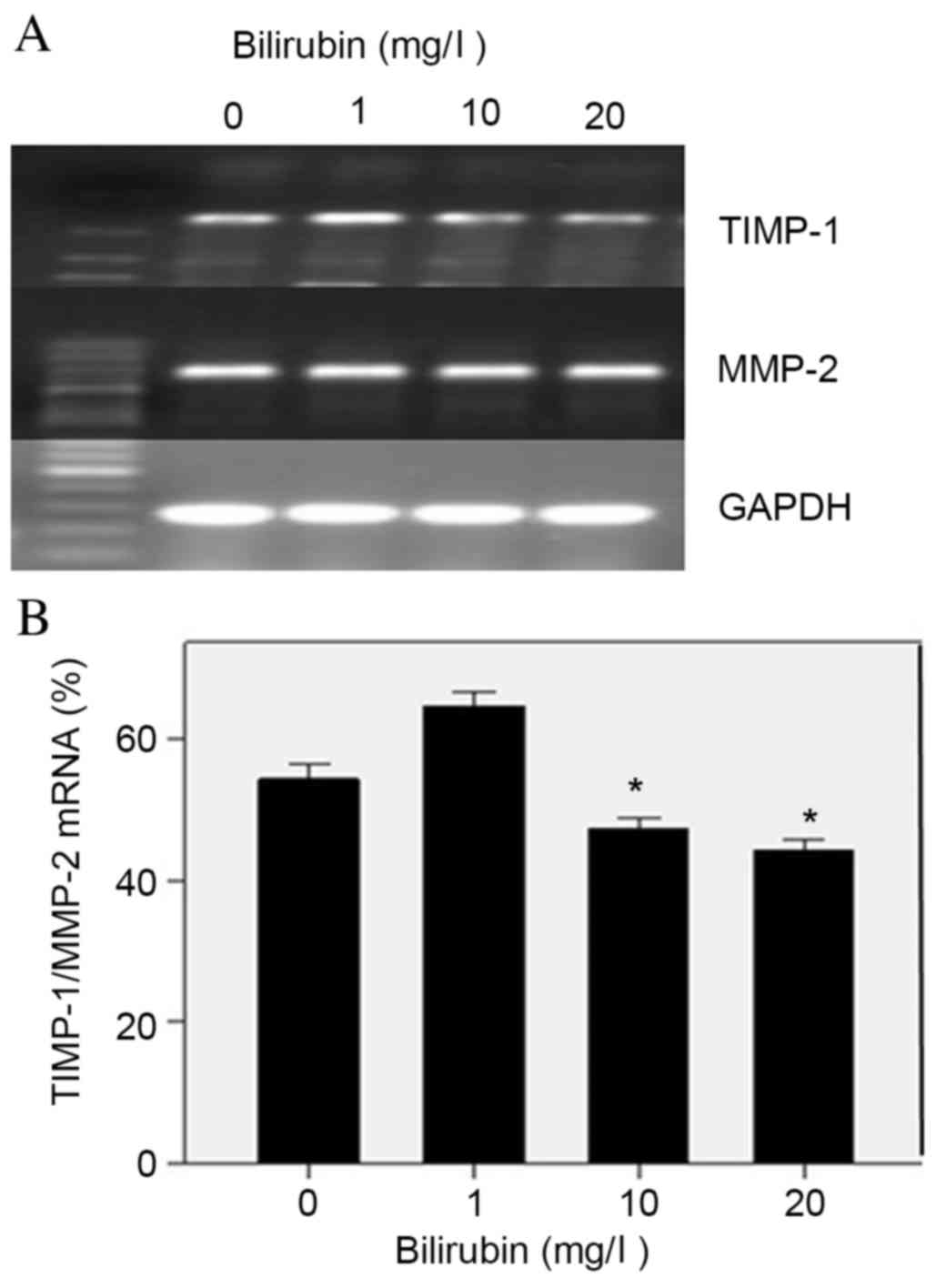
Effect of bilirubin on ECM
remodeling-associated gene expression
Upon activation, HSCs express a combination of MMPs
and TIMPs to remodel the ECM and facilitate the progression of
liver fibrosis. With increased concentrations of bilirubin in HSC
culture, the mRNA expression levels of MMP-2 remained relatively
unaltered, as indicated by RT-PCR (Fig. 4A); however, the mRNA ratio of
TIMP-1/MMP-2 decreased significantly, from 54 to 44% (F=73.4;
P=0.047; Fig. 4B). TIMP-1 may
inhibit the degradation of fibrillary collagen, therefore these
findings indicated that bilirubin may promote ECM degradation
during HSC activation.
Effect of bilirubin on HSC
proliferation
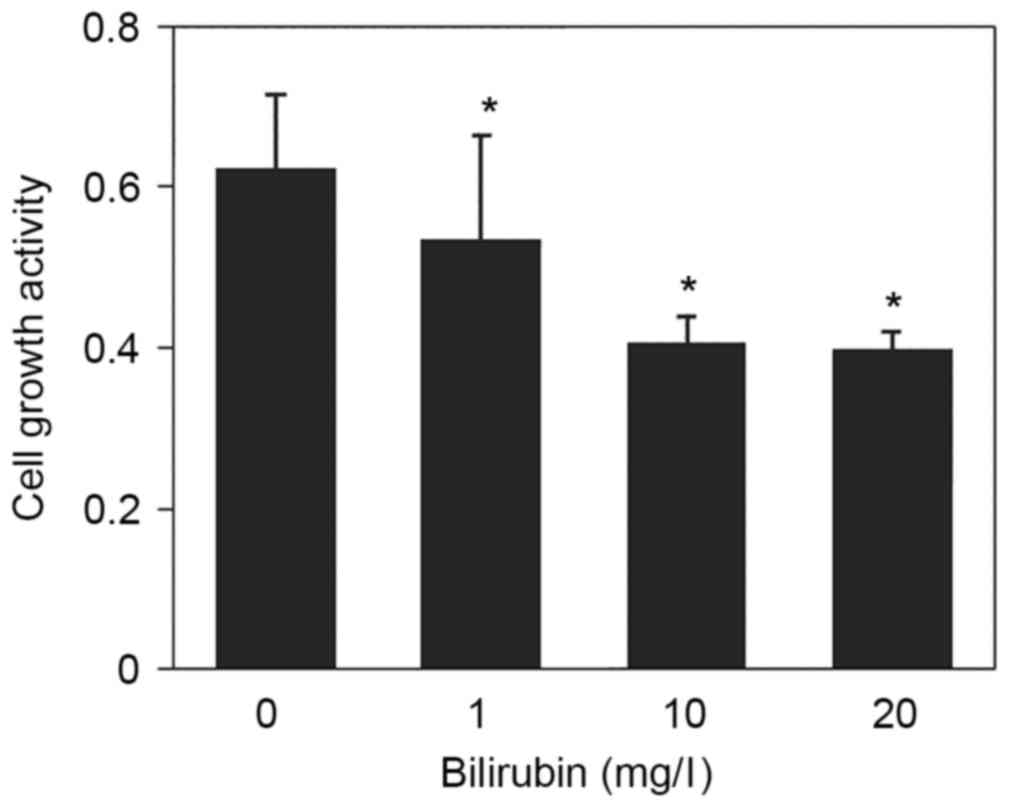
Cell proliferation is an important component of HSC
activation that increases the number of active cells. The effect of
bilirubin on HSC proliferation was examined by CCK-8 assay.
Following treatment with 0, 1, 10 and 20 mg/l bilirubin, HSC
proliferation activity was 0.624±0.092, 0.536±0.127, 0.407±0.033
and 0.399±0.022, respectively (F=13.454; P=0.041; Fig. 5). These results indicated that
increased bilirubin appeared to have an inhibitory effect on HSC
proliferation.
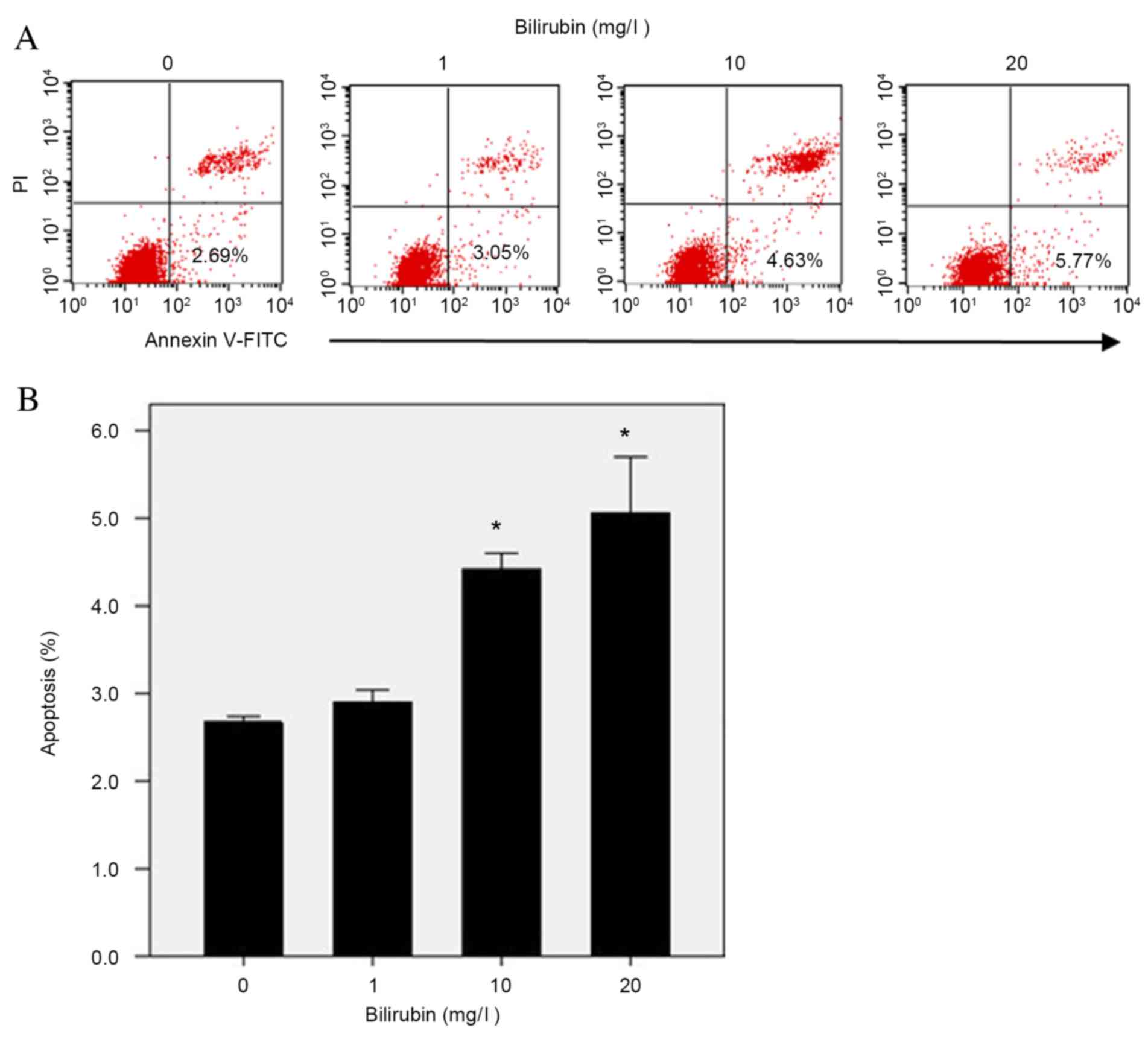
Effect of bilirubin on HSC
apoptosis
Apoptosis of HSCs may account for the decrease in
activated HSCs during the resolution phase of hepatic fibrosis. As
bilirubin concentrations increased from 0 to 20 mg/l, the
percentage of apoptotic HSCs increased from 2.69 to 5.77% (Fig. 6A). Repeated assays demonstrated
that bilirubin treatment resulted in increased apoptosis of HSCs in
a dose-dependent manner (F=34.731; P=0.033; Fig. 6B).
Discussion
To the best of our knowledge, this is the first
study to demonstrate that bilirubin treatment at physiological
concentrations attenuates the activation of rat HSCs in
vitro. Bilirubin treatment exerted numerous effects on HSCs,
including reduced ROS production, decreased protein expression
levels of the HSC activation marker α-SMA and the ECM remodeling
gene TIMP-1, inhibited proliferation and increased apoptosis. These
characteristics indicated inhibition of HSC activation.
HSC activation is a pleiotropic and tightly
regulated response that occurs in a reproducible sequence. It may
be divided into two phases: Initiation and perpetuation (3). The initiation phase is associated
with rapid alterations in gene expression in response to
stimulations from liver injury, including increased expression
levels of the contractile protein α-SMA, and upregulation of MMP-2
and TIMP-1 to remodel the ECM. The perpetuation phase is a dynamic
process, which consists of cellular events that amplify the
activated phenotype, including HSC proliferation. Apoptosis of HSCs
is typical during fibrosis resolution, which facilitates clearance
of the activated HSCs (17). The
results of the present study revealed that bilirubin treatment
altered the gene expression and growth rate of HSCs, thus impacting
the initiation and perpetuation phases of HSC activation.
Furthermore, bilirubin treatment promoted HSC apoptosis, and may
influence the resolution of liver fibrosis.
The underlying molecular mechanisms of
bilirubin-mediated downregulation of HSC activation involve its
antioxidant activity. The primary source of ROS in activated HSCs
is the plasma membrane-associated nicotinamide adenine dinucleotide
phosphate (NADPH) oxidase (18).
HSC cultures from NADPH oxidase-deficient mice were demonstrated to
exhibit reduced ROS production, and following bile duct ligation,
these mice were protected from liver injury and underwent fibrosis
to a lesser degree (12).
Additionally, bilirubin has been revealed to be a potent inhibitor
of NADPH oxidase (19,20). Thus, the inhibition of HSC
activation by bilirubin in the present study may have been mediated
via inhibition of NADPH oxidase activity in HSCs.
Bilirubin is the breakdown product of heme, present
in RBCs. The degradation of heme in humans is catalyzed by the
enzyme heme oxygenase (HO), which is the rate-limiting step in the
formation of bilirubin (21).
Consistent with the present findings, induction of HO expression in
human hepatic myofibroblasts had an antifibrogenic effect,
including inhibition of proliferation and procollagen expression,
which was ascribed to the increased levels of bilirubin (22). Additionally, these results indicate
that bilirubin may attenuate HSC activation in rats and humans.
Furthermore, adeno-associated virus-mediated HO-1 gene transfer was
revealed to suppress the progression of micronodular cirrhosis in
rats (23), suggesting that these
in vitro data may be applicable in vivo, and that
bilirubin may be beneficial for the treatment of hepatic
fibrosis.
Lanone et al (24) demonstrated the potent antioxidant
effects of bilirubin, which appeared to protect against endotoxic
shock in rats by inhibiting NADPH oxidase. However, their study was
conducted in genetically modified jaundiced rats, which limited its
clinical application and significance. Visible jaundice typically
occurs when total bilirubin concentration is >25 mg/l. The
results of the present study were generated in assays using
bilirubin at physiological concentrations, which demonstrated its
dose-dependent effect. Therefore, although cytotoxic at high
concentrations, bilirubin may potentially contribute to
antifibrotic therapies within its physiological levels.
In conclusion, a previous study have suggested that
specific homologs of bilirubin, combined with additional
therapeutic agents, may be beneficial for the clinical treatment of
hepatic fibrosis (19). The
present study demonstrated that low concentrations of bilirubin may
inhibit the activation of isolated HSCs, including ROS production,
fibroblast transition, ECM remodeling, cell proliferation and
apoptosis. Further in vivo studies investigating the effect
of bilirubin on HSC activation and hepatic fibrosis may reveal
potential novel strategies for the use of bilirubin in
antifibrogenic therapy.
Acknowledgements
The present study was supported in part by the
Zhejiang Province Key Surgery Projects (Zhejiang High-Tech; grant.
no. 2008-255) and Zhejiang Provincial Natural Science Foundation
(grant no. LY16H030014).
Glossary
Abbreviations
Abbreviations:
|
HSC
|
hepatic stellate cell
|
|
ROS
|
reactive oxygen species
|
|
SMA
|
smooth muscle actin
|
|
ECM
|
extracellular matrix
|
|
RBCs
|
red blood cells
|
|
FBS
|
fetal bovine serum
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
DCFH-DA
|
2′,7′-dichlorofluorescin diacetate
|
|
FITC
|
fluorescein isothiocyanate
|
|
CCK-8
|
Cell Counting Kit-8
|
|
RT-PCR
|
reverse transcription-polymerase chain
reaction
|
|
MMP-2
|
matrix metalloproteinase-2
|
|
TIMP-1
|
tissue inhibitor of matrix
metalloproteinase-1
|
|
HO
|
heme oxygenase
|
References
|
1
|
Geerts A: History, heterogeneity,
developmental biology, and functions of quiescent hepatic stellate
cells. Semin Liver Dis. 21:311–335. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brandão DF, Ramalho LN, Ramalho FS,
Zucoloto S, Martinelli Ade L and Silva Ode C: Liver cirrhosis and
hepatic stellate cells. Acta Cir Bras. 21 Suppl 1:S54–S57. 2006.
View Article : Google Scholar
|
|
3
|
Li JT, Liao ZX, Ping J, Xu D and Wang H:
Molecular mechanism of hepatic stellate cell activation and
antifibrotic therapeutic strategies. J Gastroenterol. 43:419–428.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Atzori L, Poli G and Perra A: Hepatic
stellate cell: A star cell in the liver. Int J Biochem Cell Biol.
41:1639–1642. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tomaro ML and Batlle AM: Bilirubin: Its
role in cytoprotection against oxidative stress. Int J Biochem Cell
Biol. 34:216–220. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Washington K, Wright K, Shyr Y, Hunter EB,
Olson S and Raiford DS: Hepatic stellate cell activation in
nonalcoholic steatohepatitis and fatty liver. Hum Pathol.
31:822–828. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stocker R, Yamamoto Y, McDonagh AF, Glazer
AN and Ames BN: Bilirubin is an antioxidant of possible
physiological importance. Science. 235:1043–1046. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu TW, Fung KP, Wu J, Yang CC and Weisel
RD: Antioxidation of human low density lipoprotein by unconjugated
and conjugated bilirubins. Biochem Pharmacol. 51:859–862. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamaguchi T, Horio F, Hashizume T, Tanaka
M, Ikeda S, Kakinuma A and Nakajima H: Bilirubin is oxidized in
rats treated with endotoxin and acts as a physiological antioxidant
synergistically with ascorbic acid in vivo. Biochem Biophys Res
Commun. 214:11–19. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hansen TW: Bilirubin production,
breast-feeding and neonatal jaundice. Acta Paediatr. 90:716–717.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gitto E, Reiter RJ, Karbownik M, Tan DX,
Gitto P, Barberi S and Barberi I: Causes of oxidative stress in the
pre- and perinatal period. Biol Neonate. 81:146–157. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bataller R, Schwabe RF, Choi YH, Yang L,
Paik YH, Lindquist J, Qian T, Schoonhoven R, Hagedorn CH, Lemasters
JJ and Brenner DA: NADPH oxidase signal transduces angiotensin II
in hepatic stellate cells and is critical in hepatic fibrosis. J
Clin Invest. 112:1383–1394. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhong Z, Froh M, Wheeler MD, Smutney O,
Lehmann TG and Thurman RG: Viral gene delivery of superoxide
dismutase attenuates experimental cholestasis-induced liver
fibrosis in the rat. Gene Ther. 9:183–191. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ahmad A and Ahmad R: Resveratrol mitigate
structural changes and hepatic stellate cell activation in
N'-nitrosodimethylamine-induced liver fibrosis via restraining
oxidative damage. Chem Biol Interact. 221:1–12. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu FX, Teng YY, Zhu QD, Zhang QY and Tang
YH: Inhibitory effects of capsaicin on hepatic stellate cells and
liver fibrosis. Biochem Cell Biol. 92:406–412. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu F, Su L, Ji S, Zhang S, Yu P, Zheng Y
and Zhang Q: Inhibition of hepatic stellate cell activation and
liver fibrosis by fat-specific protein 27. Mol Cell Biochem.
369:35–43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Elsharkawy AM, Oakley F and Mann DA: The
role and regulation of hepatic stellate cell apoptosis in reversal
of liver fibrosis. Apoptosis. 10:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi SS, Sicklick JK, Ma Q, Yang L, Huang
J, Qi Y, Chen W, Li YX, Goldschmidt-Clermont PJ and Diehl AM:
Sustained activation of Rac1 in hepatic stellate cells promotes
liver injury and fibrosis in mice. Hepatology. 44:1267–1277. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McCarty MF, Barroso-Aranda J and Contreras
F: Genistein and phycocyanobilin may prevent hepatic fibrosis by
suppressing proliferation and activation of hepatic stellate cells.
Med Hypotheses. 72:330–332. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang F, Roberts SJ, Datla Sr and Dusting
GJ: NO modulates NADPH oxidase function via heme oxygenase-1 in
human endothelial cells. Hypertension. 48:950–957. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kikuchi G, Yoshida T and Noguchi M: Heme
oxygenase and heme degradation. Biochem Biophys Res Commun.
338:558–567. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li L, Grenard P, Nhieu JT, Julien B,
Mallat A, Habib A and Lotersztajn S: Heme oxygenase-1 is an
antifibrogenic protein in human hepatic myofibroblasts.
Gastroenterology. 125:460–469. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsui TY, Lau CK, Ma J, Glockzin G, Obed A,
Schlitt HJ and Fan ST: Adeno-associated virus-mediated heme
oxygenase-1 gene transfer suppresses the progression of
micronodular cirrhosis in rats. World J Gastroenterol.
12:2016–2023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lanone S, Bloc S, Foresti R, Almolki A,
Taillé C, Callebert J, Conti M, Goven D, Aubier M, Dureuil B, et
al: Bilirubin decreases nos2 expression via inhibition of NAD(P)H
oxidase: Implications for protection against endotoxic shock in
rats. FASEB J. 19:1890–1892. 2005.PubMed/NCBI
|