Introduction
Diabetes has become one of the three major chronic
diseases, along with hypertension and coronary heart disease, that
seriously endanger human health (1). Diabetic complications, comprising
macroangiopathy and microangiopathy, involve multiple organs,
including the heart, brain, kidney and eyes, and are the primary
underlying causes of increased mortality and disability rates in
patients with diabetes. The basic pathological process of diabetes
is atherosclerosis (2). It has
been reported (3,4) that the level of advanced glycation
end products (AGEs) in vivo was clearly associated with the
severity of diabetic complications and atherosclerosis.
AGEs are formed following long-term exposure to high
levels of sugar by a series of reactions and structural
rearrangements of non-enzymatic glycosylation and lipid oxidation.
While the glycosylation reaction occurs slowly in normal organisms,
the level of AGEs gradually increases with age (5,6).
However, during the aging process, particularly in the case of
long-term high blood glucose, the glycosylation reaction and
subsequent formation of AGEs increases. Thus, it is acknowledged
that this reaction is an inducing factor of diabetic complications
(7). In physiological conditions,
AGEs are degraded to soluble polypeptides by the mononuclear
macrophage system via endocytosis or extracellular protein
degradation systems, and are primarily cleared by the kidneys
(8). However, in pathological
conditions, particularly diabetic complications, AGEs yet to be
eliminated may stimulate the secretion of cytokines from macrophage
and mesangial cells, thus leading to vascular hyperplasia,
mesangial proliferation and glomerular hypertrophy, which are
associated with the development of diabetic nephropathy (9). In addition, AGEs may modify numerous
additional proteins, leading to a number of diseases. Glycosylated
high density lipoprotein cholesterol decreases the ability to
reverse cholesterol transport, as well as increasing the oxidation
of low density lipoprotein cholesterol by reducing the activity of
paraoxonase, which promotes the development of vessel wall damage
(10,11). In addition, AGEs combine with
receptors for advanced glycation end products (RAGE), which leads
to the activation of multiple intracellular signaling pathways and
serves a key role in the pathogenesis of several diseases,
including atherosclerosis (8),
diabetes complications (12),
osteoporosis (13) and cancer
(14,15).
In diabetes complications or atherosclerosis,
increasing evidence suggests that endothelial dysfunction may be a
key factor. Endothelial dysfunction leads to inflammation,
oxidative stress and cell death, which result in vascular
remodeling and subsequent vascular diseases. It has been reported
(16–18) that AGEs bind to RAGE to induce
oxidative stress, inflammation and apoptosis that lead to
endothelium damage. Therefore, exploring the potential mechanism of
AGEs-induced endothelial dysfunction is clearly significant for the
prevention and treatment of diabetic vascular damage.
Mitochondria exist in the majority of cells and
possess their own genome, termed mitochondrial DNA (mtDNA), and a
double-membrane organization that is required to produce energy for
conducting cellular aerobic respiration. In mammals, mtDNA encodes
13 key structural subunits required for the catalytic activity of
four out of five oxidative phosphorylation (OXPHOS) enzyme
complexes I, III, IV and V (19).
The OXPHOS enzyme complexes serve a critical role in electron
transfer in the respiratory chain (20). In addition to providing adenosine
5′-triphosphate (ATP), mitochondria may serve as an important
source of reactive oxygen species (ROS) and regulate various
cellular process, including cell proliferation (21), cell signal transduction and
apoptotic cell death (22).
Mitochondrial-derived ROS (mROS) appear to serve a cell signaling
function when maintained at low levels. However, under pathological
stresses, the production of excessive mROS may contribute to
decreased mitochondrial quality via several interrelated
mechanisms, including mitochondrial calcium overload, increased
levels of ROS, endoplasmic reticulum stress and the accumulation of
aggregated proteins. That oxidative damage impairs the quality of
the mitochondrial population has been demonstrated by the increased
harm to mtDNA, decreased membrane potential and a diminished
bioenergetic reserve capacity that may be conducive to damage of
the complexes (20,23–25).
Nevertheless, cells take immediate measures in response to the
accumulation of impaired mitochondria. According to previous
studies (26,27), two main pathways are involved in
this process. First, intramitochondrial proteases enable
degradation of the oxidized proteins, including ATP-stimulated
mitochondrial Lon protease (27).
The mitochondrial ATPases associated with various cellular
activities have been reported to serve a vital role in preventing
mtDNA escaping from mitochondria to the nucleus ensuring
appropriate fission and fusion, in addition to expeditious
proteolysis of non-assembled inner membrane proteins (27). Second, in response to more
extensive mitochondrial damage, elevated mitochondrial
fragmentation and decreased fusion may separate damaged
mitochondrial proteins, lipids and DNA from functional components,
and the entire damaged organelle is processed by mitophagy
(26). Previously, Kizhakekuttu
et al (28) reported that
mitochondrial dysfunction serves a central role in endothelial
dysfunction in diabetes mellitus, which was evidenced by lower
mitochondrial O2 consumption, mitochondrial membrane
potential, glutathione/glutathione disulfide ratio and higher mROS
production in an additional study (29). Furthermore, 4-hydroxynonenal, one
of the oxidized lipids, contributes to mytocyte injury by
decreasing the bioenergetic reserve capacity (20). Together these results indicate that
mitochondrial energetic metabolism may serve an essential role in
cellular dysfunction. Therefore, the purpose of the present study
was to explore whether AGEs may induce the dysfunction of human
umbilical vein endothelial cells (HUVECs) through mitochondrial
dysfunction and to determine the effect of AGEs on mitochondrial
aerobic respiration and glycolysis.
In summary, the primary aim was to explore the
effect of AGEs on the proliferation of HUVECs and to assess whether
mitochondrial metabolism may serve a key role in this process, as
well as the potential underlying mechanisms involved.
Materials and methods
Materials
A Seahorse metabolic flux analyzer (Seahorse
Bioscience; Agilent Technologies, Inc., Santa Clara, CA, USA),
LSM510 confocal microscope (Zeiss AG, Thornwood, NY, USA),
MicroChemi 4.2 (DNR Bio-Imaging Systems, Ltd., Jerusalem, Israel)
and bovine serum albumin (BSA) and AGEs-BSA (Calbiochem; Merck
KGaA, Darmstadt, Germany) were used in the present study and
obtained from their respective manufacturers. All materials and
reagents for the extracellular flux assays were purchased from
Seahorse Bioscience (Agilent Technologies, Inc.). All antibodies,
staining and additional reagents used in the present study were
obtained from Sigma-Aldrich (Merck KGaA) unless otherwise
specified.
Cell culture and treatment
Primary HUVECs (S200-05N; Sigma-Aldrich; Merck KGaA)
were cultured at 37°C in a humidified atmosphere and 5%
CO2 in Dulbecco's modified Eagle's medium (DMEM;
HyClone; GE Healthcare Life Sciences, Logan, UT, USA) containing
10% fetal bovine serum (FBS; HyClone; GE Healthcare Life Sciences).
HUVECs were incubated for 3 days until 70–80% confluence, before
they were starved in serum-free Basal Medium Eagle (B9638;
Sigma-Aldrich; Merck KGaA) for 24 h. Cells treated with BSA served
as a control. Cells in the AGEs-treated group were incubated with
AGEs-BSA (100 µg/ml) for varying durations (12, 24 and 48 h), while
cells in the control group were treated with BSA (100 µg/ml) for 24
h.
Cell viability assays
For cell viability assays, cells were plated in
96-well dishes (1×104 cells/well) and incubated
overnight. The following day, cells were treated with AGEs in media
containing 10% FBS and incubated for a further 12, 24 and 48 h.
Cell viability was assessed using the MTT assay (Chemicon; EMD
Millipore, Billerica, MA, USA) according to the manufacturer's
protocol. The absorbance was measured with a microplate reader
(Bio-Rad, Hercules, CA, USA) at 570 nm.
Growth curve assays using the
real-time cell analyzer (RTCA)
Growth curve assays were performed in real-time and
in quadruplicate using the xCELLigence system (ACEA Biosciences,
Inc., San Diego, CA, USA). RTCA E-plates (ACEA Biosciences, Inc.)
were seeded with 5,000 cells/well. After all the chambers were set
up, the RTCA E-plates were put into the xCELLigence instrument in
an incubator at 37°C and 5% CO2. Cell growth was
reported as the cell index, which was a dimensionless, relative
measure of impedance reflecting the number of viable adherent
cells, with a consistent logarithmic association with cell number.
Cell index was recorded automatically every 15 min and monitored
continuously for 48 h.
XF Cell Mito Stress test using the
XF24 Extracellular Flux analyzer
An XF24 Analyzer (Seahorse Bioscience; Agilent
Technologies, Inc.) was used to measure bioenergetic function in
HUVECs. The XF24 possesses a transient 2 µl chamber in specialized
microplates that allows for the measurement of the oxygen
consumption rate (OCR) and extracellular acidification rate (ECAR)
or proton production rate in real-time (30). The cells were seeded at a density
of 20,000 cells/well. Following adherence for 4 h, AGEs or BSA was
added to the microplates for co-incubation with cells for 24 h, and
1 µM oligomycin, 0.5 µM carbonyl cyanide-4-(trifluoromethoxy)
phenylhydrazone (FCCP) and 0.5 µM rotenone/antimycin A were
subsequently added. The sensor cartridge was hydrated in XF
Calibrant at 37°C in a non-CO2 incubator overnight. The
culture medium was refreshed at 1 h prior to all bioenergetic
assays using unbuffered DMEM (pH 7.4) supplemented with 4 mM
L-glutamine (Seahorse Bioscience; Agilent Technologies, Inc.).
To test mitochondrial respiration, a XF Cell Mito
Stress Test kit (Seahorse Bioscience; Agilent Technologies, Inc.)
was used according to the manufacturer's protocol. Briefly, 1 µM
oligomycin (final concentration), 0.5 µM FCCP (final concentration)
and 0.5 µM rotenone and antimycin A (final concentration) were
subsequently added to the microplates. This enabled determination
of the basal level of oxygen consumption, ATP-linked oxygen
consumption, non-ATP-linked oxygen consumption, the maximal
respiration capacity and the non-mitochondrial oxygen consumption.
A total of three basal OCR measurements were recorded prior to the
injection of oligomycin. The decreased level of OCR represented
oligomycin-sensitive OCR due to its inhibition of ATP synthase
(complex V). FCCP, an uncoupling protein, was then injected and the
FCCP-stimulated OCR was used to calculate spare respiratory
capacity, which was defined as the difference between maximal
respiration and basal respiration. The third injection was a
mixture of rotenone (a complex I inhibitor) and antimycin A (a
complex III inhibitor). This combination inhibited mitochondrial
respiration completely, and thus no oxygen was further consumed by
cytochrome c oxidase. The remaining OCR measurement obtained
following this treatment was primarily non-mitochondrial and may
have been due to cytosolic oxidase enzymes.
XF Glycolysis Stress Test using the
XF24 Extracellular Flux Analyzer
The XF Glycolysis Stress Test (Seahorse Bioscience;
Agilent Technologies, Inc.) was used to assess glycolysis function
in cells, which was conducted using the XF24 Analyzer. By directly
measuring ECAR, the kit provided a standard method to assess the
following key parameters of glycolysis flux: Glycolysis, glycolytic
capacity and glycolytic reserve, in addition to non-glycolytic
acidification. Cells were seeded at a density of 20,000 cells/well.
Cells were first incubated in pyruvate-free glycolytic assay medium
for 1 h prior to the first injection of a saturated concentration
of glucose (final concentration: 10 mM). The cells catabolize
glucose into pyruvate via the glycolysis pathway, producing ATP,
nicotinamide-adenine dinucleotide (reduced form), water and
protons. The discharge of protons into surrounding medium leads to
a sudden increase in ECAR, which was used to define the basal
glycolytic capacity. The second injection was oligomycin (final
concentration: 1 µM), which may divert energy production to
glycolysis by restricting mitochondrial ATP production.
Consequently, the sharp increase in ECAR indicates the level of
glycolytic capacity. The final injection was 2-deoxy-glucose (2-DG;
final concentration: 50 mM), which is a glucose analog that
inhibits glycolysis through competitive binding to glucose
hexokinase; the first enzyme in the glycolytic pathway. The
resulting decrease in ECAR confirmed that the ECAR produced in the
experiment was caused by glycolysis. The gap between glycolytic
capacity and glycolysis was defined as the glycolytic reserve. The
ECAR prior to glucose injection is referred to as non-glycolytic
acidification, and may occur due to additional processes in the
cell.
Measurement of mitochondrial membrane
potential
Tetraethylrhodamine (TMRE) and MitoTracker Red were
used to detect changes in mitochondrial membrane potential, as
previously described (31,32). Briefly, cells were seeded into a
6-wells plate at a density of 5,000/well. Then cells were cultured
in the presence of either 100 µg/ml BSA or 100 µg/ml AGEs for 24 h.
The old culture media was removed and cells were incubated with 100
nM TMRE or 200 nM MitoTracker Red for 30 min at 37°C and 5%
CO2. In the last 5 min, cells were incubated with
Hoechst 33,342 (final concentration: 8 µg/ml) for 5 min at 37°C and
5% CO2 to stain the nuclei. Following washing with PBS,
HUVECs were analyzed under a fluorescence microscope.
Western blot analysis
Cells were lysed in lysis buffer (20 mM Tris-HCl,
150 mM NaCl, 2 mM EDTA and 1% Triton X-100) containing a protease
inhibitor cocktail (Sigma-Aldrich; Merck KGaA). Cell protein
extracts were quantified using a bicinchoninic acid protein assay
kit (Sigma-Aldrich; Merck KGaA). Equal quantities of protein (20
µg) were separated by 12% SDS-PAGE and transferred to
polyvinylidene difluoride (PVDF) membranes (EMD Millipore,
Billerica, MA, USA). Then the PVDF membranes were blocked with 5%
nonfat milk in Tris-buffered solution (TBS) for 1.5 h at the room
temperature. After washing with 1X TBS (three times, 15 min/time),
the membranes were incubated with primary antibodies against OXPHOS
(ab110413; 1:1,000; Abcam, Cambridge, UK) or GAPDH (AB2302;
1:1,000; EMD Millipore, Billerica, MA, USA) at 4°C overnight. After
washing with 1X TBS (three times, 15 min/time), the membranes were
incubated with horseradish peroxidase-conjugated anti-mouse or
anti-rabbit secondary antibodies (AP181R for anti-mouse and AP187R
for anti-rabbit; 1:10,000; EMD Millipore) for 1.5 h at room
temperature. Finally, enhanced chemiluminescence solutions (GE
Healthcare Life Sciences, Chalfont, UK) was used to detect
immunoreactive binding. The band intensity was quantified with
Image J software version 1.47 (NIH, Bethesda, MD, USA).
Statistical analysis
All data were obtained from >3 independent
experiments. Comparisons between two groups were analyzed using the
unpaired Student's t-test. Comparisons among multiple groups were
analyzed using one-way analysis of variance with Bonferroni post
hoc tests where applicable. All the statistical analyses were
performed using SPSS 17.0 (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
AGEs inhibited the proliferation of
HUVECs
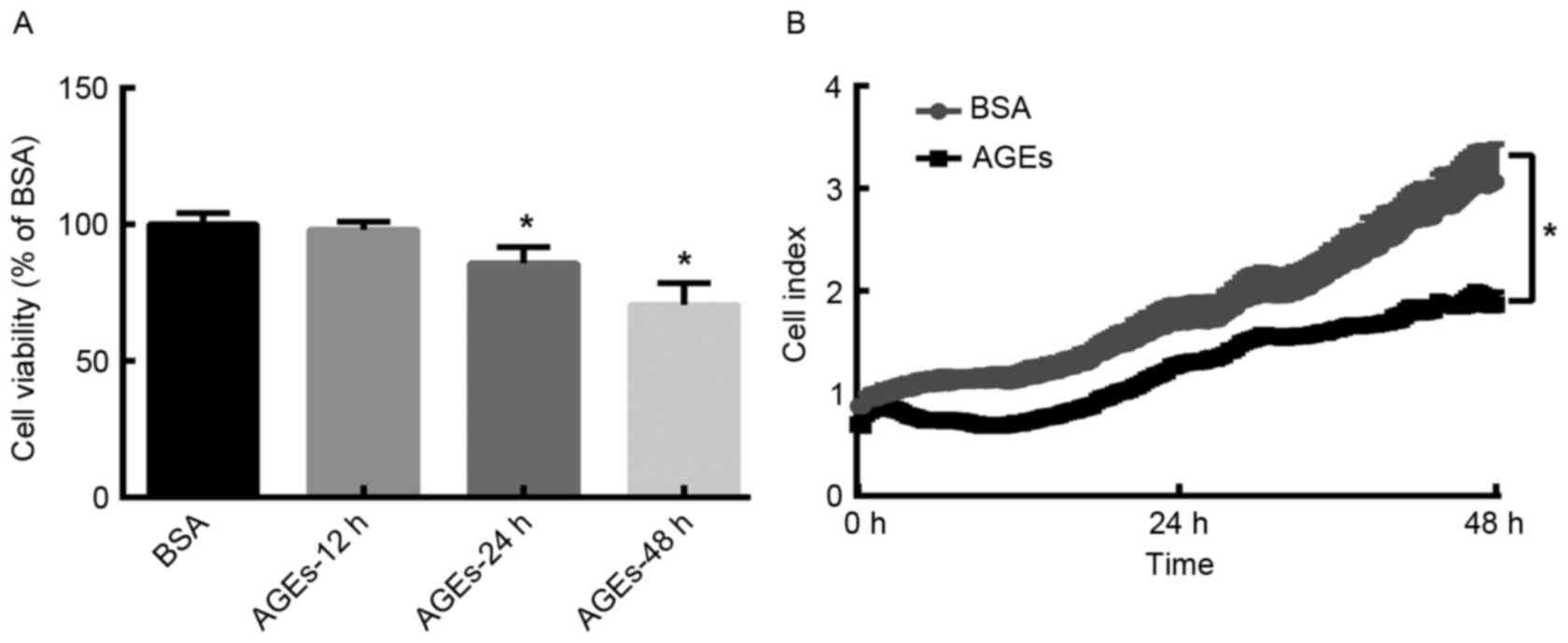
In order to determine the effect of AGEs on the
growth of HUVECs, HUVECs were treated with AGEs (100 µg/ml) or BSA
(100 µg/ml) for 24 h and cell proliferation was assessed using MTT
and RTCA assays. MTT assay analysis demonstrated that the viability
of HUVECs decreased in a time-dependent manner following AGEs
treatment (Fig. 1A). Similarly,
the RTCA proliferation assay demonstrated that the cell index was
significantly lower in AGEs (100 µg/ml)-group when compared with
BSA-group following treatment for 48 h (Fig. 1B). These results suggest that AGEs
reduces the proliferation of HUVECs in a time-dependent manner.
Effect of AGEs on mitochondrial
aerobic metabolism
In order to investigate the specific negative
effects of AGEs on mitochondrial aerobic metabolism in HUVECs, the
XF Cell Mito Stress assay was employed. HUVECs were treated with
AGEs for 24 h prior to exposure to 1 µM oligomycin, 0.5 µM FCCP and
0.5 µM rotenone and antimycin A at various time points. As
demonstrated in Fig. 2, AGEs
reduced the OCR prior to the injection of oligomycin, suggesting
that AGEs prohibited mitochondrial aerobic respiration. In
addition, HUVECs treated with AGEs exhibited an increase in basal
oxygen consumption and proton leakage when compared with the
BSA-treated cells (Fig. 2C). By
contrast, a significant reduction in maximal respiration capacity,
spare respiration capacity and non-mitochondrial respiration was
observed in the AGEs-treated cells when compared with the
BSA-treated cells (Fig. 2C).
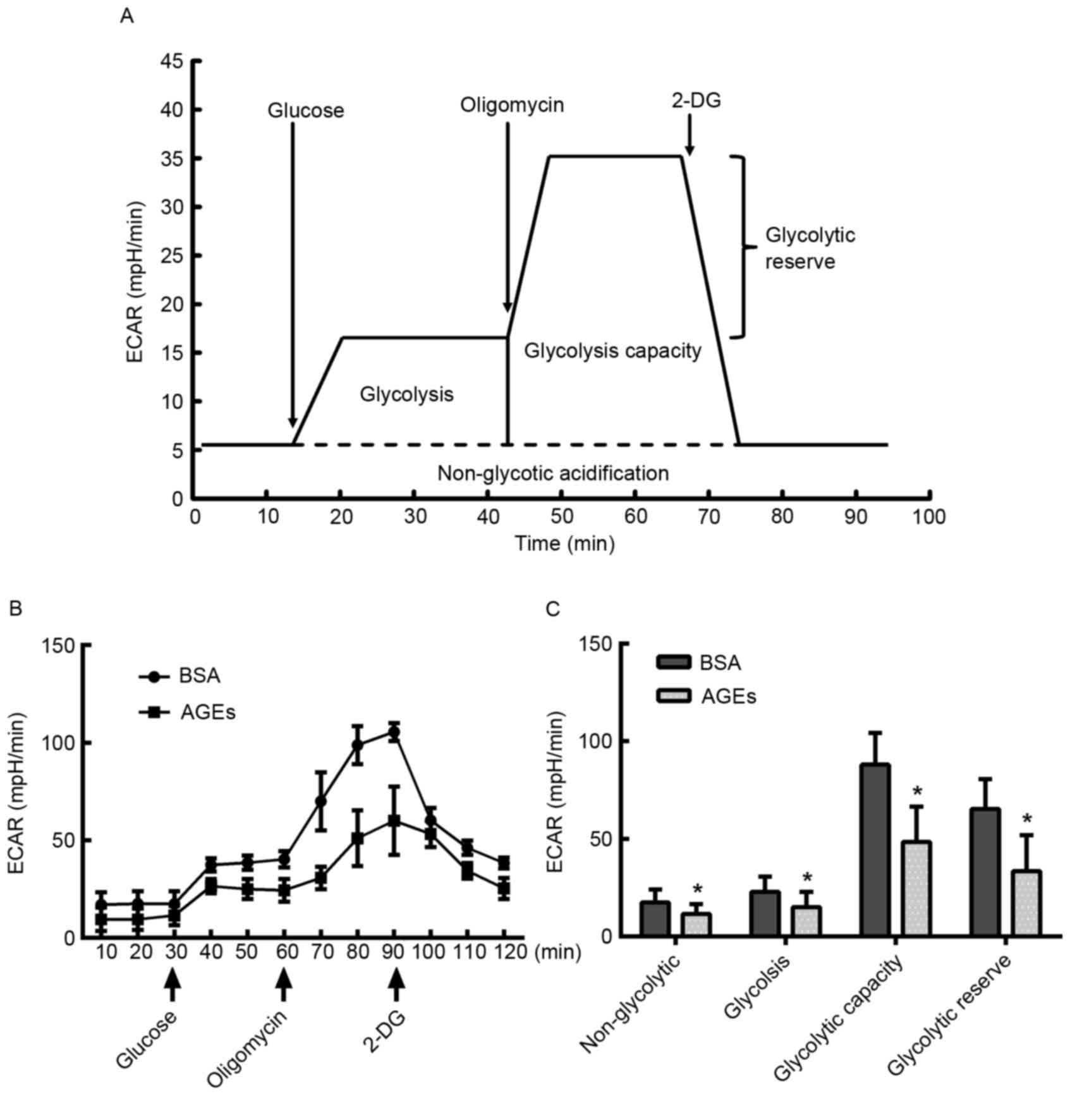
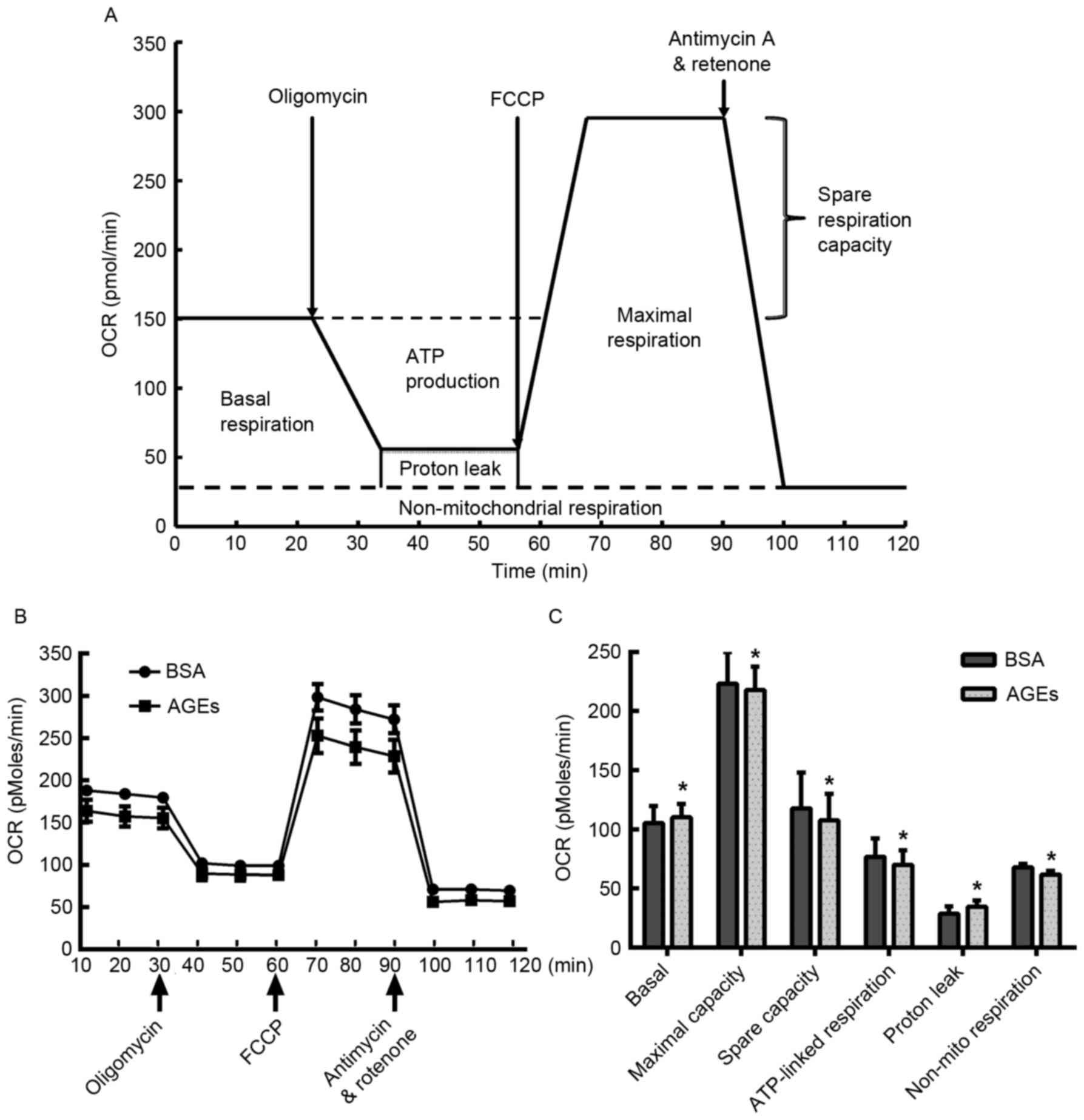 | Figure 2.Measurement of mitochondrial aerobic
respiration profile using the XF24 Extracellular Flux Analyzer. (A)
Schematic of the XF Mito Stress Test used to determine the OCR. (B)
HUVECs were seeded in the Seahorse Bioscience microplates (20,000
cells/well). Following adherence for 4 h, AGEs or BSA was added to
the microplates for co-incubation with cells for 24 h, and 1 µM
oligomycin, 0.5 µM FCCP and 0.5 µM rotenone/antimycin A were
subsequently added. (C) Individual parameters for respiration,
including basal respiration, maximal respiration, spare respiration
capacity, ATP-linked oxygen consumption, proton leakage and
non-mito respiration in HUVECs. Each data point represents an OCR
measurement. Data are presented as the mean ± standard deviation
(n=4). *P<0.05 vs. BSA group. OCR, oxygen consumption rate;
HUVECs, human umbilical vein endothelial cells; AGEs, advanced
glycation end products; BSA, bovine serum albumin; FCCP, carbonyl
cyanide-4-(trifluoromethoxy) phenylhydrazone; ATP, adenosine
5′-triphosphate; Non-mito, non-mitochondrial. |
Effect of AGEs on glycolytic
function
In addition to the OCR, the XF Cell Mito Stress
assay facilitated the measurement of protons that were produced by
HUVEC cells, which is an indicator of lactate production and may
therefore be used as an index of glycolysis (30). HUVECs were treated with AGEs for 24
h prior to exposure to 10 mM glucose, 1 µM oligomycin and 50 mM
2-DG at various time points, as demonstrated in Fig. 3A. AGEs demonstrated an inhibitory
effect on glycolytic function (Fig.
3B). The key parameters of glycolysis, glycolytic capacity,
glycolytic reserve and non-glycolytic acidification were decreased
in the AGEs group compared with the BSA-treated control group,
indicating that AGEs inhibited glycolysis (Fig. 3C).
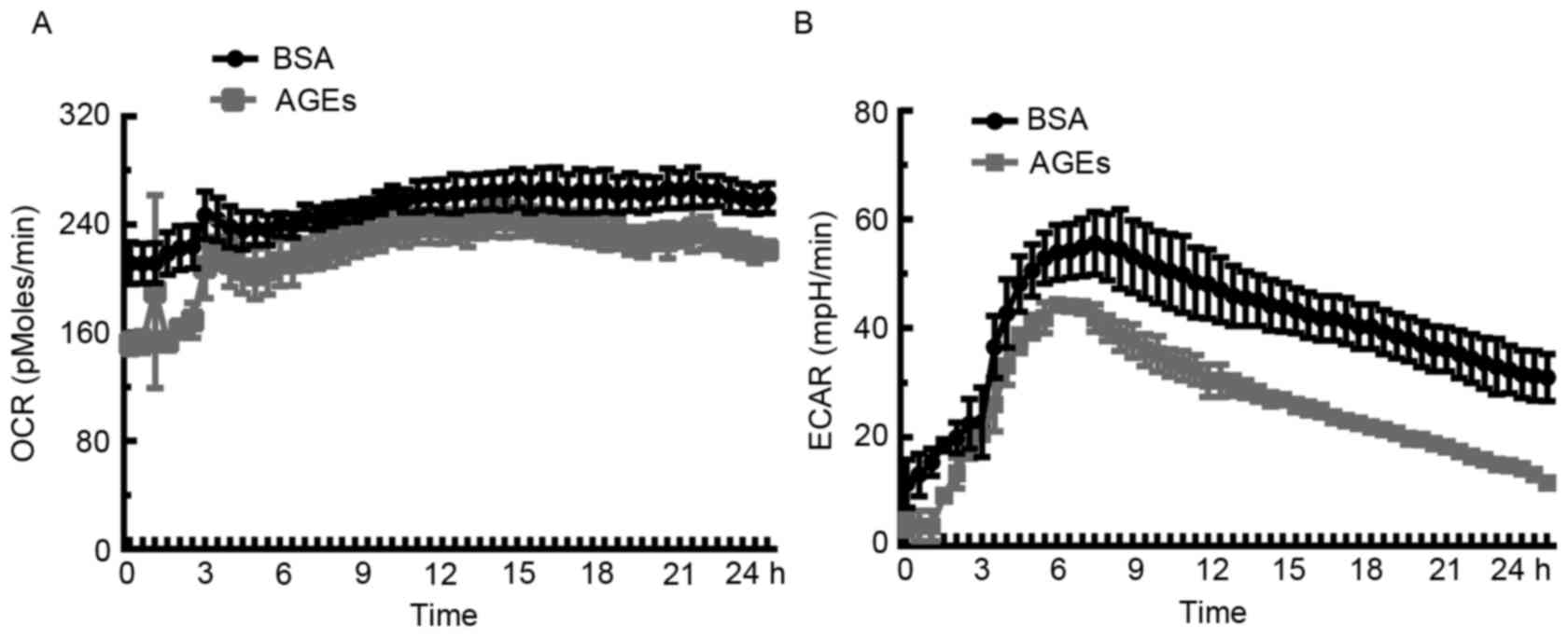
Inhibitory effect of AGEs on OCR and
ECAR in HUVECs
In order to assess the influence of AGEs on
mitochondrial bioenergetic metabolism and anaerobic glycolysis,
extracellular flux analysis was used to determine rates of
O2 consumption. Cells were incubated with AGEs (100
µg/ml) or BSA (100 µg/ml) in the specialized microplates, and the
XF24 assay was performed to measure real-time OCR and ECAR over 24
h. As demonstrated in Fig. 4A, the
OCR decreased in response to AGEs when compared with BSA over the
course of the analysis. Unexpectedly, ECAR in the AGEs-treated
group was lower compared with the BSA-treated cells (Fig. 4B), which suggests that significant
cellular damage or even cell death may occur in HUVECs exposed to
AGEs.
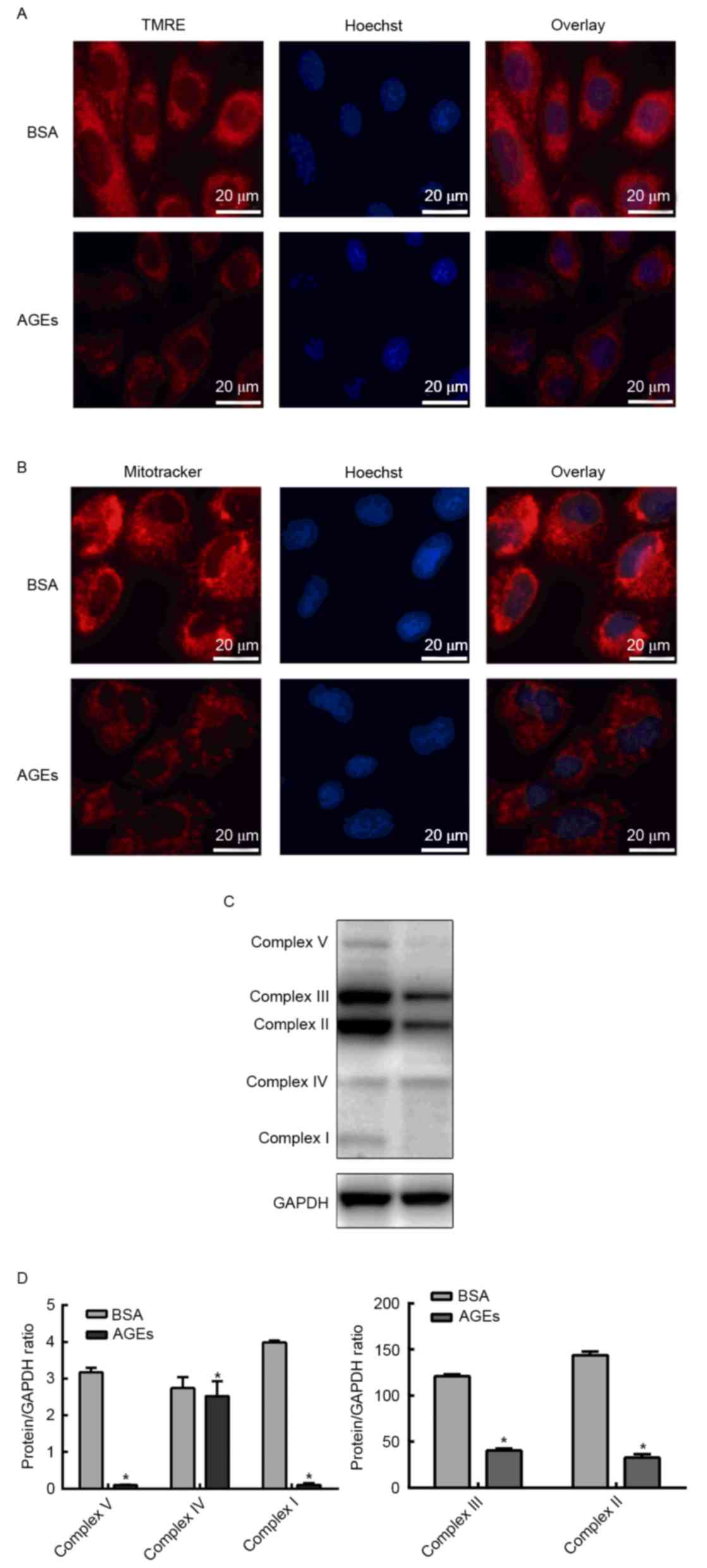
Effect of AGEs on the mitochondrial
membrane potential and mitochondrial respiration chain complex
Since AGEs exhibited an inhibitory effect on aerobic
metabolism, the next aim of the present study was to investigate
whether AGEs affected mitochondrial function. In order to determine
whether AGEs affected the mitochondrial membrane potential, cells
were labeled with TMRE or MitoTracker Red and HUVEC nuclei were
counterstained with Hoechst stain. TMRE and MitoTracker Red
staining demonstrated that the mitochondrial membrane potential
declined following treatment with AGEs (Fig. 5A and B). In order to investigate
the influence of AGEs on mitochondrial respiration chain complexes,
western blotting was performed to detect the protein expression
levels of complexes I–V. Fig. 5C and
D demonstrates that the expression levels of complexes I–V were
significantly reduced in the AGEs group when compared with the BSA
group (Fig. 5C and D).
Discussion
The present study utilized a newly-emerging assay
for the determination of mitochondrial function in intact HUVECs.
To the best of the authors' knowledge, the results of the current
study demonstrate for the first time the precise energy metabolism
and mitochondrial dysfunction elicited by AGEs in endothelial
cells.
AGEs serve a detrimental role in the pathogenesis of
diabetic complications and atherosclerosis (5,33).
The majority of studies (6,34–36)
have demonstrated that AGEs induce harmful effects during
endothelial dysfunction, including inhibited proliferation,
apoptosis, increased inflammatory response and endoplasmic
reticulum and oxidative stress, via various signaling pathways. An
increasing number of studies [reviewed in (37)] have focused on mitochondrial
dysfunction induced by AGEs in endothelial cells. However, few
studies have investigating the effect of AGEs on mitochondrial
function and the precise alterations in energy metabolism in HUVECs
have been conducted. In the present study, extracellular flux
analysis, which is a recent mainstream method for measuring
mitochondrial function in cells and tissues, was used to monitor
the mitochondrial function of intact HUVECs. The results revealed
that AGEs significantly inhibit the viability and proliferation of
HUVECs potentially via an energy deficiency due to decreasing
mitochondrial aerobic respiration and glycolysis.
Mitochondria are the metabolic powerhouses of the
cell, providing ATP for inducing reactions and maintaining core
metabolites for the fundamental survival of cells. Mitochondria
regulate various cellular processes, including proliferation,
apoptosis and ROS generation (21,22).
Under pathological conditions, several interacting mechanisms
facilitate a decline in mitochondrial quality, as evidenced by the
decreased mitochondrial potential, increased mtDNA damage and
destruction of bioenergetic reserve capacity (38). Defects in mitochondrial biogenesis
and dynamics serve a detrimental role in the bioenergy supply and
appear to be a contributor to endothelial cell dysfunction and the
pathogenesis of cardiovascular diseases (39,40).
In the present study, the effect of AGEs on the mitochondrial
aerobic metabolism and mitochondrial membrane potential profiles
were analyzed in order to investigate the mechanism of AGEs-induced
cell dysfunction associated with mitochondrial dysfunction. A
previous review (41) confirmed
that oxidative stress serves a fundamental role in AGEs-induced
cell death. The results of the present study demonstrated that
AGEs-induced oxidative stress elevated proton leakage thus
resulting in a decline of mitochondrial membrane potential. In
addition, the results demonstrated that AGEs may damage the OXPHOS
system encoded by mtDNA. It is possible that the impaired
mitochondrial respiration chain in AGEs-treated HUVECs was unable
to transfer electrons to oxygen, which is the major factor in the
production of ATP, resulting in the observed increase in basal
respiration capacity, ATP-linked oxygen consumption and spare
respiration capacity.
Of particular note, endothelial cells are known to
possess a relatively low mitochondrial content and depend primarily
on glycolysis (42). As energy
deficiency occurs in cells, a high glycolytic flux may yield
increased ATP in a shorter period than OXPHOS in the presence of an
unlimited glucose supply (43).
Previous studies (20,23) demonstrated that stimuli factors
such as ROS and 4-hydroxynonenal significantly augmented the
glycolysis response to the decreased OCR in myocardial cells or
bovine aortic endothelial cells. Unexpectedly, the present study
demonstrated that AGEs negatively affected the glycolysis response.
However, the latent molecular mechanisms underlying the observed
AGEs-induced glycolytic dysfunction require further investigation.
The present study hypothesized that AGEs may affect the activity or
expression of crucial rate-limiting enzymes involved in
glycolysis.
In conclusion, the results of the present study
demonstrated that AGEs inhibited the viability and proliferation of
intact HUVECs by negatively affecting mitochondrial aerobic
respiration and glycolysis. These results may serve as a foundation
for the further study of AGEs-induced endothelial dysfunction,
which may provide a novel perspective for exploring therapeutic
strategies for diabetic vascular complications.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81470417) and the
Natural Science Foundation of Liaoning Province (grant no.
2013021090).
References
|
1
|
Yisahak SF, Beagley J, Hambleton IR and
Narayan KM: IDF Diabetes Atlas: Diabetes in North America and the
Caribbean: An update. Diabetes Res Clin Pract. 103:223–230. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aso Y, Inukai T, Tayama K and Takemura Y:
Serum concentrations of advanced glycation endproducts are
associated with the development of atherosclerosis as well as
diabetic microangiopathy in patients with type 2 diabetes. Acta
Diabetol. 37:87–92. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kiuchi K, Nejima J, Takano T, Ohta M and
Hashimoto H: Increased serum concentrations of advanced glycation
end products: A marker of coronary artery disease activity in type
2 diabetic patients. Heart. 85:87–91. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Genuth S, Sun W, Cleary P, Sell DR, Dahms
W, Malone J, Sivitz W and Monnier VM: DCCT Skin Collagen Ancillary
Study Group: Glycation and carboxymethyllysine levels in skin
collagen predict the risk of future 10-year progression of diabetic
retinopathy and nephropathy in the diabetes control and
complications trial and epidemiology of diabetes interventions and
complications participants with type 1 diabetes. Diabetes.
54:3103–3111. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu L, Zang P, Feng B and Qian Q:
Atorvastatin inhibits the expression of RAGE induced by advanced
glycation end products on aortas in healthy Sprague-Dawley rats.
Diabetol Metab Syndr. 6:1022014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Adamopoulos C, Piperi C, Gargalionis AN,
Dalagiorgou G, Spilioti E, Korkolopoulou P, Diamanti-Kandarakis E
and Papavassiliou AG: Advanced glycation end products upregulate
lysyl oxidase and endothelin-1 in human aortic endothelial cells
via parallel activation of ERK1/2-NF-κB and JNK-AP-1 signaling
pathways. Cell Mol Life Sci. 73:1685–1698. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goldin A, Beckman JA, Schmidt AM and
Creager MA: Advanced glycation end products: Sparking the
development of diabetic vascular injury. Circulation. 114:597–605.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kishikawa H, Mine S, Kawahara C, Tabata T,
Hirose A, Okada Y and Tanaka Y: Glycated albumin and cross-linking
of CD44 induce scavenger receptor expression and uptake of oxidized
LDL in human monocytes. Biochem Biophys Res Commun. 339:846–851.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nangaku M, Miyata T, Sada T, Mizuno M,
Inagi R, Ueda Y, Ishikawa N, Yuzawa H, Koike H, van Ypersele de
Strihou C and Kurokawa K: Anti-hypertensive agents inhibit in vivo
the formation of advanced glycation end products and improve renal
damage in a type 2 diabetic nephropathy rat model. J Am Soc
Nephrol. 14:1212–1222. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thomas MC, Baynes JW, Thorpe SR and Cooper
ME: The role of AGEs and AGE inhibitors in diabetic cardiovascular
disease. Curr Drug Targets. 6:453–474. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Machado AP, Pinto RS, Moysés ZP,
Nakandakare ER, Quintão EC and Passarelli M: Aminoguanidine and
metformin prevent the reduced rate of HDL-mediated cell cholesterol
efflux induced by formation of advanced glycation end products. Int
J Biochem Cell Biol. 38:392–403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Goh SY and Cooper ME: Clinical review: The
role of advanced glycation end products in progression and
complications of diabetes. J Clin Endocrinol Metab. 93:1143–1152.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Steenvoorden MM, Huizinga TW, Verzijl N,
Bank RA, Ronday HK, Luning HA, Lafeber FP, Toes RE and DeGroot J:
Activation of receptor for advanced glycation end products in
osteoarthritis leads to increased stimulation of chondrocytes and
synoviocytes. Arthritis Rheum. 54:253–263. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fuentes MK, Nigavekar SS, Arumugam T,
Logsdon CD, Schmidt AM, Park JC and Huang EH: RAGE activation by
S100P in colon cancer stimulates growth, migration, and cell
signaling pathways. Dis Colon Rectum. 50:1230–1240. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Leclerc E, Fritz G, Weibel M, Heizmann CW
and Galichet A: S100B and S100A6 differentially modulate cell
survival by interacting with distinct RAGE (receptor for advanced
glycation end products) immunoglobulin domains. J Biol Chem.
282:31317–31331. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim S and Kwon J: Actin cytoskeletal
rearrangement and dysfunction due to activation of the receptor for
advanced glycation end products is inhibited by thymosin beta 4. J
Physiol. 593:1873–1886. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Del Turco S, Navarra T, Gastaldelli A and
Basta G: Protective role of adiponectin on endothelial dysfunction
induced by AGEs: A clinical and experimental approach. Microvasc
Res. 82:73–76. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin J, Tang Y, Kang Q, Feng Y and Chen A:
Curcumin inhibits gene expression of receptor for advanced
glycation end-products (RAGE) in hepatic stellate cells in vitro by
elevating PPARγ activity and attenuating oxidative stress. Br J
Pharmacol. 166:2212–2227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hill BG, Benavides GA, Lancaster JR Jr,
Ballinger S, Dell'Italia L, Jianhua Z and Darley-Usmar VM:
Integration of cellular bioenergetics with mitochondrial quality
control and autophagy. Biol Chem. 393:1485–1512. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hill BG, Dranka BP, Zou L, Chatham JC and
Darley-Usmar VM: Importance of the bioenergetic reserve capacity in
response to cardiomyocyte stress induced by 4-hydroxynonenal.
Biochem J. 424:99–107. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mitra K, Wunder C, Roysam B, Lin G and
Lippincott-Schwartz J: A hyperfused mitochondrial state achieved at
G1-S regulates cyclin E buildup and entry into S phase. Proc Natl
Acad Sci USA. 106:11960–11965. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dranka BP, Hill BG and Darley-Usmar VM:
Mitochondrial reserve capacity in endothelial cells: The impact of
nitric oxide and reactive oxygen species. Free Radic Biol Med.
48:905–914. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Higdon AN, Benavides GA, Chacko BK, Ouyang
X, Johnson MS, Landar A, Zhang J and Darley-Usmar VM: Hemin causes
mitochondrial dysfunction in endothelial cells through promoting
lipid peroxidation: The protective role of autophagy. Am J Physiol
Heart Circ Physiol. 302:H1394–H1409. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ballinger SW, Patterson C, Knight-Lozano
CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R,
Hunter GC, et al: Mitochondrial integrity and function in
atherogenesis. Circulation. 106:544–549. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee J, Giordano S and Zhang J: Autophagy,
mitochondria and oxidative stress: Cross-talk and redox signalling.
Biochem J. 441:523–540. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hori O, Ichinoda F, Tamatani T, Yamaguchi
A, Sato N, Ozawa K, Kitao Y, Miyazaki M, Harding HP, Ron D, et al:
Transmission of cell stress from endoplasmic reticulum to
mitochondria: Enhanced expression of Lon protease. J Cell Biol.
157:1151–1160. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kizhakekuttu TJ, Wang J, Dharmashankar K,
Ying R, Gutterman DD, Vita JA and Widlansky ME: Adverse alterations
in mitochondrial function contribute to type 2 diabetes
mellitus-related endothelial dysfunction in humans. Arterioscler
Thromb Vasc Biol. 32:2531–2539. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hernandez-Mijares A, Rocha M,
Rovira-Llopis S, Bañuls C, Bellod L, de Pablo C, Alvarez A,
Roldan-Torres I, Sola-Izquierdo E and Victor VM: Human
leukocyte/endothelial cell interactions and mitochondrial
dysfunction in type 2 diabetic patients and their association with
silent myocardial ischemia. Diabetes Care. 36:1695–1702. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ferrick DA, Neilson A and Beeson C:
Advances in measuring cellular bioenergetics using extracellular
flux. Drug Discov Today. 13:268–274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gottlieb E, Armour SM, Harris MH and
Thompson CB: Mitochondrial membrane potential regulates matrix
configuration and cytochrome c release during apoptosis. Cell Death
Differ. 10:709–717. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lei T, Guo N, Tan MH and Li YF: Effect of
mouse oocyte vitrification on mitochondrial membrane potential and
distribution. J Huazhong Univ Sci Technolog Med Sci. 34:99–102.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luft VC, Duncan BB, Schmidt MI, Chambless
LE, Pankow JS, Hoogeveen RC, Couper DJ and Heiss G: Carboxymethyl
lysine, an advanced glycation end product, and incident diabetes: A
case-cohort analysis of the ARIC Study. Diabet Med. 33:1392–1398.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sang HQ, Gu JF, Yuan JR, Zhang MH, Jia XB
and Feng L: The protective effect of Smilax glabra extract on
advanced glycation end products-induced endothelial dysfunction in
HUVECs via RAGE-ERK1/2-NF-κB pathway. J Ethnopharmacol.
155:785–795. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rempel LC, Finco AB, Maciel RA, Bosquetti
B, Alvarenga LM, Souza WM, Pecoits-Filho R and Stinghen AE: Effect
of PKC-β signaling pathway on expression of MCP-1 and VCAM-1 in
different cell models in response to advanced glycation end
products (AGEs). Toxins (Basel). 7:1722–1737. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Solé M, Miñano-Molina AJ and Unzeta M:
Cross-talk between Aβ and endothelial SSAO/VAP-1 accelerates
vascular damage and Aβ aggregation related to CAA-AD. Neurobiol
Aging. 36:762–775. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tang X, Luo YX, Chen HZ and Liu DP:
Mitochondria, endothelial cell function, and vascular diseases.
Front Physiol. 5:1752014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brand MD and Nicholls DG: Assessing
mitochondrial dysfunction in cells. Biochem J. 435:297–312. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shenouda SM, Widlansky ME, Chen K, Xu G,
Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, et
al: Altered mitochondrial dynamics contributes to endothelial
dysfunction in diabetes mellitus. Circulation. 124:444–453. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ong SB, Subrayan S, Lim SY, Yellon DM,
Davidson SM and Hausenloy DJ: Inhibiting mitochondrial fission
protects the heart against ischemia/reperfusion injury.
Circulation. 121:2012–2022. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Schleicher E and Friess U: Oxidative
stress, AGE, and atherosclerosis. Kidney Int Supp. S17–S26. 2007.
View Article : Google Scholar
|
|
42
|
Groschner LN, Waldeck-Weiermair M, Malli R
and Graier WF: Endothelial mitochondria-less respiration, more
integration. Pflugers Arch. 464:63–76. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Eelen G, de Zeeuw P, Simons M and
Carmeliet P: Endothelial cell metabolism in normal and diseased
vasculature. Circ Res. 116:1231–1244. 2015. View Article : Google Scholar : PubMed/NCBI
|