Introduction
Pyelonephritis, one of the most serious forms of
urinary tract infection (UTI), is an infection of the upper urinary
tract involving bacterial invasion of the renal parenchyma, and is
usually caused by the ascent of bacteria from the bladder to the
renal medulla via the ureters (1,2). Up
to a quarter of the hospitalizations for UTI involve infection of
the kidney or pyelonephritis, causing serious conditions including
sepsis and septic shock (3).
Neutrophil infiltration is a characteristic pathological change in
pyelonephritis. Neutrophils have been implicated in the
antibacterial defense of the urinary tract, and neutrophil
recruitment to the infected urinary tract is initiated when
bacteria stimulate the epithelial cells to secrete chemokines and
express chemokine receptors (4).
Although neutrophils are essential to host defense, they have also
been implicated in the pathology of many inflammatory conditions
and tissue injury (5). Neutrophil
recruitment is an important early step in controlling tissue
infections or injury, and it promotes early and robust inflammation
(6).
Since neutrophil recruitment can cause severe tissue
damage, understanding neutrophil trafficking mechanisms is
important to attenuate neutrophil-mediated damage. Several
neutrophil chemoattractants have been characterized, of which CXCL2
also known as macrophage inflammatory protein-2 (MIP-2) is
especially important (7). MIP-2 is
a member of a family of cytokines that play roles in inflammatory,
immune, and wound healing responses (8). MIP-2 is required for peripheral
neutrophil migration across the epithelium into the urine and can
be produced not only by non-immune cells such as epithelial cells
and fibroblasts (9), but also by
immune cells including dendritic cells (10). Several signaling pathways such as
PI3K and NF-κB/ERK are associated with MIP-2 production during
inflammatory condition (11,12).
p38 MAPK, one of three distinct families of MAPKs (p42/44 or ERK
kinase, JNK kinase, and p38 kinase), is a critical enzyme for
cytokine TNF production and is currently targeted for
anti-inflammatory therapy (13).
p38 MAPK was significantly activated and up-regulated by both acute
and chronic cigarette smoke exposure in C57BL/6 mouse model with
obstructive pulmonary disease. Inhibition of p38 MAPK signaling
pathway profoundly attenuated cigarette smoke induced lung
inflammation, which was evidenced by the reduced infiltration of
neutrophils and concentration of MIP-2 in the lung after
intra-peritoneally administration of p38 MAPK inhibitor (14). These data are in accordance with
the earlier discovery or inhibition of p38 MAPK prevents neutrophil
chemoattractant production and blocks chemotaxis of neutrophils
(15).
Escherichia coli (E. coli) is the most common
pathogen for pyelonephritis, and the severity of pyelonephritis
caused by E. coli is due to the expression of a wide range
of virulence factors. Many virulence factors play key roles in the
pathogenicity of these E. coli strains termed uropathogenic
E. coli (UPEC) (16–18).
TcpC, a novel virulence factor of extraintestinal pathogenic E.
coli, inhibits toll-like receptor (TLR) and MyD88-specific
signaling, thus impairs the innate immune response, promotes
bacterial survival and increases the severity of UTIs in humans and
mice (19–21). TcpC is common in the most virulent
UPEC strains, it can also directly interact with the NACHT
leucin-rich repeat PYD protein 3 (NLRP3) inflammasome and
caspase-1, hereby inhibiting the activation of NLRP3 inflammasome
and caspase-1, leading to the reduction of IL-1β production
(22). However, the influence of
TcpC on MIP-2 production by kidney cells remains elucidative. In
the present study, we showed that TcpC could promote MIP-2
production and neutrophil recruitment in renal cells via p38
activation, which might contribute to the pathogenesis of
pyelonephritis.
Materials and methods
Mice and reagents
C57BL/6 mice (H-2b), 8–10 weeks of age,
were purchased from SLC Laboratory Animal Co., Ltd. (Shanghai,
China), and were housed in specific pathogen-free conditions. All
the animal procedures were performed according to the proper used
and care of laboratory animals by the Institutional Committee. The
primary antibodies against p38, p-p38 (Thr180/Tyr182), p-JNK1/2,
p-ERK1/2, PI3K, β-actin, and horseradish peroxidase (HRP)-labeled
secondary anti-mouse and anti-rabbit antibodies were purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). The p38 MAPK
inhibitor SB203580 was purchased from Selleck Chemicals (Houston,
TX, USA) and dissolved in dimethyl sulfoxide (DMSO) (10 mM stock
solution) and stored at −20°C. TcpC expressing uropathogenic E.
coli strain CFT073 (TcpCwt) was kindly provided by
Professor Jian-Guo Xu (State Key Laboratory for Infectious Disease
Prevention and Control, National Institute for Communicable Disease
Control and Prevention, China). The tcpC-knock out CFT073
strain (TcpC−/−) was constructed by λ red homologous
recombination as described in our previous published paper
(23). Rabbit anti-TcpC polyclonal
antibody (IgG) was produced in our laboratory. Rabbit IgG isotype
control was purchased from Beyotime Biotechnology (Shanghai,
China).
Cell culture
Human embryonic kidney cell line (HEK-293) was
purchased from Shanghai Institute of Biochemistry and Cell Biology
(Shanghai, China), and the genotypes were authenticated by DNA
fingerprinting. HEK-293 was grown in DMEM medium containing 10%
fetal bovine serum (FBS) plus 2 mM glutamine and 50 units/ml
penicillin in a humidified atmosphere of 5% CO2 at
37°C.
Transwell co-culture
Co-culture of HEK-293 cells with wild-type E.
coli CFT073 (TcpCwt) and TcpC−/− was
performed in transwell system (Corning, NY, USA) as previously
described (24). Briefly, 1 ml
HEK-293 cell suspension (5.0×105 cells) was added to the
lower compartment of the transwell, and 0.2 ml containing different
numbers of TcpCwt or TcpC−/− was added to the
upper compartment of the transwell (0.4 µm transwell filters). To
conform the influence of TcpCwt on the MIP-2 production
was caused by the TcpC secreted by TcpCwt,
5.0×105 HEK-293 cells were separately co-cultured in
transwell system with or without TcpCwt at multiplicity
of infection (MOI)=1 in the presence or absence of 10 µg/ml of
rabbit anti-TcpC polyclonal antibodies or rabbit IgG isotype
control for 15 h. To examine the influence of p38 MAPK inhibitor on
the production of MIP-2, HEK-293 cells were pretreated with
SB203580 (1.25 µM) or solvent DMSO for 1 h and then co-cultured
with TcpCwt or TcpC−/− for 15 h. Culture of
HEK-293 cells without bacteria served as the blank control.
Mouse pyelonephritis model
TcpCwt and TcpC−/− were grown
in LB medium and harvested by centrifugation at 4,000 × g for 5 min
and resuspended in 1 ml of LB medium to a final concentration of
1×1011 CFU/ml. Female C57BL/6 mice of 8 to 10 weeks of
age were anesthetized with Avertin [40 mg of 2,2,2-tribromoethanol
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) dissolved in 1 ml
of tertamyl alcohol (0.01 ml/g body weight intraperitoneally)] and
infected by transurethral instillation of 0.1 ml of the bacterial
suspension or LB medium using a flexible polyethylene catheter. The
infected mice were sacrificed 3 days later and the kidneys were
obtained for histological analysis and detection of MIP-2.
Histological analysis
Paraformaldehyde-fixed and paraffin-embedded kidneys
were sectioned at 4 µm and stained with hematoxylin and eosin using
the standard method. These sections were observed with a light
microscopy to determine the histological changes in kidneys from
the control, TcpCwt and TcpC−/−-treated
mice.
Detection of MIP-2 in the culture
supernatants and in the homogenates of kidney
Concentrations of MIP-2 in the culture supernatants
and kidney homogenates were determined by using commercially
available ELISA kits (R&D Systems, Minneapolis, MN, USA and
Abcam, Cambridge, MA, USA, respectively) according to the
manufacturer's instructions. To make homogenates of kidney, a 200
mg portion of the left kidney of the mouse from different groups
was homogenized in 1 ml PBS. After centrifugation at 8,000 × g for
10 min at 4°C, the supernatant was collected and stored at −20°C
for measurement of MIP-2.
Cell lysate preparation and western
blot analysis
Proteins were extracted with lysis buffer (50 mM
Tris-HCl, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 0.5% deoxycholic acid,
0.02% sodium azide, 1% NP-40, 2.0 µg/ml aprotinin, 1 mM
phenylmethylsulfonylfluoride). The lysates were centrifuged at
10,000 × g for 30 min at 4°C, the supernatants were transferred to
a new tube, and the protein concentration was determined. Protein
samples were fractionated on 8 to 15% Tris-glycine gels, followed
by proteins transfer onto a PVDF membrane (Millipore, Bedford, MA,
USA). The membranes were blocked with 5% non-fat dry milk and then
probed with primary antibodies (dilution range, 1:500-1:1,000),
followed by HRP-labeled secondary antibodies at a 1:5,000 dilution.
Antibody binding was visualized with a chemiluminescent substrate
and visualized on autoradiography film.
Neutrophil isolation and chemotaxis
assay
Neutrophils were isolated as described (25). Briefly, venous blood of healthy
volunteers was collected on anticoagulant 3.8% sodium citrate
solution (blood to sodium citrate is 9:1 in volume), centrifuged
500 × g for 15 min and the resulting platelet-rich plasma was
discarded. The leucocyte-rich upper layer was then aspirated,
placed on top of a Percoll (Sigma-Aldrich) step gradient (60 and
75% Percoll in PBS) and centrifuged at 2,000 rpm for 15 min. The
enriched neutrophil population was recovered at the interface
between 60 and 75% Percoll. The purified neutrophils were
resuspended in RPMI 1640 containing 10% FBS medium and used freshly
for migration assays. Neutrophil migration was carried out in a
transwell system. Briefly, cell culture inserts (8 µm pore size)
were used to form dual compartments (upper and bottom chamber) in a
24-well culture plate. Freshly purified human peripheral blood
neutrophils were loaded into the upper chamber (cell culture
insert) and their migration was initiated by the addition of the
MIP-2 containing culture supernatants to the bottom chamber in a
24-well plate. Neutrophil migration was carried out for 12 h at
37°C and 5% CO2. The migrated neutrophils were collected
from the bottom chambers and counted with a hemacytometer.
Neutrophil migration to the supernatants of HEK-293 cells treated
with TcpCwt was set as 100%.
Statistical analysis
All the experiments were performed at least three
times, and results were expressed as mean ± SD. Two-tailed
Student's t-test was used to determine the significance of the
differences between the experimental conditions. Differences were
considered significant at P<0.05.
Results
TcpC plays an important role in the
pathogenesis of pyelonephritis
Since neutrophils are involved in the pathology of
various inflammatory conditions (25), and neutrophil infiltration is a
characteristic pathological change of acute pyelonephritis
(10,19), we examined the histological changes
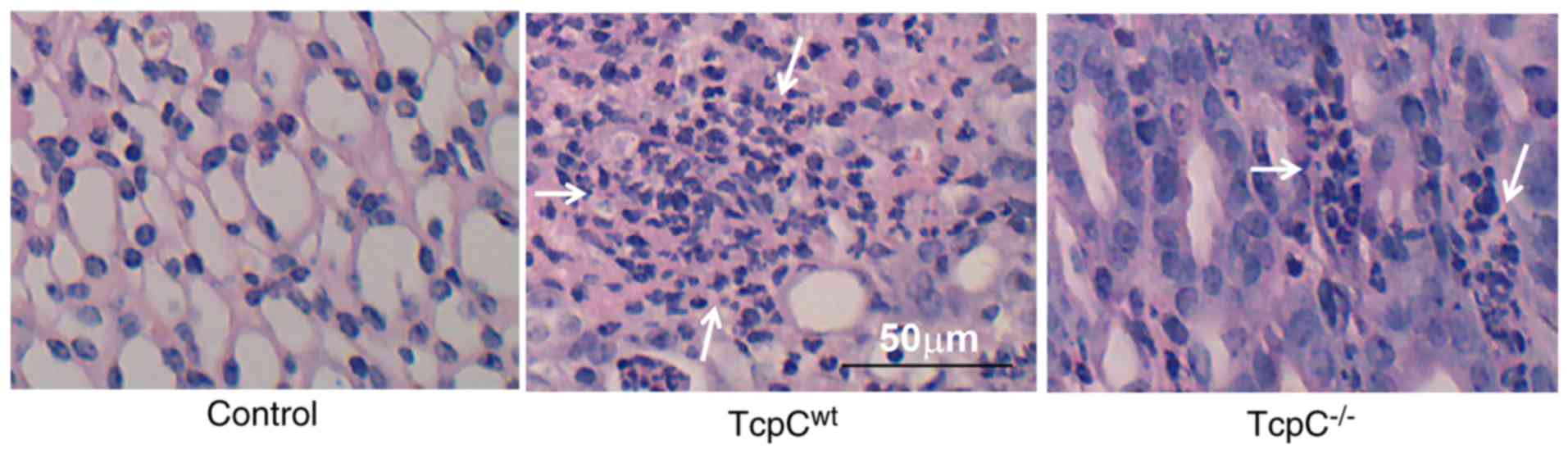
of kidneys from mice models with pyelonephritis. As shown in
Fig. 1, in accordance with
previous reports (10,19), few neutrophils were seen in kidneys
of C57BL/6 mice infected by TcpC−/−, but large numbers
of neutrophils were infiltrated in kidneys from mice infected by
TcpCwt, indicating TcpC plays an important role in the
pathogenesis of pyelonephritis.
TcpC promoted MIP-2 production in vivo
and in vitro
Because MIP-2 exhibits potent neutrophil chemotactic
activity and plays a major role in mediating the neutrophilic
inflammation (9), we detected the
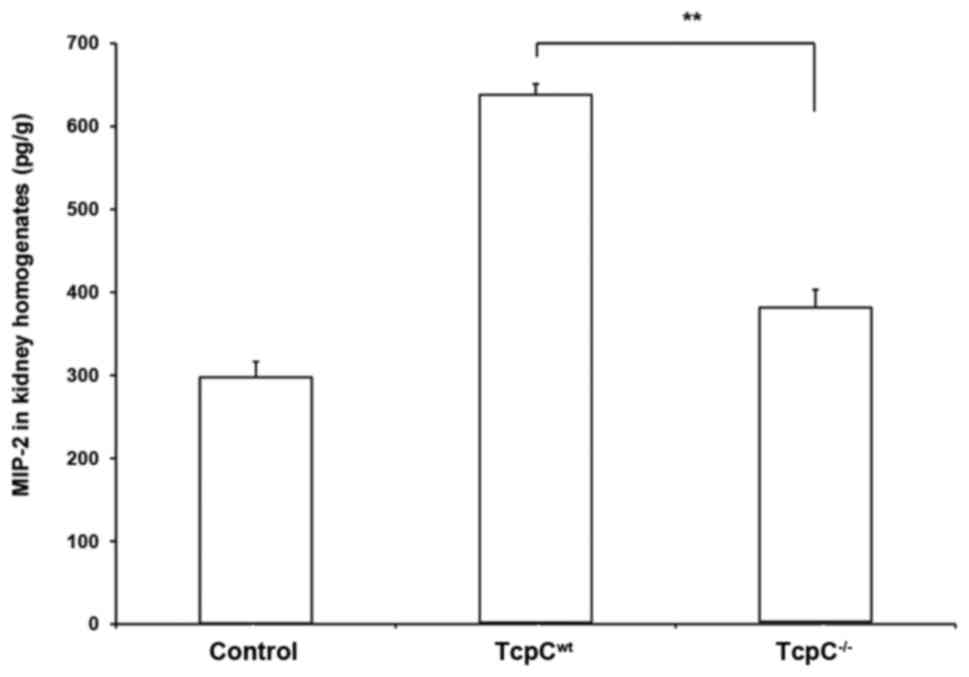
levels of MIP-2 in kidney homogenates of pyelonephritis mouse
models caused by TcpCwt or TcpC−/−. A marked
MIP-2 in vivo response to infection of TcpCwt but
a weak response to infection of TcpC−/− in the C57BL/6
mice can be observed (Fig. 2).
Concentration of MIP-2 in kidney homogenates of
TcpCwt-caused pyelonephritis was significantly higher
than that in kidney homogenates of TcpC−/−-caused
pyelonephritis (P<0.01). In order to examine the in vitro
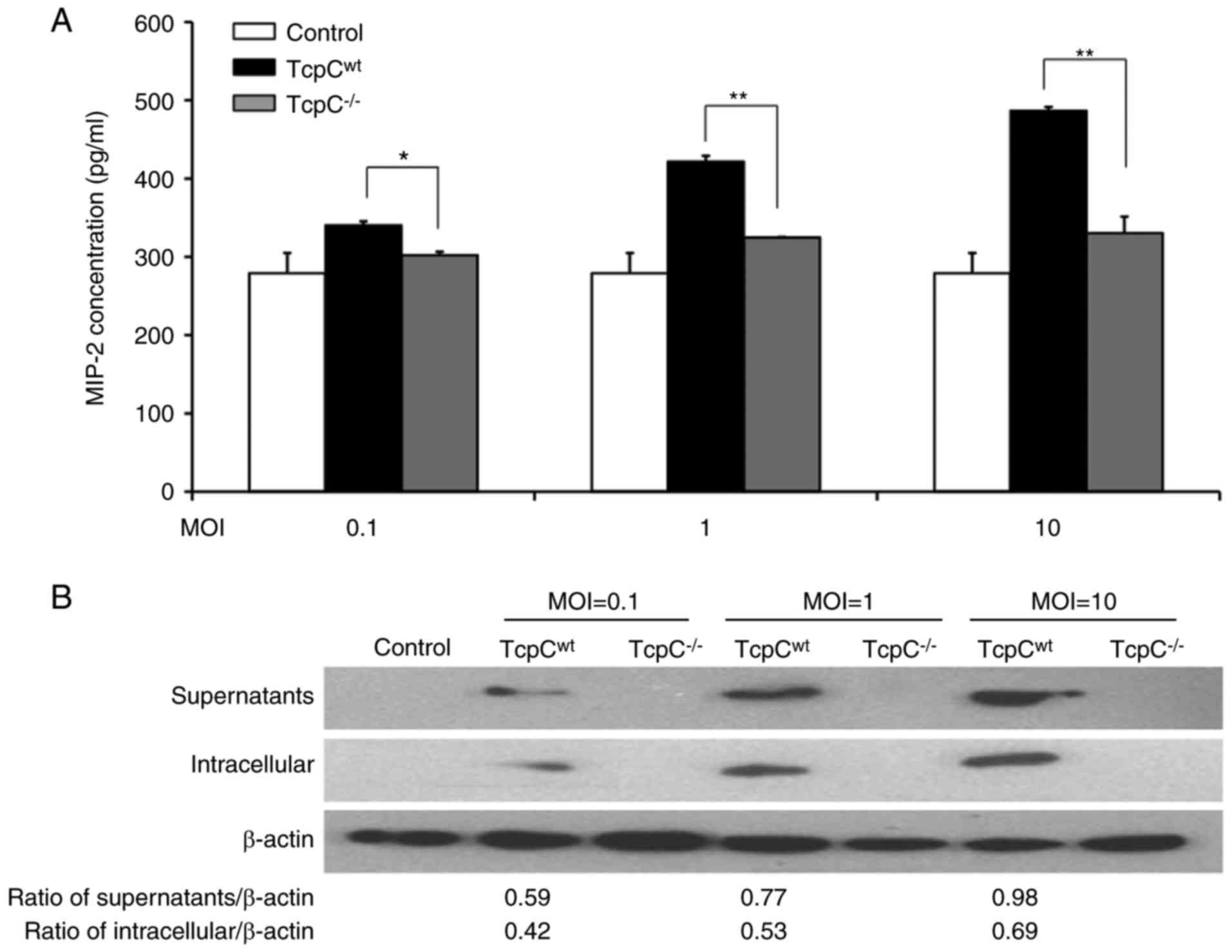
influence of TcpC on the production of MIP-2 by kidney cells,
HEK-293 cells were separately co-cultured with TcpCwt or
TcpC−/− at different MOI in transwell for 15 h and
concentrations of MIP-2 in supernatants were detected. As shown in
Fig. 3A, TcpC dose-dependently
promoted MIP-2 production in HEK-293 cells, concentration of MIP-2
in TcpCwt group was profoundly higher than that in the
TcpC−/− group (P<0.01). To confirm this enhanced
production of MIP-2 in TcpCwt-treated HEK-293 cells was
really caused by TcpC, we, at first, analyzed TcpC in the culture
supernatants and in the cells of the co-cultured system by western
blot analysis. As demonstrated in Fig.
3B, TcpC was found only in both the supernatants and in the
cells of the TcpCwt group, while no TcpC was detected in
the groups of control and TcpC−/−. Furthermore, the
quantity of TcpC in the group of TcpCwt was increased
along with the increase of MOI. Then, we observed the influence of
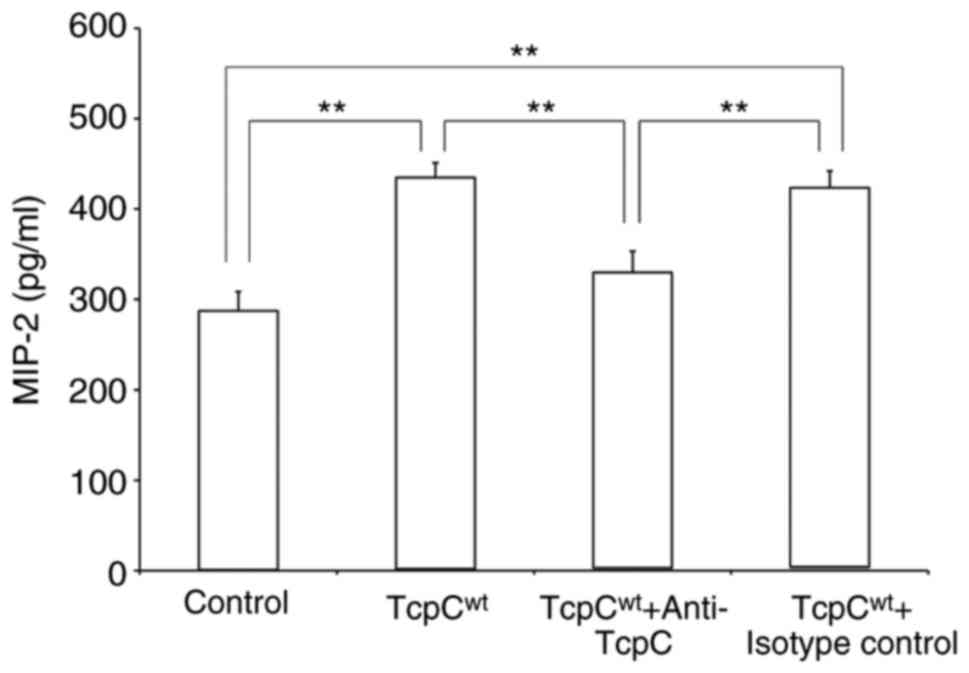
TcpC neutralization on MIP-2 production by HEK-293 cells
co-cultured with TcpCwt (Fig. 4). In the presence of 10 µg/ml
rabbit anti-TcpC polyclonal antibodies, the TcpCwt
induced MIP-2 production in HEK-293 was almost completely
abrogated, while the rabbit IgG isotype control had no effect on
TcpCwt-induced MIP-2 production, suggesting that TcpC
was the crucial factor which caused the difference in MIP-2
production between the TcpCwt and TcpC−/−
groups. These results indicated that TcpC secreted by
TcpCwt promoted kidney cells to produce MIP-2 both in
vitro and in vivo.
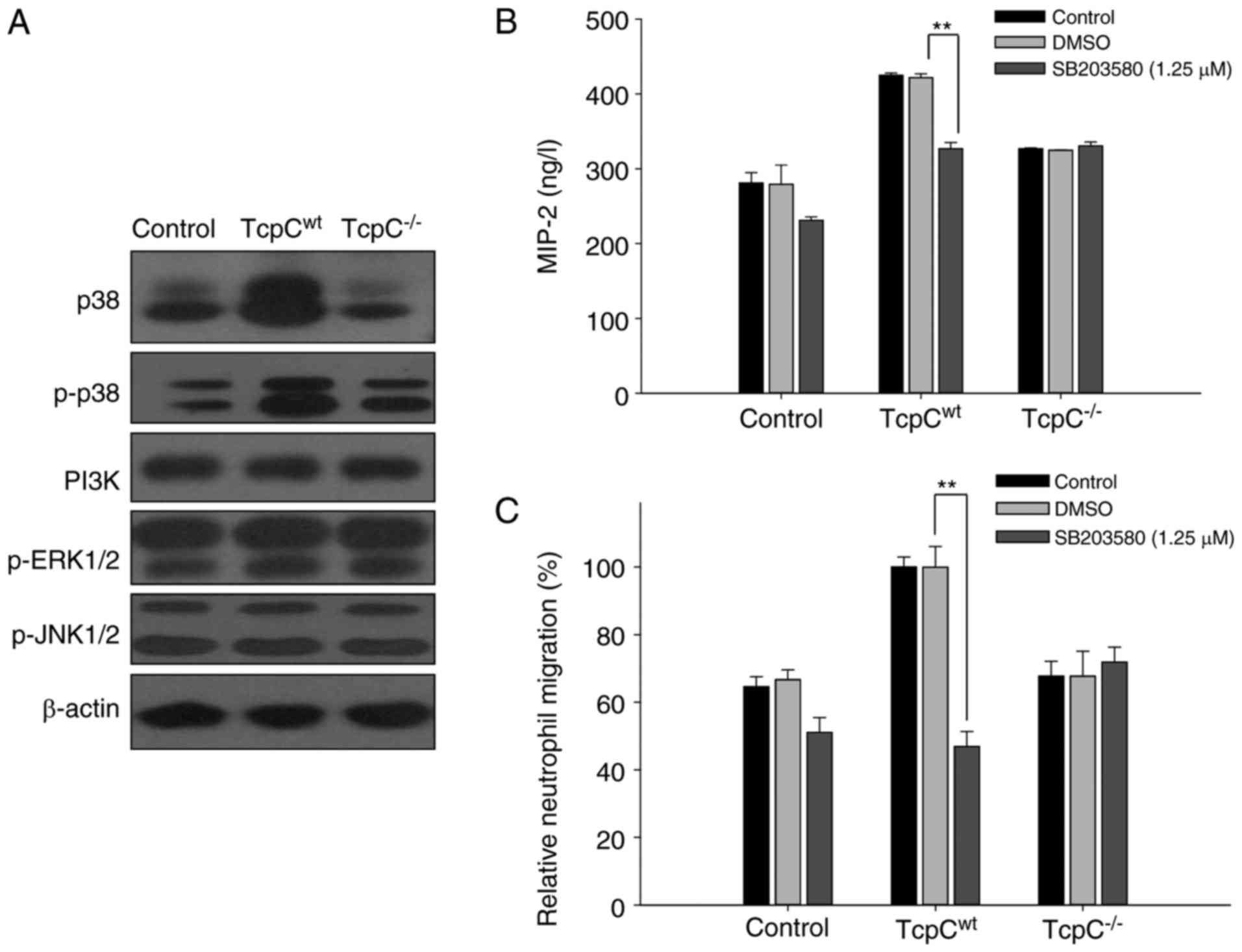
TcpC activated p38 MAPK pathway in
HEK-293 cells
To confirm the signal transduction pathway of TcpC
induced production of MIP-2 in HEK-293 cells, we detected PI3K and
MAPK signaling pathways in TcpCwt or
TcpC−/−-treated HEK-293 cells. As shown in Fig. 5A, TcpC−/− treatment had
no effect on p38 MAPK pathway, while TcpCwt treatment
resulted in the activation of p38 MAPK in HEK-293 cells, as
indicated by a simultaneous increase of p38 and p-p38
(Thr180/Tyr182). No significant changes of p-JNK1/2, p-ERK1/2 and
PI3K proteins in TcpCwt or TcpC−/−-treated
HEK-293 cells were observed (Fig.
5A), indicating that JNK, ERK and PI3K signaling pathways might
not be involved in TcpC induced MIP-2 production by HEK-293
cells.
Inhibition of p38 MAPK blocked
TcpC-induced MIP-2 production in HEK-293 cells
To further analyze whether phosphorylation and
activation of p38 MAPK was required for TcpC-mediated MIP-2
production, we tested the effect of p38 MAPK inhibitor SB203580 on
the secretion of MIP-2 induced by TcpC in HEK-293 cells. The
secretion of MIP-2 induced by TcpCwt was consistently
inhibited (P<0.01) by ~25%, when HEK-293 cells were
pre-incubated with p38 MAPK inhibitor SB203580 (Fig. 5B). Unsurprisingly, the difference
in MIP-2 production between the groups of Control and
TcpC−/− with p38 MAPK inhibitor SB203580 can be seen,
suggesting that factors other than TcpC and p38 might also be
involved in MIP-2 production. In accordance with the effect of
SB203580 on MIP-2 production by TcpCwt-treated HEK-293
cells, neutrophil chemotaxis mirrored the same trend. Neutrophil
chemotaxis to MIP-2 containing supernatants of
TcpCwt-treated HEK-293 cells was blocked by ~45% if the
HEK-293 cells were pretreated with the p38 MAPK inhibitor SB203580
(Fig. 5C). These data suggested
that in vitro inhibition of p38 MAPK could abrogate MIP-2
production induced by TcpCwt in HEK-293 cells.
Discussion
Pyelonephritis is the most severe form of UTI
(26), and it is mainly caused by
infection of UPEC (16). With the
increasing frequency of antibiotic resistance among uropathogens
(27), further understanding of
the pathogenesis of pyelonephritis is beneficial to the treatment
of the disease.
The characteristic pathological change of bacterial
pyelonephritis is neutrophil infiltration (10,19).
Neutrophils are the double-edged sword in many inflammatory
diseases (28). On one hand,
neutrophils are the essential effector cells of the innate immune
response, forming the first line of defense against bacterial and
fungal pathogens (29).
Pharmacological or genetic inhibition of neutrophil
migration/activation has been shown to drastically impair the
antibacterial defense, resulting in poor bacterial clearance and
drastic tissue pathology (30). On
the other hand, neutrophils are attracted in large numbers to
infection site and cause tissue damage during inflammatory
responses (31). It was
demonstrated that neutrophils contributed significantly to tissue
damage in acute disease processes, such as acute lung injury and
spinal cord injuries, as well as in chronic disease processes, such
as rheumatoid arthritis and asthma (32). TcpC is clinically relevant as a
virulence factor in some strains of UPEC that cause severe
pyelonephritis (19). In our
study, we have observed that large numbers of neutrophils were
infiltrated in kidneys from mouse pyelonephritis models caused by
TcpCwt, but not by TcpC−/−. These results
demonstrated that TcpC secreted by E. coli facilitated
neutrophil recruitment and inflammatory response, which might be
one of the mechanisms by which TcpC contribute to the kidney injury
and pathogenesis of pyelonephritis.
Because MIP-2 is one of the most important
chemokines that contribute significantly to the influx of
neutrophils and their activation (33,34),
we, at first, detected MIP-2 concentrations in kidney homogenates
of mouse models with pyelonephritis. In accordance with
histological examinations that showed large numbers of neutrophils
infiltration in the group of TcpCwt, we demonstrated
that TcpCwt induced greater MIP-2 response than did
TcpC−/− in the kidneys, indicating that the TcpC
producing E. coli might induce, at least in part, neutrophil
recruitment via modulation of MIP-2. MIP-2, like many other
chemokines, can be produced by a variety of cell types, including
macrophages, epithelial cells, and fibroblasts (35) as well as kidney DCs (10). MIP-2 production was triggered by
infection, and kidney epitheliums and DCs are the main source of
MIP-2 in UTI models (10,36). Using a transwell separate
co-culture system, we showed that TcpCwt
dose-dependently increased MIP-2 production and the concentration
of MIP-2 in the group of TcpCwt-treated HEK293 cells was
profoundly higher than that in the TcpC−/−-treated
group, which was also supported by our western blot analyses of
TcpC both in the culture supernatants and in the cells of the
co-culture system that showed TcpC was only detected in the
TcpCwt groups and the amount of TcpC increased along
with the increase of MOI. Furthermore, polyclonal antibodies to
TcpC could abrogate the TcpCwt induced production of
MIP-2 in HEK293 cells, confirming that this difference in MIP-2
production between the TcpCwt-treated HEK293 cells and
TcpC−/−-treated group was really caused by TcpC secreted
from the wild-type CFT073. Thus, our data showed that UPEC-derived
TcpC could induce MIP-2 production both in vitro and in
vivo.
Then, we examined which signaling pathway was
involved in MIP-2 production induced by TcpC in HEK-293 cells. The
MAPK family, including ERK1/2, p38 MAPK, and JNK, has been shown to
play key roles in mediating signals triggered by cytokines, growth
factors, stress and phagocytosis, and is involved in various
cellular functions (37,38). PI3K signaling pathway has been
identified as playing central roles in neutrophil chemotaxis and
MIP-2 production (39,40). To determine the signaling pathway
involved in TcpC-induced MIP-2 production, we examined MAPK and
PI3K activation in HEK-293 cells treated by TcpCwt and
TcpC−/−. Changes in JNK, ERK and PI3K family proteins in
TcpCwt or TcpC−/−-treated HEK-293 cells were
detected by western blot analysis, but no activation of ERK1/2,
JNK1/2 and PI3K pathway was observed. Interestingly, our data
showed that TcpCwt treatment resulted in the activation
of p38 MAPK in HEK-293 cells, as indicated by a simultaneous
increase of p38 and p-p38 (Thr180/Tyr182), while TcpC−/−
treatment had no effect on p38 MAPK pathway. Furthermore,
inhibition of p38 MAPK by SB203580 could block TcpC induced MIP-2
production in HEK-293 cells, which was further demonstrated by
neutrophil migration assays. Therefore, our data suggested that p38
MAPK was involved in TcpC-induced MIP-2 production by HEK-293
cells.
In conclusion, we presented evidence showing that
E. coli CFT073 derived TcpC could induce, through p38 MAPK
signaling pathway, MIP-2 production by kidney cells both in
vitro and in vivo, which might contribute to the
generation of characteristic pathological changes, or neutrophil
infiltration in the kidneys of pyelonephritis. Our data provided
not only novel evidence to further clarify the role of TcpC in the
pathogenesis of pyelonephritis, but also further evidence to
clarify the pathogenicity of UPEC.
Acknowledgements
The authors gratefully acknowledge financial support
from grants of National Natural Science Foundation of China
(81302806, 81671613), Scientific Research Foundation of Zhejiang
Health Bureau (2013KYA149), Science and Technology Development
Program of Hangzhou (20140633B39, 20150633B44) and Innovation
Project for High Level Overseas Returnees in Hangzhou.
Glossary
Abbreviations
Abbreviations:
|
UTI
|
urinary tract infection
|
|
UPEC
|
uropathogenic Escherichia coli
|
|
TcpC
|
toll/interleukin-1 receptor
domain-containing protein encoded by E. coli
|
|
MIP-2
|
macrophage inflammatory protein-2
|
|
TcpCwt
|
TcpC-secreting wild-type UPEC CFT073
strain
|
|
TcpC−/−
|
tcpC knock-out UPEC CFT073 strain
|
|
MAPK
|
mitogen-activated protein kinase
|
|
NLRP3
|
NACHT leucin-rich repeat PYD protein
3
|
References
|
1
|
Pinson AG, Philbrick JT, Lindbeck GH and
Schorling JB: Oral antibiotic therapy for acute pyelonephritis: A
methodologic review of the literature. J Gen Intern Med. 7:544–553.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tambo M, Okegawa T, Shishido T,
Higashihara E and Nutahara K: Predictors of septic shock in
obstructive acute pyelonephritis. World J Urol. 32:803–811. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Foxman B: Epidemiology of urinary tract
infections: Incidence, morbidity, and economic costs. Dis Mon.
49:53–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haraoka M, Hang L, Frendéus B, Godaly G,
Burdick M, Strieter R and Svanborg C: Neutrophil recruitment and
resistance to urinary tract infection. J Infect Dis. 180:1220–1229.
1999. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bhansali RS, Yeltiwar RK and Bhat KG:
Assessment of peripheral neutrophil functions in patients with
localized aggressive periodontitis in the Indian population. J
Indian Soc Periodontol. 17:731–736. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hang L, Frendeus B, Godaly G and Svanborg
C: Interleukin-8 receptor knockout mice have subepithelial
neutrophil entrapment and renal scarring following acute
pyelonephritis. J Infect Dis. 182:1738–1748. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Montecucco F, Lenglet S, Braunersreuther
V, Godaly G, Burdick M, Strieter R and Svanborg C: Single
administration of the CXC chemokine-binding protein Evasin-3 during
ischemia prevents myocardial reperfusion injury in mice.
Arterioscler Thromb Vasc Biol. 30:1371–1377. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Driscoll KE, Hassenbein DG, Howard BW,
Isfort RJ, Cody D, Tindal MH, Suchanek M and Carter JM: Cloning,
expression, and functional characterization of rat MIP-2: A
neutrophil chemoattractant and epithelial cell mitogen. J Leukoc
Biol. 58:359–364. 1995.PubMed/NCBI
|
|
9
|
Driscoll KE: TNFalpha and MIP-2: Role in
particle-induced inflammation and regulation by oxidative stress.
Toxicol Lett. 112–113:177–183. 2000. View Article : Google Scholar
|
|
10
|
Tittel AP, Heuser C, Ohliger C, Knolle PA,
Engel DR and Kurts C: Kidney dendritic cells induce innate immunity
against bacterial pyelonephritis. J Am Soc Nephrol. 22:1435–1441.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang YY, Xia MZ, Wang H, Liu XJ, Hu YF,
Chen YH, Zhang C and Xu DX: Cadmium selectively induces MIP-2 and
COX-2 through PTEN-mediated Akt activation in RAW264.7 cells.
Toxicol Sci. 138:310–321. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tateno N, Matsumoto N, Motowaki T, Suzuki
K and Aratani Y: Myeloperoxidase deficiency induces MIP-2
production via ERK activation in zymosan-stimulated mouse
neutrophils. Free Radic Res. 47:376–385. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Janssens S and Beyaert R: Functional
diversity and regulation of different interleukin-1
receptor-associated kinase (IRAK) family members. Mol Cell.
11:293–302. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marumo S, Hoshino Y, Kiyokawa H, Tanabe N,
Sato A, Ogawa E, Muro S, Hirai T and Mishima M: p38
mitogen-activated protein kinase determines the susceptibility to
cigarette smoke-induced emphysema in mice. BMC Pulm Med. 14:792014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schnyder-Candrian S, Quesniaux VF, Di
Padova F, Maillet I, Noulin N, Couillin I, Moser R, Erard F,
Vargaftig BB, Ryffel B and Schnyder B: Dual effects of p38 MAPK on
TNF-dependent bronchoconstriction and TNF-independent neutrophil
recruitment in lipopolysaccharide-induced acute respiratory
distress syndrome. J Immunol. 175:262–269. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tarchouna M, Ferjani A, Ben-Selma W and
Boukadida J: Distribution of uropathogenic virulence genes in
Escherichia coli isolated from patients with urinary tract
infection. Int J Infect Dis. 17:e450–e453. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Blum-Oehler G, Dobrindt U, Janke B, Nagy
G, Piechaczek K and Hacker J: Pathogenicity islands of
uropathogenic E. coli and evolution of virulence. Adv Exp
Med Biol. 485:25–32. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Derakhshandeh A, Firouzi R, Motamedifar M,
Boroojeni A Motamedi, Bahadori M, Arabshahi S, Novinrooz A and
Heidari S: Distribution of virulence genes and multiple
drug-resistant patterns amongst different phylogenetic groups of
uropathogenic Escherichia coli isolated from patients with
urinary tract infection. Lett Appl Microbiol. 60:148–154. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cirl C, Wieser A, Yadav M, Duerr S,
Schubert S, Fischer H, Stappert D, Wantia N, Rodriguez N, Wagner H,
et al: Subversion of Toll-like receptor signaling by a unique
family of bacterial Toll/interleukin-1 receptor domain-containing
proteins. Nat Med. 14:399–406. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Snyder GA, Cirl C, Jiang J, Chen K,
Waldhuber A, Smith P, Römmler F, Snyder N, Fresquez T, Dürr S, et
al: Molecular mechanisms for the subversion of MyD88 signaling by
TcpC from virulent uropathogenic Escherichia coli. Proc Natl
Acad Sci USA. 110:6985–6990. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yadav M, Zhang J, Fischer H, Huang W,
Lutay N, Cirl C, Lum J, Miethke T and Svanborg C: Inhibition of TIR
domain signaling by TcpC: MyD88-dependent and independent effects
on Escherichia coli virulence. PLoS Pathog. 6:e10011202010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Waldhuber A, Puthia M, Wieser A, Cirl C,
Dürr S, Neumann-Pfeifer S, Albrecht S, Römmler F, Müller T, Zheng
Y, et al: Uropathogenic Escherichia coli strain CFT073
disrupts NLRP3 inflammasome activation. J Clin Invest.
126:2425–2436. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang C, Zhou JL, Fang J, Zhang DY, Wang
BM, Chen RL and Pan JP: TcpC induces apoptosis of human vascular
endothelial cells and its mechanisms. Zhejiang Da Xue Xue Bao Yi
Xue Ban. 42:492–497. 2013.(In Chinese). PubMed/NCBI
|
|
24
|
Zhang DY, Lin YQ, He F, Fang J, Zhang C,
Wang BM and Pan JP: TcpC induces apoptosis of macrophages through
promoting ROS production. Zhejiang Da Xue Xue Bao Yi Xue Ban.
42:486–491. 2013.(In Chinese). PubMed/NCBI
|
|
25
|
Cox G: Glucocorticoid treatment inhibits
apoptosis in human neutrophils. Separation of survival and
activation outcomes. J Immunol. 154:4719–4725. 1995.PubMed/NCBI
|
|
26
|
Ragnarsdóttir B and Svanborg C:
Susceptibility to acute pyelonephritis or asymptomatic bacteriuria:
Host-pathogen interaction in urinary tract infections. Pediatr
Nephrol. 27:2017–2029. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gupta K, Hooton TM, Naber KG, Wullt B,
Colgan R, Miller LG, Moran GJ, Nicolle LE, Raz R, Schaeffer AJ, et
al: International clinical practice guidelines for the treatment of
acute uncomplicated cystitis and pyelonephritis in women: A 2010
update by the infectious diseases society of America and the
European society for microbiology and infectious diseases. Clin
Infect Dis. 52:e103–e120. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Smith JA: Neutrophils, host defense, and
inflammation: A double-edged sword. J Leukoc Biol. 56:672–686.
1994.PubMed/NCBI
|
|
29
|
Segal AW: How neutrophils kill microbes.
Annu Rev Immunol. 23:197–223. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Svensson M, Irjala H, Svanborg C and
Godaly G: Effects of epithelial and neutrophil CXCR2 on innate
immunity and resistance to kidney infection. Kidney Int. 74:81–90.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ng LG, Qin JS, Roediger B, Wang Y, Jain R,
Cavanagh LL, Smith AL, Jones CA, de Veer M, Grimbaldeston MA, et
al: Visualizing the neutrophil response to sterile tissue injury in
mouse dermis reveals a three-phase cascade of events. J Invest
Dermatol. 131:2058–2068. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sadik CD, Kim ND and Luster AD:
Neutrophils cascading their way to inflammation. Trends Immunol.
32:452–460. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shanley TP, Schmal H, Warner RL, Schmid E,
Friedl HP and Ward PA: Requirement for C-X-C chemokines (macrophage
inflammatory protein-2 and cytokine-induced neutrophil
chemoattractant) in IgG immune complex-induced lung injury. J
Immunol. 158:3439–3448. 1997.PubMed/NCBI
|
|
34
|
Aratani Y, Miura N, Ohno N and Suzuki K:
Role of neutrophil-derived reactive oxygen species in host defense
and inflammation. Med Mycol J. 53:123–128. 2012.(In Japanese).
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Armstrong DA, Major JA, Chudyk A and
Hamilton TA: Neutrophil chemoattractant genes KC and MIP-2 are
expressed in different cell populations at sites of surgical
injury. J Leukoc Biol. 75:641–648. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hang L, Haraoka M, Agace WW, Leffler H,
Burdick M, Strieter R and Svanborg C: Macrophage inflammatory
protein-2 is required for neutrophil passage across the epithelial
barrier of the infected urinary tract. J Immunol. 162:3037–3044.
1999.PubMed/NCBI
|
|
37
|
Kurosaka K, Takahashi M and Kobayashi Y:
Activation of extracellular signal-regulated kinase 1/2 is involved
in production of CXC-chemokine by macrophages during phagocytosis
of late apoptotic cells. Biochem Biophys Res Commun. 306:1070–1074.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kawaguchi M, Onuchic LF and Huang SK:
Activation of extracellular signal-regulated kinase (ERK)1/2, but
not p38 and c-Jun N-terminal kinase, is involved in signaling of a
novel cytokine, ML-1. J Biol Chem. 277:15229–15232. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Heit B, Liu L, Colarusso P, Puri KD and
Kubes P: PI3K accelerates, but is not required for, neutrophil
chemotaxis to fMLP. J Cell Sci. 121:205–214. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zampetaki A, Mitsialis SA, Pfeilschifter J
and Kourembanas S: Hypoxia induces macrophage inflammatory
protein-2 (MIP-2) gene expression in murine macrophages via
NF-kappaB: The prominent role of p42/p44 and PI3 kinase pathways.
FASEB J. 18:1090–1092. 2004.PubMed/NCBI
|