Introduction
Dilated cardiomyopathy (DCM) is the third most
common cause of heart failure, which is characterized by
ventricular chamber enlargement and systolic dysfunction (1). Myocardial fibrosis is a pathological
entity of extracellular matrix remodeling, often leading to
increased myocardial stiffness, which may promote cardiac
dysfunction (2). Both reactive
(interstitial and perivascular) fibrosis and reparative
(replacement) fibrosis are found in DCM, indicating that
inflammation and microvascular injuries were involved in the
disease process (3). Cardiac
fibrosis is associated with an adverse prognosis and impaired
response to therapeutic interventions in selected groups of
patients with DCM (4), indicating
that anti-fibrotic therapies might be useful in improving cardiac
function of the diseased heart. However, it was limited by an
indistinct understanding of the origin of fibroblasts in the
heart.
Fibroblasts are regarded as a predominant cellular
mediator of fibrosis in the heart, which play an important role in
regulating normal cardiac function and adverse myocardial
remodeling (5). A subset of
cardiac fibroblasts is of endothelial origin via a cellular
transition which is referred to as endothelial-to-mesenchymal
transition (Endo-MT) (6). Endo-MT
is a process by which endothelial cells disaggregate, change shapes
and migrate into the surrounding tissue, accompanied by loss of
endothelial cell markers like CD31 and vascular endothelial
cadherin (VE-cadherin), and up-regulation of mesenchymal markers
like smooth muscle actin (αSMA) and fibroblast specific protein-1
(FSP1). It was first described in embryonic heart development,
where endocardium cells loss its expression and become the
mesenchymal cells that form the matrix of atrioventricular cushion
and give rise to cardiac valve and septa (7). A plethora of evidence indicated that
Endo-MT mediated the development of fibrosis in multiple diseases,
including cardiac infarction (8),
hyperglycemia (9–10), chronic pulmonary hypertension
(11), tumor progression (12–13)
and kidney injury (14).
Compelling evidence reveal that endothelial cells, as a subset
origin of cardiac fibroblasts, were involved in the progression of
cardiac fibrosis via Endo-MT (6).
Given that endothelial cells were reported to
contribute to pool of organ myofibroblasts, Endo-MT was
investigated to explore its potential role in the progression of
DCM in the present study.
Materials and methods
Patient population
Forty consecutive patients with idiopathic dilated
cardiomyopathy with heart failure (mean age 50.3±11.6 years, 30
M/10F) were recruited for the study. Patients with abnormal renal
and hepatic functions were excluded, as endothelial functions were
affected by renal and liver diseases. Patients with ischemic
cardiomyopathy were not included in the study to eliminate other
possible concomitant factors in the endothelial dysfunction of
cardiac tissue. Left ventricular samples were obtained from these
DCM patients for heart transplantation. Control heart samples were
collected at forensic autopsy of road traffic accident fatality
from ten persons of corresponding to age and sex (7 males and 3
females, mean age 42.7±20.5y), excluding cardiovascular, liver and
kidney diseases. The present study conformed to the principles
outlined in the Declaration of Helsinki and procurement of tissue
for research. The study protocol was approved by the Ethic
Committee of Zhongshan Hospital affiliated to Fudan University.
Histopathological analysis
The excised heart specimens were fixed with 10%
phosphate buffered solution (PBS) formalin and embedded in
paraffin. Sections (4-µm-thick) were stained with hematoxylin-eosin
(HE) for the measurement of the myofibrils distribution. Deposition
of collagen was analyzed by Sirius Red staining (SR). Each sample
slice was photographed (HE ×200 magnification; SR ×40
magnification) under the microscope (Olympus BX51; Olympus, Tokyo,
Japan). All photos were analyzed with image-Pro Plus 6.0 analyzing
software (Media Cybernetics, Bethesda, MD, USA) by computer.
Detection of serum procollagen type I
carboxy-terminal propeptide (PICP) and procollagen type III
amino-terminal propeptide (PIIINP)
PICP and PIIINP were measured from blood samples
collected from DCM patients before surgery and healthy subjects.
All samples were centrifuged immediately at 2,000 rpm for 10 min
and store at −70°C until assay. PICP and PIIINP were measured by
enzymelinked immunosorbent assay (ELISA) (E90570Hu and E90963Hu;
USCN Life Sciences Inc., China).
Immunohistochemistry
Cardiac specimens were fixed with 10% PBS formalin,
and 4-um-thick paraffin-embedded tissue sections were prepared.
Immunostaining was performed using primary antibodies against FSP-1
(ab27957, rabbit polyclonal; Abcam, Cambridge, UK), αSMA (ab5694,
rabbit polyclonal; Abcam), CD31 (ab24950, mouse polyclonal; Abcam),
VE-cadherin (ab7047, mouse polyclonal; Abcam) according to standard
protocols. To block the activity of endogenous peroxidase, sections
were immersed in 0.3% hydrogen peroxidase in methanol for 20 min at
room temperature. After pretreatment with blocking goat serum,
sections were incubated overnight at 4°C with individual primary
antibodies as FSP (1:100), αSMA (1:100) CD31 (1:1,000) and
VE-cadherin (1:50). Then sections were incubated with secondary
antibodies conjugated to peroxidase-labeled polymer, and then were
washed and developed using diaminobenzidine (DAB) as the chromogen.
Brown yellow staining of cytoplasmic membrane was considered
positive binding. Semi-quantitative assessment of overall positive
area was performed on randomly chosen high-power fields (HPF) in
each section with a Leica-Qwin multipurpose color image processor
(Leica, Germany).
Double immunofluorescence
staining
The co-expression of endothelial markers and
mesenchymal markers were detected using confocal fluorescence
microscopy. Frozen cardiac sections were cut into 5 µm thick
sections, fixed in phosphate-buffered paraformaldehyde (PFA) at
room temperature for 30 min, and then permeabilised with 1% Triton
X-100 in PBS for 5 min. After treated with 5% BSA for 1 h, the
tissues were then incubated with two primary antibodies at 4°C
overnight. The primary antibodys were αSMA (ab5694, rabbit
polyclonal; Αbcam) and CD31 antibody (ab24950, mouse polyclonal;
Αbcam), as well as FSP-1 (ab27957, rabbit polyclonal; Αbcam) and
VE-cadherin antibody (ab7047, mouse polyclonal; Αbcam). Tissues
were incubated with a mixture of two secondary antibodies that had
been raised in different species and conjugated to different
fluochromes (i.e., rhod red-conjugated goat-anti-rabbit and
FITC-conjugated goat-anti-mouse; Jackson ImmunoResearch Labs, West
Grove, PA, USA), in 1% BSA for 1 h at room temperature in the dark,
normal rabbit IgG was used for control. After the nuclei were
stained with DAPI, slides were mounted with mounted with Mowiol
antifade reagent (Sigma-Aldrich, St. Louis, MO, USA). Confocal
images were captured using Leica TCS-SP5 laser-scanning confocal
microscope. Digital images were analyzed with LAS AF software.
Western blot analysis
Western blot analysis was performed as previously
described. Briefly, tissues were homogenized in RIPA lysis buffer
supplemented with a protease and phosphatase inhibitor cocktail
(1:100; Thermo Fisher Scientific, Waltham, MA, USA). Protein
samples were separated by 10~12% SDS-PAGE and transferred to
polyvinylidense difluoride membrane (0.22 µM, Immobilin-P,
Millipore, Billerica, MA, USA). Membranes were incubated with
primary antibodies specific for Wnt (BS1777, Rabbit polyclonal,
1:500; Bioworld Technology, Inc., St. Louis Park, MN, USA),
β-catenin (BS3730, Rabbit polyclonal, 1:500; Bioworld), and snail
(BS1853, Rabbit polyclonal, 1:500; Bioworld), followed by
incubation with horseradish peroxidase-conjugated secondary
antibodies. Proteins were visualized using the Supersignal West
Pico Chemiluminescence detection system (Pierce Chemical Co.,
Rockford, IL, USA). The arbitrary units were normalized to GAPDH
and expressed as fold induction over control values.
Statistical analysis
Data were expressed as the mean ± SEM. Statistical
significance was determined using the Student t test and the
Pearson correlation test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Clinical characteristics of the study
subjects
Clinical and echocardiographic features of patient
population were summarized in Table
I. Cardiac enlargement and impaired cardiac dysfunction were
observed in DCM patients (mean LA chamber: 44±9 mm; LVEDD 65.8±9.7
mm; LVEF 28.5±8.3%; BNP 780.4±308.9 pg/ml). Most patients had
premature ventricular contractions, among which five patients were
implanted with pacemaker (ICD, implantable
cardioverter-defibrillator) and two patients had both atrial
fibrillation and premature ventricular beat. Over 80% patients were
treated with β-blockers, whereas only 32.5% patients with ACE
inhibitors and 47.5% patients with ARB.
 | Table I.Characteristic of patient population
(DCM, n=40). |
Table I.
Characteristic of patient population
(DCM, n=40).
| Age (years) | 50.3±11.6 | Sex (M/F) | 30/10 |
|---|
| NYHA class (%) |
| Atrial
fibrillation, n (%) | 8 (25) |
| I | 0 (0) | Premature
ventricular contractions, n (%) | 36 (90) |
| II | 0 (0) | Atrioventricular
block, n (%) | 3 (7.5) |
|
III | 26 (65) | Pacemaker
implantation, n (%) | 5 (12.5) |
| IV | 14 (35) | Drug, n (%) |
|
| Echo results |
| β-blockers | 32 (80) |
| left atrium
(mm) | 44±9 | ACE inhibitors | 13 (32.5) |
| LV end-diastolic
diameter (mm) | 65.8±9.7 | ARBs | 19 (47.5) |
| LV end-systolic
diameter (mm) | 49±10.2 | CCBs | 14 (35) |
| LV ejection
fraction (%) | 28.5±8.3 | Digoxin | 23 (57.5) |
| LV posterior wall
(mm) | 8.52±0.76 | Diuretics | 30 (75) |
| Intervetricular
septum (mm) | 8.75±0.94 | BNP (pg/ml) | 780.4±308.9 |
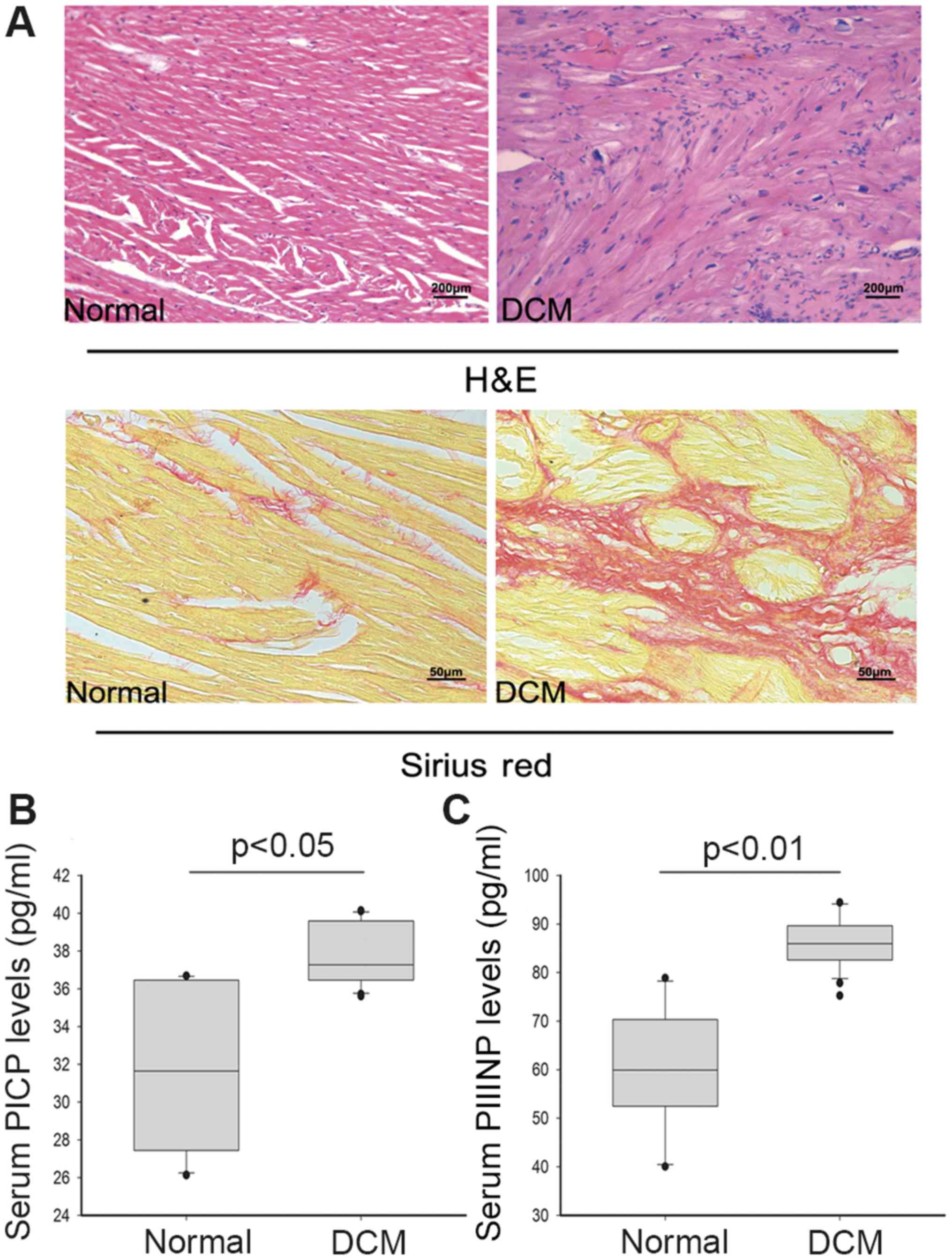
Histopathological features and serum
PICP and PIIINP levels
HE staining revealed moderate-to-severe myofibrils
disarrangement, myocytes degeneration, interstitial vacuolization
and replacement of fibrosis with or without inflammatory
infiltrates in myocardial tissue samples of DCM patients. It was
observed that interstitial fibrosis was markedly increased in
pathologic cardiac samples after Sirius red staining (Fig. 1A).
PIIINP is an extension peptide of procollagen type
III, which is cleaved off during conversion from type III
procollagen to type III collagen. Elevated serum PIIINP is believed
to reflect enhanced collagen turnover. In the present study, serum
concentration of PICP was higher in DCM patients than in the
healthy persons (mean value: 37.66 pg/ml vs. 32.03 pg/ml,
P<0.05). Moreover, the PIIINP level was significantly increased
compared to healthy group (mean value: 85.95 pg/ml vs. 60.06 pg/ml,
P<0.01) (Fig. 1B). The
concentrations of serum PICP and PIIINP represent the synthesis of
type I and III myocardial collagen respectively. The higher serum
PICP and PIIINP levels of DCM group indicated increasing collagen
production, which was consistent with the pathologic results.
Endo-MT in DCM
Sections of 40 patients and 10 donors were stained
respectively with endothelial markers and mesenchymal markers. In
agreement with previous studies (6,9,10),
CD31 and VE-cadherin were used as markers of endothelial cells, and
FSP-1 and αSMA were used as markers of myofibroblasts cells in this
study. Immunohistochemistry staining depicted that the expression
of CD31 and VE-cadherin was significantly decreased in the DCM
samples, compared with control (Fig.
2A). It was also found that no FSP-1-positive staining cells
were detected in cardiac microvascular endothelial cells of normal
healthy samples, whereas increased FSP-1-positive cells were found
in the cardiac micrangium wall of DCM (Fig. 2A, P<0.01). Similar results were
observed using αSMA staining (Fig.
2A, P<0.01). Moreover, immunohistochemistry was performed
for CD31, VE-cadherin, FSP-1 and αSMA. Compared with control,
semiquantitative analysis indicated that the CD31 and VE-cadherin
labeling indexes were significantly decreased in DCM samples (CD31,
P<0.01; VE-cadherin, P<0.01), whereas the FSP-1 and αSMA
labeling indexes were significantly increased (Fig. 2B, P<0.01).
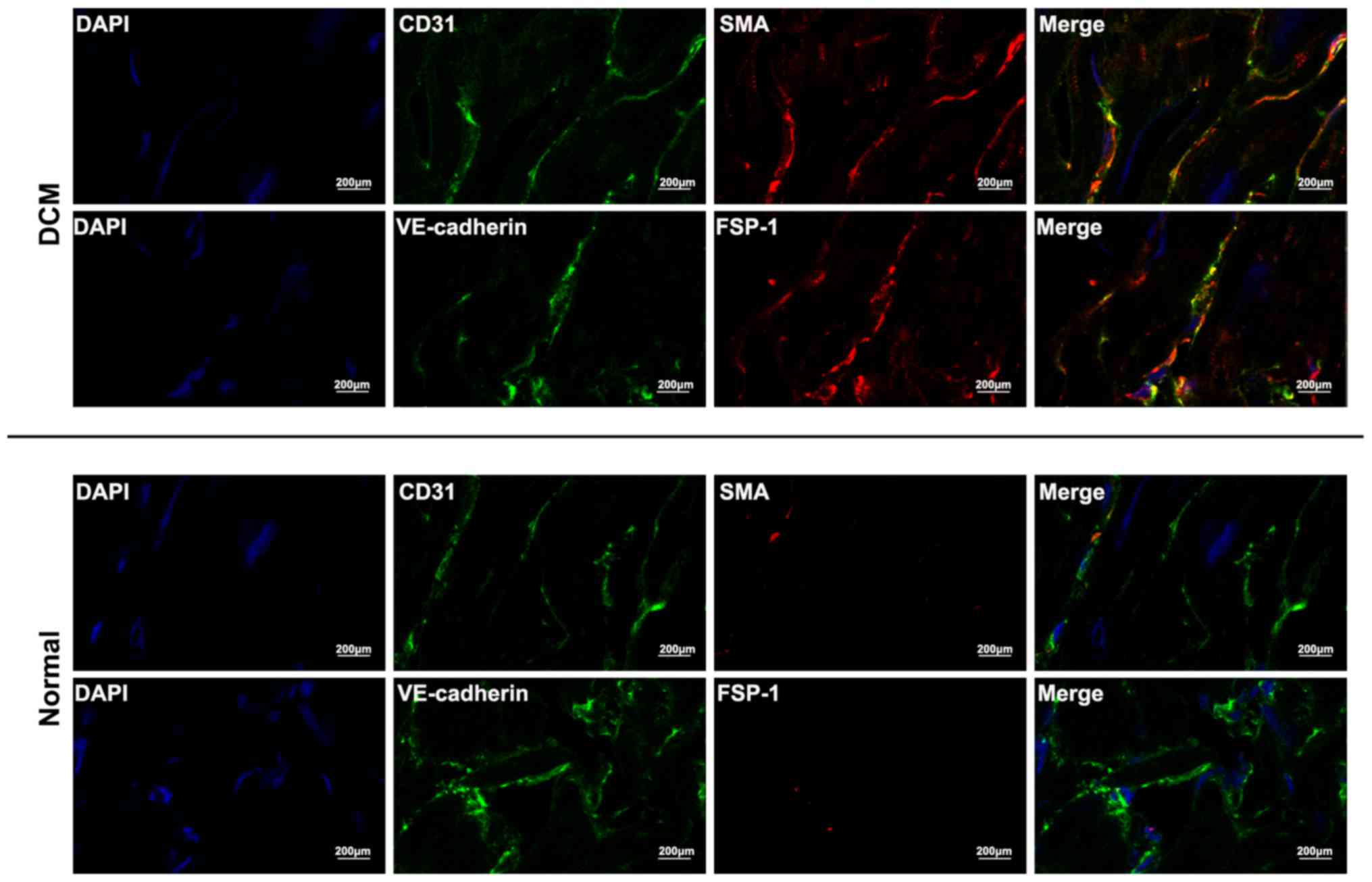
To further delineate Endo-MT, co-localization of
endothelial markers and mesenchymal markers was analyzed using
double immunofluorescence staining. An analysis of αSMA/CD31 and
FSP-1/VE-cadherin demonstrated that some cells acquired both
endothelial and mesenchymal markers, which occurred exclusively in
cardiac sections of DCM patients, whereas no such cells was
detected in normal cardiac samples (Fig. 3).
Correlation between Endo-MT and
cardiac function indexes
As depicted in Fig.
4, CD31 and VE-cadherin immunestaining labeling indexes were
respectively negatively correlated with left ventricular
end-diastolic diameter (Fig. 4A,
CD31 r=−0.82, P<0.01; VE-cadherin r=−0.73, P<0.01), while
FSP-1 and αSMA immunestaining labeling indexes were positively
associated with left ventricular chamber enlargement (Fig. 4B, αSMA r=0.65, P<0.01, FSP1
r=0.53, P<0.01) and left ventricular ejection fraction (Fig. 4D, αSMA r=−0.18, P<0.05; FSP1
r=−0.21, P<0.05). However, neither CD31 nor VE-cadherin had
statistically significant correlation with left ventricular
ejection fraction (Fig. 4C).
Moreover, neither left ventricular posterior wall (LVPW) nor
intervetricular septum (IS) had statistically significant
correlation with the CD31 or VE-cadherin indexes.
 | Figure 4.Relationship between cardiac functions
and immunostaining indexes of endothelial and mesenchymal markers
in DCM patients: Cardiac functions were demonstrated as left
ventricular end diastolic diameters (LVEDD) and left ventricular
ejection fraction (EF). CD31 and VE-cadherin immunestaining
labeling indexes were respectively negatively correlated with left
ventricular end-diastolic diameter (A) CD31 r=−0.82, P<0.01;
VE-cadherin r=−0.73, P<0.01, while FSP-1 and αSMA immunestaining
labeling indexes were positively associated with left ventricular
chamber enlargement (B) αSMA r=0.65, P<0.01, FSP1 r=0.53,
P<0.01 and left ventricular ejection fraction (D) αSMA r=−0.18,
P<0.05; FSP1 r=−0.21, P<0.05. However, neither CD31 nor
VE-cadherin has statistically significant correlation with left
ventricular ejection fraction (C) CD31 r=0.16, VE-cadherin r=0.24,
P>0.05. |
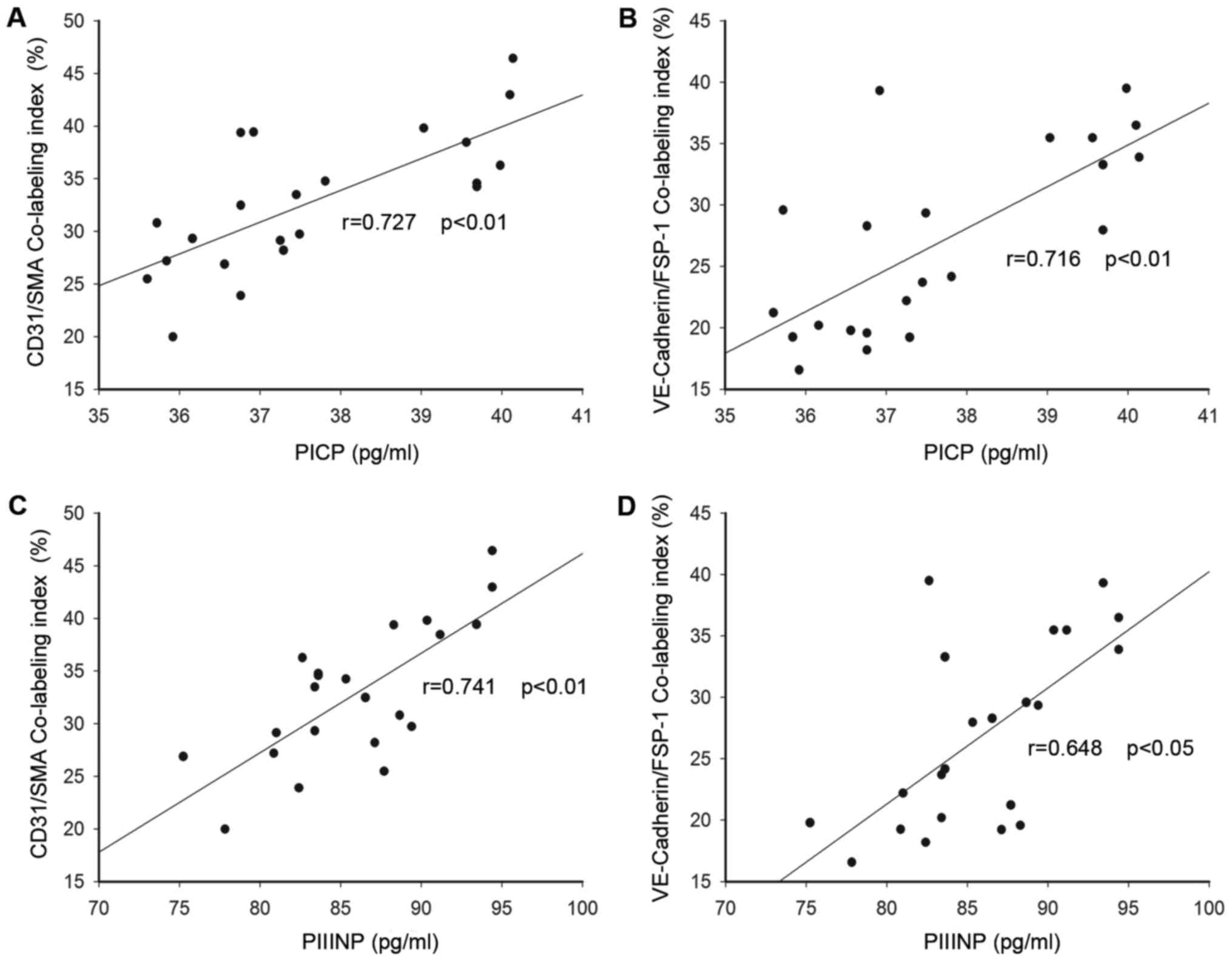
In addition, similar semiquantitative analysis was
also performed for co-localization of endothelial markers and
mesenchymal markers; and the CD31/SMA and VE-cadherin/FSP-1
co-labeling indexes were indentified. The fibroblasts coexpressed
both the VE-cadherin and the FSP1 varied from 16 to 39%, while the
CD31/SMA co-localization index was 20–46%. As depicted in Fig. 5, both circulating PICP and PIIINP
levels were positively related with the co-expression labeling
indexes (Fig. 5A: CD31/SMA
co-labeling index and PICP r=0.727, P<0.01; Fig. 5B: CD31/SMA co-labeling index and
PIIINP r=0.741, P<0.01; Fig.
5C: CD31/SMA co-labeling index and PICP r=0.727, P<0.01;
VE-Cadherin/FSP-1 co-labeling index and PICP r=0.716, P<0.01;
Fig. 5D: VE-cadherin/FSP-1
co-labeling index and PIIINP r=0.648, P<0.05). These results
indicated that Endo-MT was probably associated with myocardial
fibrosis and remodeling in the development of DCM.
Elevation of canonical Wnt signaling
family members
Canonical Wnt/β-catenin signaling pathway is an
important regulatory mechanism in embryonic cardiac progress
(15,16). To identify signaling pathway and
transcription factors that interact with the Endo-MT in human
samples, we evaluated the Wnt signaling pathway and transcription
factor snail. In this study, western blot analysis depicted that
protein levels of Wnt and β-catenin were hardly detected in normal
heart controls. However, these proteins were significantly
increased in DCM samples compared with control (Fig. 6), suggesting that canonical
Wnt/β-catenin pathway was activated in DCM samples. Snail is well
known for its capability to trigger Endo-MT. Our results
demonstrated that snail was significantly elevated in DCM samples
compared with control, suggesting that snail probably participated
in the regulation of Endo-MT for mesenchymal markers induction.
Discussion
The salient findings of this study indicated that
Endo-MT might contribute to myofibroblasts recruitment in human
DCM, characterized by decreased endothelial markers and increased
mesenchymal markers. These markers also were correlated with
cardiac function indexes and circulating levels of collagen I and
III peptides. Moreover, our data revealed that canonical Wnt
signaling pathway was involved in Endo-MT during the development of
DCM.
Endo-MT was characterized by loss of endothelial
cell markers and up-regulation of mesenchymal markers. It is now
clear that the Endo-MT can also postnatally occur in various
pathological settings, including cardiac fibrosis. Cardiac fibrosis
and myocardial remodeling was the important pathologic features of
DCM. However, the mechanism of cardiac remodeling has not been
previously addressed. In this study, our results revealed
co-expression of endothelial markers and mesenchymal markers in DCM
cardiac samples, indicating that the accumulation of myofibroblasts
originated from endothelial cells was probably implicated in
myocardial fibrosis in DCM. Intriguingly, we found that both
endothelial and mesenchymal markers were strongly correlated with
left ventricular chamber diameter, whereas only mesenchymal markers
indexes had their low correlation with LVEF.
The rate of extracellular synthesis of collagen type
I can be assessed by measuring the serum concentration of PICP,
which was formed during the extracellular processing of procollagen
type I before collagen molecules form fibers. Moreover, PIIINP was
formed during the extracellular conversion of procollagen type III
into mature fibril-forming collagen type III. The predominance of
the synthesis of collagen types I and III over their degradation
results in the accumulation within the myocardium of an excess of
collagen type I and type III fibers that characterizes fibrosis
(17). As a marker of collagen
type I synthesis, serum PICP was regarded as a marker of myocardial
fibrosis in hypertensive heart disease (18). In this study, our result indicated
that serum PICP and PIIINP was significantly elevated in DCM
patients. Moreover, co-expression labeling indexes were
respectively closely associated with PCIP and PIIIINP, which
respectively presented collagen I and III synthesis and indicated
increasing collagen production. These results indicated that
increased serum PICP and PIIINP was probably associated with
Endo-MT induced cardiac fibrosis in DCM. Together, our results
suggested that Endo-MT was associated with myocardial fibrosis,
remodeling and cardiac dysfunction in pathogenesis of DCM, which
was similar with the previous animal data that Endo-MT contributed
to the progression of cardiac fibrosis in pressure overload-induced
and diabetic-induced mice models (6,8,19,20).
The excessive formation of fibrotic tissue in the heart in turn
reduces cardiac muscle tissue, thereby decreasing cardiac function
(21).
Endo-MT has been extensively studied in generation
of cardiac valve and endocardial cushion during embryonic heart
development (7,22), which provide a useful framework for
guiding research on the Endo-MT in the postnatal pathologic
diseases. Wnt family is highly conserved during embryogenesis
(23) and required for adult
tissue maintenance (24).
Accumulating evidence indicated that Wnt signaling is required for
different aspects of cardiac development during embryonic period,
including myocardial specification, cardiac morphogenesis, and
cardiac valve formation (15–16,25).
In normal adult condition, Wnt protein keeps low level and
β-catenin is degraded by the proteosome (26), whereas Wnt signaling pathway could
be activated in pathogenesis status in order to maintain
homeostasis (24,27). Data in this study indicated that
Endo-MT might participate in the pathogenesis of DCM. Furthermore,
we found that Wnt/β-catenin pathway was significantly activated in
the DCM samples. However, it is difficult to deliberate the
increase of Wnt signal coming from Endo-MT-derived myofibroblasts
cell in the heart sample of DCM patients. Consistent with our
results, Aisagbonhi and co-workers demonstrated that the activation
of canonical Wnt signaling is a potentially important regulatory
mechanism of Endo-MT-derived myofibroblasts cells, which take part
in cardiac tissue repair and myocardial fibrosis after myocardial
infarction (8). Collectively, it
is plausible to speculate that Wnt/β-catenin is probably involved
in the process of Endo-MT during the development of DCM.
In addition, transcriptional networks mediating
Endo-MT in DCM still remain unclear. Compelling evidence
demonstrated that snail might be a crucial factor controlling
epithelial-mesenchymal transitions by repressing E-cadherin
expression (28). Moreover,
previous studies indicated that snail mediated the action of TGF-β2
induced Endo-MT (29,30). Endothelial cells that ectopically
express snail gene would adopt a fibroblastoid phenotype and
acquire invasive properties (31).
Our results demonstrated that the protein level of snail was
significantly increased in the DCM samples compared with control.
Combined with previous studies, our data suggested that snail may
play a critical role in Endo-MT during the development of DCM.
During embryogenesis, upregulation of several
molecular such as BMP-7 and HGF appeared critical for the reverse
of Endo-MT through increasing cadherin expression (32,33).
Emerging evidence demonstrated that exogenous recombinant human
BMP-7 (rh-BMP) contributed to the regression of Endo-MT in fibrotic
tissue (6). Intriguingly,
angiotensin II receptor blocker was found to relieve fibrotic
progress through blunting Endo-MT in murine models (9,34).
Simvastatin treatment decreases TGF-β1-induced Endo-MT in
vitro (35). These data open
up new avenues for the design of specific anti-fibrosis drug via
Endo-MT.
In conclusion, our results provide a novel insight
that Endo-MT was probably implicated in the pathogenesis of cardiac
fibrosis and myocardial remodeling in DCM. Endo-MT might be a
potential therapeutic target against cardiac fibrosis and
remodeling for DCM treatment.
Acknowledgements
The present was supported by grants from National
Natural Science Youth Foundation of China (No. 81300166, No.
81300096), 2013 Shanghai Outstanding Academic Leaders Plan (No.
13XD1401500) and Youth foundation of Zhongshan Hospital affiliated
to Fudan University (No. 2013ZSQN09).
References
|
1
|
Maron BJ, Towbin JA, Thiene G,
Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE and Young
JB: American Heart Association, et al: Contemporary
definitions and classification of the cardiomyopathies: An American
heart association scientific statement from the council on clinical
cardiology, heart failure and transplantation committee; quality of
care and outcomes research and functional genomics and
translational biology interdisciplinary working groups; and council
on epidemiology and prevention. Circulation. 113:1807–1816. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jellis C, Martin J, Narula J and Marwick
TH: Assessment of nonischemic myocardial fibrosis. J Am Coll
Cardiol. 56:89–97. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
de Leeuw N, Ruiter DJ, Balk AH, de Jonge
N, Melchers WJ and Galama JM: Histopathologic findings in explanted
heart tissue from patients with end-stage idiopathic dilated
cardiomyopathy. Transpl Int. 14:299–306. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schalla S, Bekkers SC, Dennert R, van
Suylen RJ, Waltenberger J, Leiner T, Wildberger J, Crijns HJ and
Heymans S: Replacement and reactive myocardial fibrosis in
idiopathic dilated cardiomyopathy: Comparison of magnetic resonance
imaging with right ventricular biopsy. Eur J Heart Fail.
12:227–231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Porter KE and Turner NA: Cardiac
fibroblasts: At the heart of myocardial remodeling. Pharmacol Ther.
123:255–278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zeisberg EM, Tarnavski O, Zeisberg M,
Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT,
Roberts AB, et al: Endothelial-to-mesenchymal transition
contributes to cardiac fibrosis. Nat Med. 13:952–961. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mjaatvedt CH, Lepera RC and Markwald RR:
Myocardial specificity for initiating endothelial-mesenchymal cell
transition in embryonic chick heart correlates with a particulate
distribution of fibronectin. Dev Biol. 119:59–67. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Aisagbonhi O, Rai M, Ryzhov S, Atria N,
Feoktistov I and Hatzopoulos AK: Experimental myocardial infarction
triggers canonical Wnt signaling and endothelial-to-mesenchymal
transition. Dis Model Mech. 4:469–483. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tang RN, Lv LL, Zhang JD, Dai HY, Li Q,
Zheng M, Ni J, Ma KL and Liu BC: Effects of angiotensin II receptor
blocker on myocardial endothelial-to-mesenchymal transition in
diabetic rats. Int J Cardiol. 162:92–99. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Widyantoro B, Emoto N, Nakayama K,
Anggrahini DW, Adiarto S, Iwasa N, Yagi K, Miyagawa K, Rikitake Y,
Suzuki T, et al: Endothelial cell-derived endothelin-1 promotes
cardiac fibrosis in diabetic hearts through stimulation of
endothelial-to-mesenchymal transition. Circulation. 121:2407–2418.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Arciniegas E, Frid MG, Douglas IS and
Stenmark KR: Perspectives on endothelial-to-mesenchymal transition:
Potential contribution to vascular remodeling in chronic pulmonary
hypertension. Am J Physiol Lung Cell Mol Physiol. 293:L1–L8. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Selek L, Dhobb M, van der Sanden B, Berger
F and Wion D: Existence of tumor-derived endothelial cells suggests
an additional role for endothelial-to-mesenchymal transition in
tumor progression. Int J Cancer. 128:1502–1503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jeong H, Ryu YJ, An J, Lee Y and Kim A:
Epithelial-mesenchymal transition in breast cancer correlates with
high histological grade and triple-negative phenotype.
Histopathology. 60:E87–E95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zeisberg EM, Potenta SE, Sugimoto H,
Zeisberg M and Kalluri R: Fibroblasts in kidney fibrosis emerge via
endothelial-to-mesenchymal transition. J Am Soc Nephrol.
19:2282–2287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Person AD, Garriock RJ, Krieg PA, Runyan
RB and Klewer SE: Frzb modulates Wnt-9a-mediated beta-catenin
signaling during avian atrioventricular cardiac cushion
development. Dev Biol. 278:35–48. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cohen ED, Tian Y and Morrisey EE: Wnt
signaling: An essential regulator of cardiovascular
differentiation, morphogenesis and progenitor self-renewal.
Development. 135:789–798. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
López B, González A, Ravassa S, Beaumont
J, Moreno MU, José San G, Querejeta R and Díez J: Circulating
biomarkers of myocardial fibrosis: The need for a reappraisal. J Am
Coll Cardiol. 65:2449–2456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Querejeta R, Varo N, López B, Larman M,
Artiñano E, Etayo JC, Ubago Martínez JL, Gutierrez-Stampa M,
Emparanza JI, Gil MJ, et al: Serum carboxy-terminal propeptide of
procollagen type I is a marker of myocardial fibrosis in
hypertensive heart disease. Circulation. 101:1729–1735. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ghosh AK, Bradham WS, Gleaves LA, De Taeye
B, Murphy SB, Covington JW and Vaughan DE: Genetic deficiency of
plasminogen activator inhibitor-1 promotes cardiac fibrosis in aged
mice: Involvement of constitutive transforming growth factor-beta
signaling and endothelial-to-mesenchymal transition. Circulation.
122:1200–1209. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Okayama K, Azuma J, Dosaka N, Iekushi K,
Sanada F, Kusunoki H, Iwabayashi M, Rakugi H, Taniyama Y and
Morishita R: Hepatocyte growth factor reduces cardiac fibrosis by
inhibiting endothelial-mesenchymal transition. Hypertension.
59:958–965. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Khan R and Sheppard R: Fibrosis in heart
disease: Understanding the role of transforming growth factor-beta
in cardiomyopathy, valvular disease and arrhythmia. Immunology.
118:10–24. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rivera-Feliciano J, Lee KH, Kong SW,
Rajagopal S, Ma Q, Springer Z, Izumo S, Tabin CJ and Pu WT:
Development of heart valves requires Gata4 expression in
endothelial-derived cells. Development. 133:3607–3618. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Donaldson IJ, Amin S, Hensman JJ, Kutejova
E, Rattray M, Lawrence N, Hayes A, Ward CM and Bobola N:
Genome-wide occupancy links Hoxa2 to Wnt-β-catenin signaling in
mouse embryonic development. Nucleic Acids Res. 40:3990–4001. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Monga SP: Role of Wnt/β-catenin signaling
in liver metabolism and cancer. Int J Biochem Cell Biol.
43:1021–1029. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brade T, Männer J and Kühl M: The role of
Wnt signalling in cardiac development and tissue remodelling in the
mature heart. Cardiovasc Res. 72:198–209. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Berendsen AD, Fisher LW, Kilts TM, Owens
RT, Robey PG, Gutkind JS and Young MF: Modulation of canonical Wnt
signaling by the extracellular matrix component biglycan. Proc Natl
Acad Sci USA. 108:17022–17027. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Luu HH, Zhang R, Haydon RC, Rayburn E,
Kang Q, Si W, Park JK, Wang H, Peng Y, Jiang W and He TC:
Wnt/beta-catenin signaling pathway as a novel cancer drug target.
Curr Cancer Drug Targets. 4:653–671. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Niessen K, Fu Y, Chang L, Hoodless PA,
McFadden D and Karsan A: Slug is a direct Notch target required for
initiation of cardiac cushion cellularization. J Cell Biol.
182:315–325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Medici D, Potenta S and Kalluri R:
Transforming growth factor-β2 promotes Snail-mediated
endothelial-mesenchymal transition through convergence of
Smad-dependent and Smad-independent signalling. Biochem J.
437:515–520. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lopez D, Niu G, Huber P and Carter WB:
Tumor-induced upregulation of Twist, snail and slug represses the
activity of the human VE-cadherin promoter. Arch Biochem Biophys.
482:77–82. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Okada H and Kalluri R: Cellular and
molecular pathways that lead to progression and regression of renal
fibrogenesis. Curr Mol Med. 5:467–474. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Okada H and Kalluri R: Recapitulation of
kidney development paradigms by BMP-7 reverses chronic renal
injury. Clin Exp Nephrol. 9:100–101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tang R, Li Q, Lv L, Dai H, Zheng M, Ma K
and Liu B: Angiotensin II mediates the high-glucose-induced
endothelial-to-mesenchymal transition in human aortic endothelial
cells. Cardiovasc Diabetol. 9:312010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tuuminen R, Syrjälä S, Krebs R, Keränen
MA, Koli K, Abo-Ramadan U, Neuvonen PJ, Tikkanen JM, Nykänen AI and
Lemström KB: Donor simvastatin treatment abolishes rat cardiac
allograft ischemia/reperfusion injury and chronic rejection through
microvascular protection. Circulation. 124:1138–1150. 2011.
View Article : Google Scholar : PubMed/NCBI
|