Introduction
Cervical cancer is a common type of reproductive
tumor with an incidence of ~50 million cases annually, 80% of which
occur in developing countries. Cervical cancer is increasing in
prevalence and the age of onset is decreasing (1). Therefore, understanding the
initiation and progression of cervical cancer is required for
prevention and the development of novel therapies.
TLRs (Toll-like receptors) are pattern recognition
receptors that regulate infection by identifying conserved
molecules of pathogenic microorganisms. Previous studies indicated
that TLRs are expressed on immune cells, in addition to normal
epithelial cells and a number of tumor cells (2,3).
TLRs contribute to mucosal immunity, and tumor initiation and
development (4,5). TLR4 is a pattern recognition receptor
that is able to recognize exogenous ligands, including
lipopolysaccharide (LPS), present in gram negative bacteria cell
walls, and is associated with tumor growth and inflammation, as
other members of the same family (6).
The cervix is exposed to bacterial flora from the
normal vaginal environment which, along with vaginal pathogens,
stimulates cervical epithelial cells repeatedly during infection.
Studies have indicated that the inflammatory response may directly
regulate cell deterioration and contribute to the formation of a
tumor microenvironment, indirectly modulating tumor initiation and
development (7,8). Pro-inflammatory cytokines and
endogenous TLR ligands produced by tissues may activate the TLR
pathway in tumor cells and promote tumor cell development, immune
escape and apoptotic resistance (9,10). A
study suggested that TLR4 may be upregulated in cervical cancer
cells compared with other TLRs (11) and that the expression of TLR4
increased markedly following simulation of cervical cancer cells
with LPS. Therefore, downregulation of TLR4 may induce apoptosis in
cervical cancer SiHa cells (12).
TLR4 may be associated with the initiation and development of
cervical cancer. An additional study demonstrated that TLR4
initiated cell activation to induce inflammation and tumors via
MyD88- and nuclear factor κB (NF-κB)-associated signaling pathways,
and that the activation of NF-κB may be involved in the initiation
and development of cancer (13).
In addition, when pathogen-associated molecular patterns bind to
TLR4, activation of the TLR4 signaling pathway is modulated by
mobile microdomains of membrane lipids known as lipid rafts
(14). The above study indicated
an interaction between lipid rafts and the TLR4 signaling pathway,
and suggested that lipid rafts may serve a role in the initiation
of the TLR4 signaling pathway.
Hypoxia inducible factor 1 (HIF-1) is a
transcription factor involved in cell adaptive adjustment under
hypoxia. HIF-1 is composed of HIF-1α and −1β subunits, and HIF-1α
maintains the stability of HIF-1 expression, which affects energy
metabolism, proliferation and apoptosis in tumor cells. HIF-1
additionally promotes tumor angiogenesis, increases tumor
invasiveness, and increases resistance to radiotherapy and
chemotherapy through its involvement in the transcriptional
regulation of a number of target genes (15). Previous studies have demonstrated
that the uncontrolled growth of cervical cancer cells may be
associated with excessive activation of HIF-1α and, therefore,
HIF-1α may be associated with cancer control mechanisms (16,17).
HIF-1 may mediate activation of telomerases in cervical cancer
cells to promote cancer progression (18). Studies have indicated that HIF-1α
can not only promote tumor growth, but also enhance tumor cell
invasion (19,20). In one study, silencing HIF-1α using
small interfering (si)RNA in cervical cancer cells downregulated
the expression of solute carrier family 2 member 1 and hexokinase
2, and reduced the glycolytic activity of tumors, promoting
apoptosis and tumor cell growth inhibition (21).
The accumulation of HIF-1 induced by bacterial LPS
in immune cells, including macrophages and monocytes, is
TLR4-dependent (22). TLR4
expression was positively associated with HIF-1α expression in
cervical cancer cells and, therefore, the TLR4 signaling pathway
may be involved in maintaining elevated HIF-1α activity to promote
cervical cancer development, although the mechanism underlying
these observations remains to be elucidated (23). In the present study, HIF-1α
expression markedly increased in cervical cancer tissues in the
presence of reactive oxygen species (ROS), and the increased ROS
was derived from intracellular reduced nicotinamide-adenine
dinucleotide phosphate (NADPH) oxidase, which caused the elevated
intracellular HIF-1α activity associated with cervical cell
malignancy (24). The above
observations may contribute to the recurrence of cancer following
treatment for cervical cancer, and previous studies indicated that
the activation of cervical epithelial membrane lipid rafts
increased the transfer of NADPH oxidase subunits to lipid rafts,
resulting in excessive activation of NADPH oxidase and increased
intracellular ROS levels (25,26).
Therefore, TLR4 may promote increased HIF-1α activity in cervical
cancer cells due to lipid raft-mediated activation of the NADPH
oxidase pathway.
To study the TLR4-mediated elevated activity of
HIF-1α, different pathways in cervical cells were selectively
inhibited using ST2825, pyrrolidine dithiocarbamate (PDTC) and
methy1-β-cyclodextrin (MβCD). ST2825 is a specific inhibitor of
myeloid differentiation factor 88 (MyD88), which inhibits the
dimerization of MyD88, and prevents the activation and transduction
of the TLR signal, thereby inhibiting the TLR signaling pathway
(27). PDTC is an inhibitor of the
NF-κB signaling pathway, preventing degradation of the inhibitor of
NF-κB subunit α (IκB), activation of NF-κB and translocation of
NF-κB to the nucleus (28). MβCD
has a strong affinity for cholesterol, and is able to remove it
from cells, and dysregulate lipid raft integrity and function
(29).
The mechanisms underlying the TLR4-mediated
activation of HIF-1α via lipid rafts and redox events remain to be
elucidated. Therefore, the present study aimed to elucidate these
mechanisms and alterations in NADPH oxidase activity in response to
the activation of TLR4 signaling. In the present study ROS levels
in cervical cancer cells were measured following deregulation of
the TLR4 signaling pathway and lipid raft function.
Materials and methods
Materials
Cervical carcinoma SiHa cells were purchased from
Saiqi Biological Engineering Company (Shanghai, China). Fetal
bovine serum (FBS), Minimum Essential Medium (MEM) and western blot
analysis kits were purchased from Thermo Fisher Scientific, Inc.
(Waltham, MA, USA). Immunohistochemistry staining diaminobenzidine
(DAB) kits were purchased OriGene Technologies, Inc. (Beijing,
China). MTT assay kits, protein quantification kits and ROS
detection reagent kits were purchased from Beyotime Institute of
Biotechnology (Jiangsu, China). The immunofluorescent staining kit
was manufactured by OriGene Technologies, Inc. HiPerFect
transfection reagent was manufactured by Qiagen China Co., Ltd.
(Shanghai, China). Rabbit anti-human HIF-1α monoclonal antibody
(cat. no. ab51608) and mouse anti-human TLR4 monoclonal antibodies
(cat. no. ab22048), and horseradish peroxidase (HRP)-labeled goat
anti-rabbit immunoglobulin (Ig)G (cat. no. ab6721) were purchased
from Abcam (Cambridge, UK). DyLight™ 488-labeled goat
anti-rabbit immunoglobulin (Ig)G (cat. no. ZF-0511) and
DyLight™ 594-labeled goat anti-mouse IgG (cat. no.
ZF-0513) were from OriGene Technologies, Inc.; HIF-1α rabbit and
β-actin rabbit polyclonal antibodies (cat. nos. PB0245 and BA2305)
were purchased from Wuhan Boster Biological Technology, Ltd.
(Wuhan, China). NADPH, lucigenin, LPS, PDTC, MβCD and ST2825 were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). TLR4
siRNA (5-GGACCTCTCTCAGTGTCAA-3) was designed and synthesized by
Shanghai Jima Industrial Co., Ltd. (Shanghai, China).
Cell cultures
The cervical cancer SiHa cells were cultured in MEM
(4 ml) containing 10% FBS at 5% CO2, 37°C and saturated
humidity. Medium was replaced daily. When cells reached 80%
confluency they were selected for subsequent experiments.
Treatment groups
SiHa cells were randomized into 6 groups: i) The
untreated control group was normally cultured without treatment or
stimulation; ii) the LPS control group was treated with 500 ng/ml
LPS for 24, 48 or 72 h; iii) lipid raft intervention, termed the
MβCD+LPS group was treated with MβCD [5 mmol/l; dilution with MEM]
and pre-incubated for 1 h prior to adding 500 ng/ml LPS; iv) the
ST2825+LPS group was treated with ST2825 (5 mmol/l; dilution with
MEM) and pre-incubated for 1 h prior to adding 500 ng/ml LPS; v)
the siTLR4+LPS group was transfected with TLR4 siRNA using
HiPerFect transfection reagent (when cells reached 50% confluency,
transfection was confirmed and 500 ng/ml LPS was added; and vi) the
NF-κB signal pathway intervention group, termed the PDTC+LPS group,
was treated with 20 mmol/l PDTC for 1 h prior to the administration
of 500 ng/ml LPS. SiHa cells in the logarithmic growth phase were
used in all experiments.
MTT assay
Cells (1×104) were plated into 96-well
culture plates with 100 µl MEM and each group was allocated 5
wells. During the logarithmic growth phase, treatments were added
to each group and culture was continued for 1–4 days. Following
growth of adherent cells, 10 µl MTT (10 mg/ml) was added at 0, 24,
48 and 72 h. After adding MTT, cells continued to be cultured for
another 4 h and then MTT activity was measured. Dimethyl sulfoxide
(DMSO; 200 µl) was added to each well following removal of the cell
culture medium. Blank wells were used for zero adjustment following
agitation of the plates for 10 min. Optical density (OD) values
were measured for all groups at a wavelength of 570 nm, using a
microplate reader. Experiments were repeated 3 times. Cell growth
inhibition curves were constructed; incubation time was plotted on
the x-axis and OD values were plotted on the y-axis.
Soft agar colony forming
experiment
Modified Thayer Martin (MTM) culture medium (6 ml)
was combined with an equal volume of 1% low melting agarose. A 0.6%
bottom agar was prepared, and 4 ml was spread over each 60 mm
diameter plate and incubated at 4°C for 10 min. Following
coagulation of the bottom agar, the MTM-agarose mixture and 1,000
cells/ml single cell suspensions in each group after treatments
were rapidly and thoroughly mixed, and 1 ml mixture was spread on
the bottom agar. Following solidification at room temperature,
culture was continued for 14 days in a 37°C incubator with 5%
CO2. Cell growth was subsequently observed and colony
forming cells in each culture dish were counted. Each visible cell
cluster (>500 cells) was counted as one colony.
Verification of HIF-1α expression via
immunocytochemistry
Cells were treated as previously described. PBS was
used instead of the primary antibody as a negative control.
Following treatments, cells were washed with 0.01 mol/l PBS three
times, fixed with 4% paraformaldehyde at room temperature for 10
min, and washed three times with 0.01 mol/l PBS. Cells were treated
with 3% H2O2 for 20 min and blocked with 10%
blocking serum (AR0009; Boster Biological Technology, Pleasanton,
CA, USA) for 30 min at room temperature. Subsequently, rabbit
anti-human HIF-1α antibody (1:200) was added and cells were
incubated at 4°C overnight. Following washing with PBS three times,
HRP-labeled goat anti-rabbit antibody (1:300) was added and the
plate was incubated at 37°C for 1 h. The plates were stained using
the immunohistochemistry DAB kit and observed under a fluorescence
microscope with magnification ×200.
Western blot analysis of HIF-1α
expression
Cells were grouped and treated as described above
and were harvested for protein extraction using a RIPA lysis
solution (Beyotime Institute of Biotechnology), quantified using
the bicinchoninic acid method, separated by 8% SDS-PAGE and
transferred to polyvinylidene fluoride membranes. The mass of
protein loaded per lane was 30 µg. Membranes were blocked with 5%
skimmed milk for 1 h at room temperature. Rabbit polyclonal
anti-HIF-1α (1:1,000) and anti-β-actin (1:1,000) were added and
incubated at 4°C overnight. Subsequently, HRP-labeled goat
anti-rabbit IgG (1:300) was added and membranes were incubated at
37°C for 1 h. The results were observed by an Odyssey infrared
imaging system (LI-COR). Target protein expression was quantified
by image J (k 1.45; National Institutes of Health, Bethesda, MD,
USA) as gray values of the target protein/β-actin protein. The
experiment was repeated three times.
ROS measurement with
dichloro-dihydro-fluorescein diacetate (DCFH-DA)
ROS levels were measured using an intracellular
peroxide-sensitive fluorescent DCFH-DA probe. SiHa cells were
seeded on 96-well plates with 100 µl culture medium, and cells were
treated as described. ROS was measured at 0, 12, 24, 36 and 48 h.
Inoculated cells were washed twice with Earle's Balanced Salt
Solution (Thermo Fisher scientific, Inc.) and incubated with 25 µM
DCFH-DA probe at 37°C for 30 min, and washed twice with Earle's
Balanced Salt Solution. Fluorescence intensity was immediately
measured at a wavelength of (excitation, 485 nm; emission, 520 nm)
using a spectrofluorometer (FLUOstar Optima Microplate Reader; BMG
Labtech GmbH, Ortenberg, Germany).
NADPH oxidase activity
SiHa cells were seeded on 96-well plates with 100 µl
MEM and treated as described above. In each group NADPH was assayed
at 0, 12, 24, 36 and 48 h following treatments as described in the
section ‘Treatment groups’. At each time point, cells were
collected using 4°C cold fresh PBS using a cell scraper. The cell
suspension was centrifuged at 2,500 × g at 4°C for 5 min. Cells
were harvested for protein extraction using a RIPA lysis solution
(Beyotime Institute of Biotechnology) and then were taken for
protein quantification as described. Subsequently, 50 µl cellular
protein, 5 µl DMSO, 5 µmol/l lucigenin and 50 mm/l Tiron (172553;
Sigma-Aldrich; Merck KGaA) were added to 96-well plates in the
dark. A total of 100 mol/l NADPH was added to initiate the
reaction. NADPH oxidase activity was measured using a
chemiluminescence detector and expressed as relative light units/mg
protein.
Cell immunofluorescence staining
Cells were treated as described and plated on 5×5 mm
slides in 35 mm diameter culture dishes. When integration reached
60% confluency, the medium was removed by suction, and slides were
washed with PBS and fixed with 1 ml 4% polyformaldehyde for 20 min
in the room temperature. Cells were treated with 0.5% TritonX-100
for 20 min and blocked with 5% FBS at room temperature for 60 min.
Rabbit anti human HIF-1α (1:100) and mouse anti-human TLR4 (1:100)
monoclonal antibodies were added and incubated overnight at 4°C.
The following day, DyLight 594-labeled goat anti-mouse IgG (1:40)
and DyLight 488-labeled goat anti-rabbit IgG (1:30) were added and
the slides were incubated at 37°C for 2 h. Nuclei were stained with
DAPI (C1002; Beyotime Institute of Biotechnology). Quenching was
achieved using an anti-fluorescence quenching agent and a
fluorescent microscope (magnification, ×200) was used to observe
and capture images.
Statistical analysis
Data were analyzed using SPSS software (version
13.0; SPSS, Inc., Chicago, IL, USA). Data are presented as the mean
± standard deviation for the indicated replicates (N=3 for each
experiment) and P<0.05 was considered to indicate a
statistically significant difference. Comparisons of two sample
means were performed using a 2-sample Student's t-test. Differences
among three or more groups were evaluated using one-way analysis of
variance, followed by LSD post hoc test.
Results
Cell growth
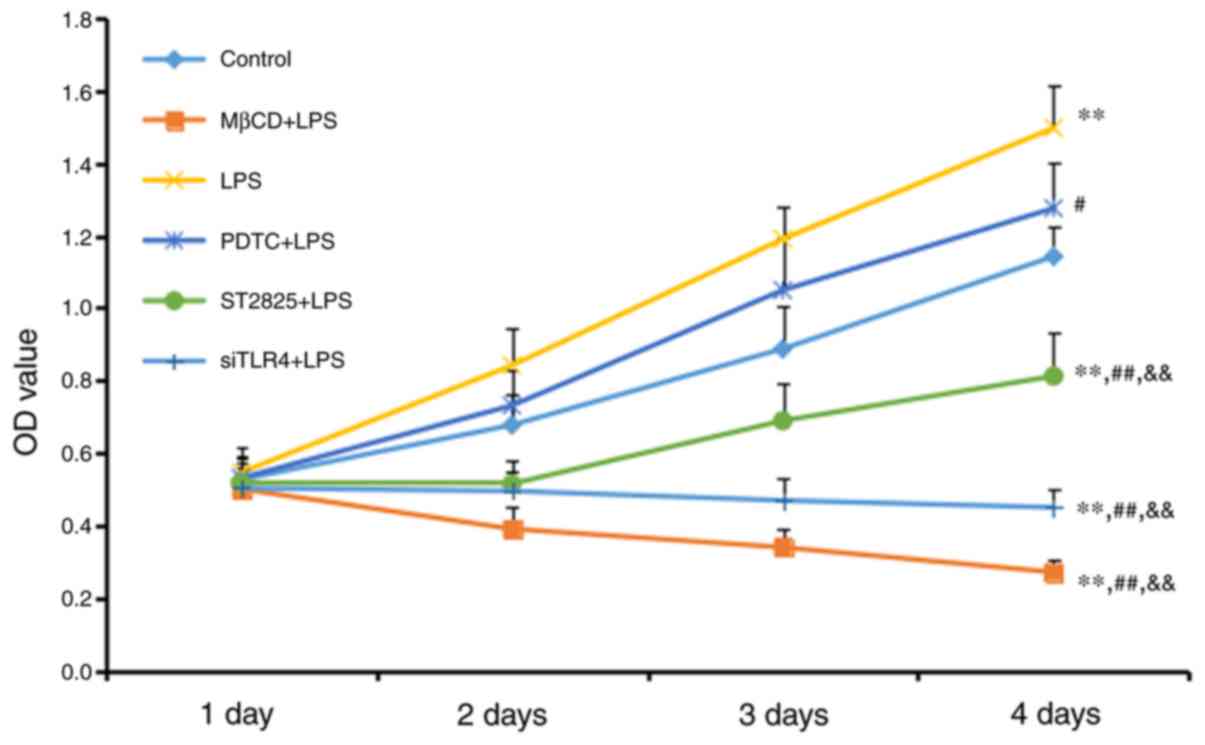
An MTT assay was used to measure cell growth
(Fig. 1). Cell growth increased in
the LPS group compared with the control group (P<0.01), although
no significant difference was observed between the LPS+PDTC and
control groups. Cell growth decreased in all other treatment other
groups compared with the control group (all P<0.01). Compared
with the LPS group, cell activity was reduced in the PDTC+LPS group
(P<0.05), and the ST2825+LPS, siTLR4+LPS and MβCD+LPS groups
(all P<0.01). Cell growth increased in the PDTC+LPS group
compared with the ST2825+LPS, siTLR4+LPS and MβCD+LPS groups (all
P<0.01). Cell growth inhibition was time-dependent, as all
significant differences were observed 4 days following treatment.
LPS stimulated cervical cancer cell growth. This growth was reduced
following inhibition of TLR4 and MyD88 signaling, and reduced lipid
raft functionality. Inhibition of NF-κB signaling additionally
reduced SiHa cell growth compared with the LPS group, although this
inhibition was decreased compared with the effect included by
inhibition of TLR4 and MyD88 signaling, and reduced lipid raft
functionality.
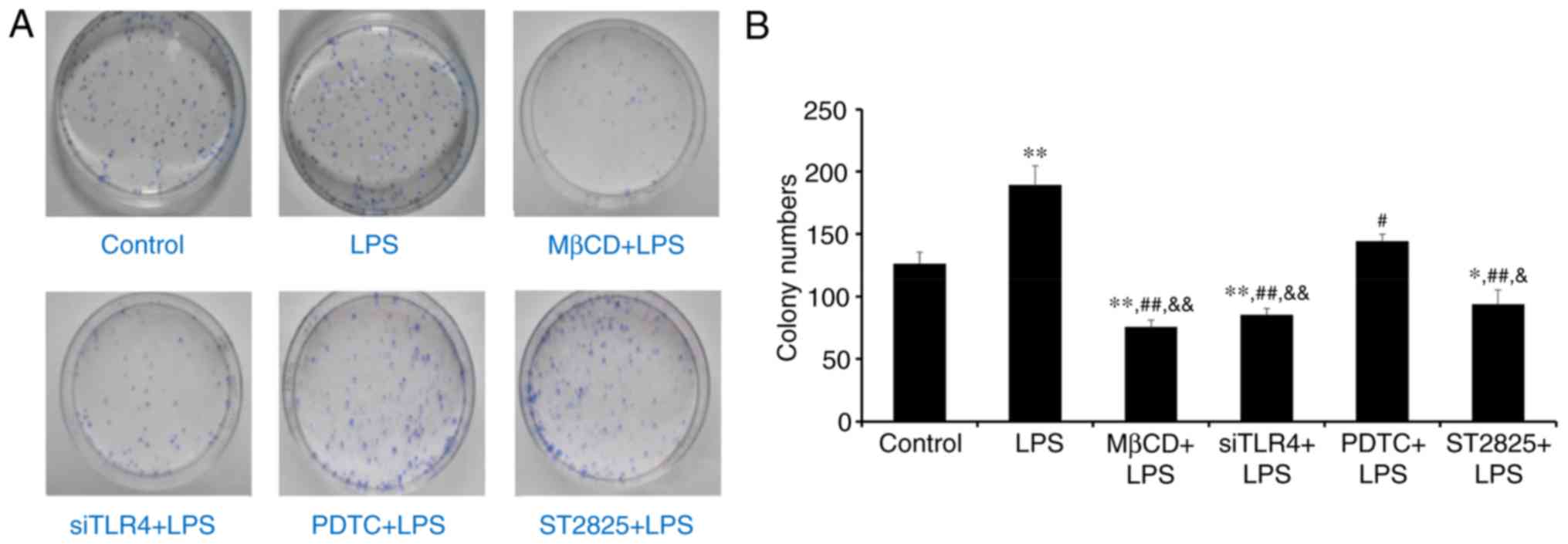
Colony forming experiment
Colony formation was assessed in SiHa cells in soft
agar following 2 weeks of culture. Visible cell clusters (>500
cells) were counted as standard colonies. Images of colonies are
presented in Fig. 2A. The LPS
group produced the most colonies (Fig.
2B). Compared with controls, treatment with LPS significantly
promoted cell growth (P<0.01), while LPS+ST2825, siTLR4 and MβCD
inhibited cell growth. Compared with the LPS group, PDTC+LPS
(P<0.05) and LPS co-administered with ST2825, siTLR4 and MβCD
(all P<0.01) inhibited colony formation. Compared with the
PDTC+LPS group, colony numbers decreased following treatment with
ST2825+LPS (P<0.05) and LPS co-treatment with siTLR4 and MβCD
(P<0.01). Therefore, LPS stimulated cervical cancer cell growth.
Inhibition of TLR4, MyD88, and NF-κB signaling and lipid raft
functionality inhibited growth of cervical cancer cells treated
with LPS. However, inhibition of TLR4 and MyD88 signaling, and
disruption of lipid raft functionality, demonstrated more
significant effects compared with inhibition of NF-κB.
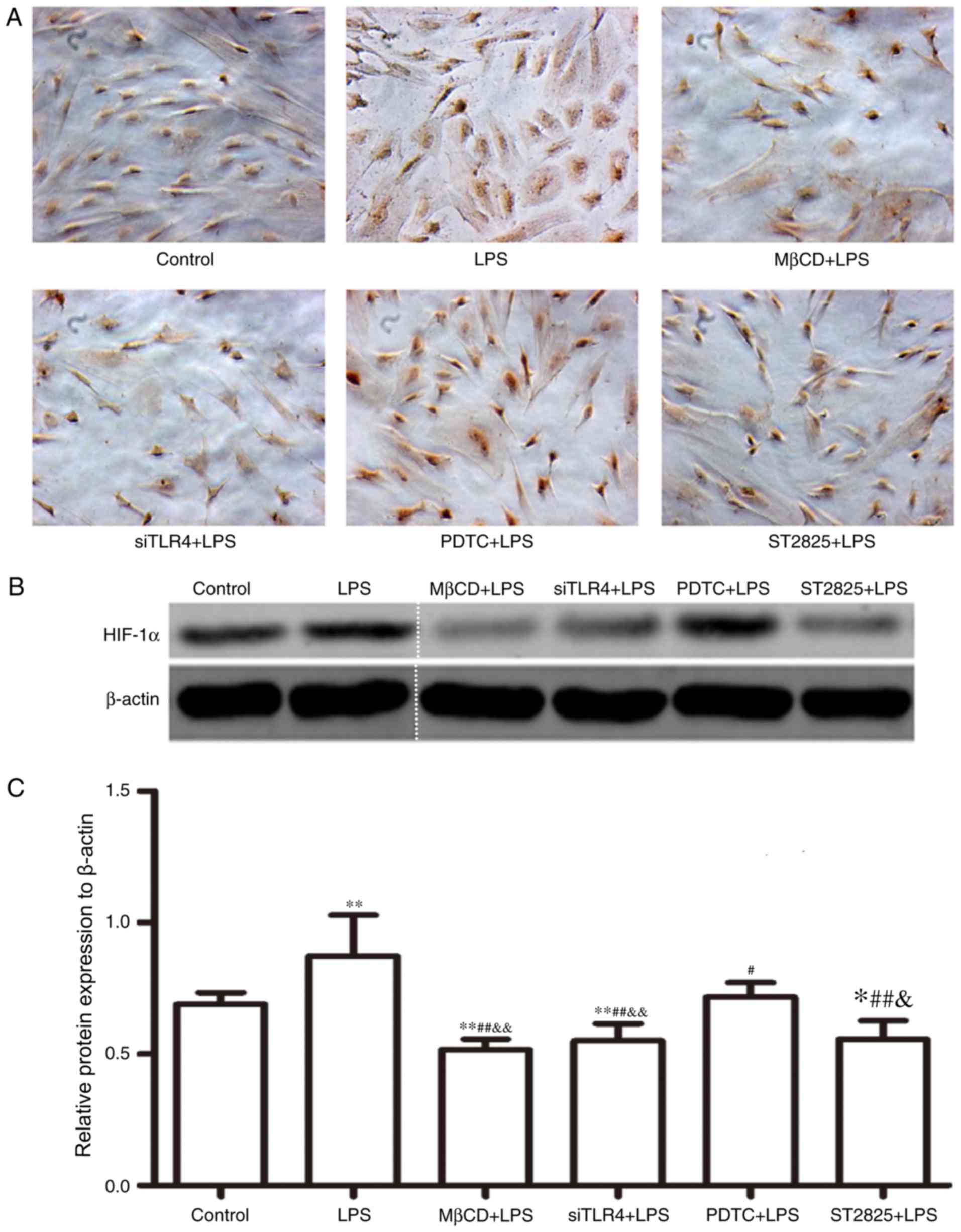
Western blot analysis of HIF-1α
expression
HIF-1α expression was assayed in SiHa cells for 24 h
and immunohistochemical data were collected (Fig. 3A). Western blotting images are
presented in Fig. 3B. Compared
with the control group, HIF-1α expression increased following
treatment with LPS (P<0.01) and decreased when LPS was
co-administered with ST2825 (P<0.05), siTLR4 and MβCD (both
P<0.01). Compared with the LPS treatment, HIF-1α expression
decreased when LPS was administered following pre-treatment with
PDTC (P<0.05), ST2825, siTLR4 and MβCD (P<0.01). HIF-1α
expression was lowest in the MβCD+LPS group. Compared with the
PDTC+LPS treatment group, HIF-1α expression decreased in the
LPS+ST2825 (P<0.05), siTLR4 and MβCD (both P<0.01) treatment
groups. Therefore, the above data suggested that the stimulation of
TLR4 signaling induced the expression of HIF-1α and the inhibition
of TLR4 and MyD88 signaling, and disruption of lipid raft
functionality inhibited HIF-1α expression. Inhibition of the NF-κB
signaling pathway decreased HIF-1α expression, although not to the
same extent as inhibition of TLR4 and MyD88 signaling and lipid
raft disruption.
Lucigenin luminescent assay for NADPH
oxidase activity
NADPH oxidase activity was determined in SiHa cells,
treated as previously described, at 0, 12, 24, 36, and 48 h
(Fig. 4). NADPH oxidase activity
increased over time in all groups. Compared with the control group,
treatment with LPS and PDTC+LPS enhanced NADPH oxidase activity at
24 h (P<0.05). NADPH oxidase activity decreased when LPS was
administered with ST2825, siTLR4 and MβCD. Compared with the LPS
groups, NADPH oxidase activity decreased when LPS was
co-administered with ST2825, siTLR4 and MβCD, although no
significant difference was observed in the PDTC+LPS group. Compared
with the PDTC+LPS group, NADPH oxidase activity in LPS+ST2825,
siTLR4 and MβCD decreased. Therefore, stimulation of TLR4 signaling
increased NADPH oxidase activity and inhibition of lipid raft
functionality, and TLR4 and MyD88 signaling inhibited NADPH oxidase
activity. However, inhibition of NF-κB signaling activation did not
alter NADPH oxidase activity, compared with the levels observed in
the LPS group.
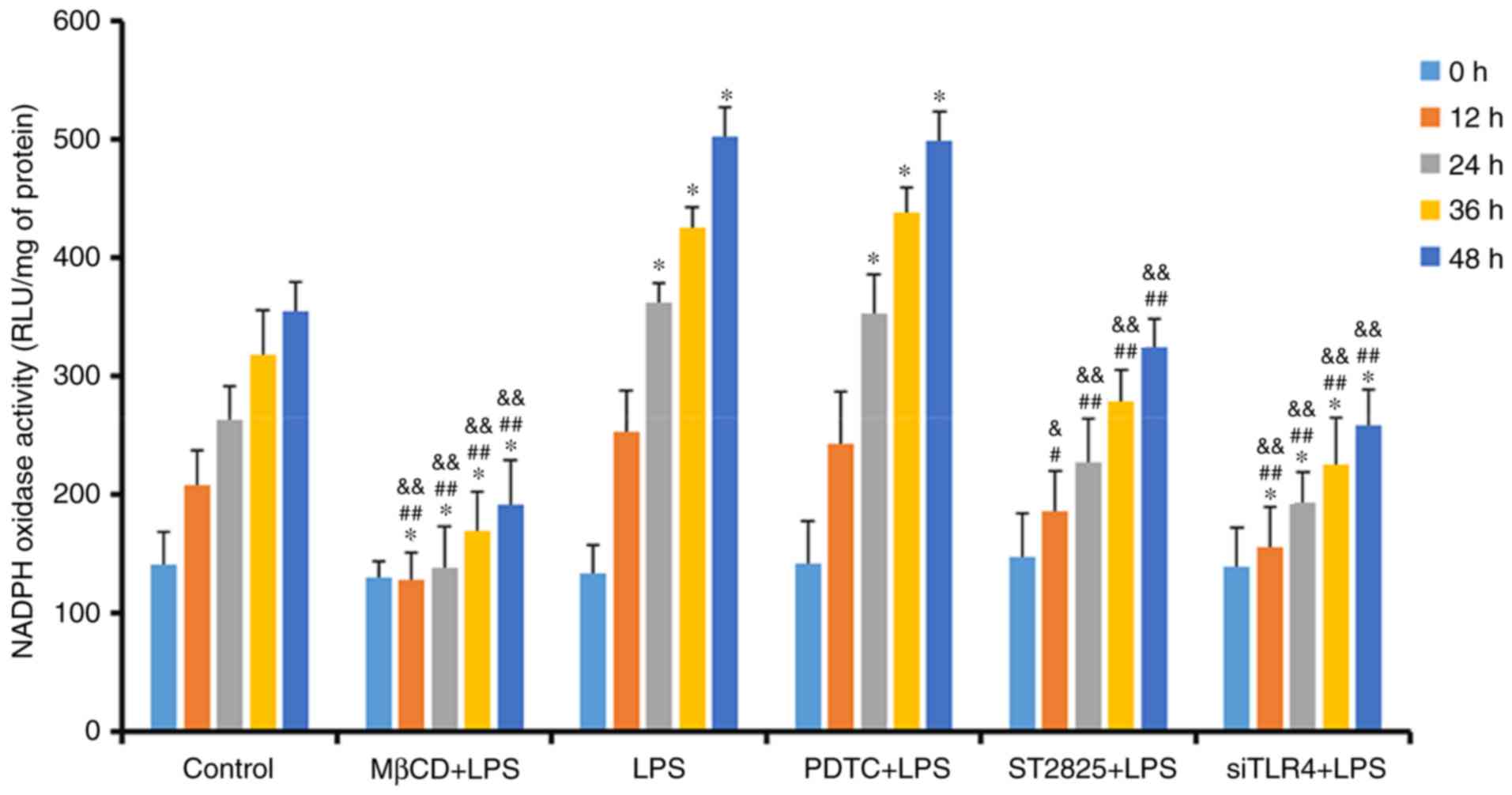 | Figure 4.Detection of NADPH oxidase activity
in treatment groups at 0, 12, 24, 36 and 48 h. *P<0.05 and
**P<0.01 vs. the control group at 0, 12, 24, 36 and 48 h.
#P<0.05, ##P<0.01 vs. the LPS group at
0, 12, 24, 36 and 48 h. &P<0.05 and
&&P<0.01 vs. the PDTC+LPS group at 0, 12, 24,
36 and 48 h. LPS, lipopolysaccharide; si, small interfering RNA;
TLR4, Toll-like receptor 4; MβCD, methyl-β-cyclodextrin; PDTC,
ammonium pyrrolidinedithiocarbamate; nicotinamide-adenine
dinucleotide phosphate; NADPH, nicotinamide-adenine dinucleotide
phosphate; RLU, relative light units. |
DCFH-DA detection of ROS activity
ROS activity alterations in SiHa cells treated with
various interventions were assayed using a DCFH-DA probe, following
0, 12, 24, 36, and 48 h of treatment (Fig. 5). ROS content in the MβCD+LPS group
decreased over time and there was an initial decrease followed by a
gradual increase in the siTLR4+LPS group. ROS in the remaining
groups increased over time. Compared with the control group, ROS
expression was enhanced following treatment with LPS and PDTC+LPS,
and was decreased when LPS was administered following pretreatment
with ST2825, siTLR4 and MβCD (all P<0.05). Compared with the LPS
group, ROS activity decreased in the LPS+ST2825, siTLR4 and MβCD
groups, and was not significantly different compared with the
PDTC+LPS treatment. Compared with the PDTC+LPS treatment, ROS in
SiHa cells decreased in the LPS+ST2825, siTLR4 and MβCD groups (all
P<0.01). Therefore, stimulation of TLR4 signaling promoted the
generation of ROS in cervical cancer cells, and the inhibition of
lipid raft functionality, and the TLR4 and MyD88 signaling
pathways, inhibited ROS production. Inhibition of NF-κB signaling
exerted no significant effect on intracellular ROS levels compared
with the LPS group.
 | Figure 5.ROS fluorescence intensity of each
group detected at 0, 12, 24, 36 and 48 h. *P<0.05 and
**P<0.01 vs. the control group at 0, 12, 24, 36 and 48 h.
##P<0.01, vs. the LPS group at 0, 12, 24, 36 and 48
h. &&P<0.01 vs. the PDTC+LPS group at 0, 12,
24, 36 and 48 h. LPS, lipopolysaccharide; si, small interfering
RNA; TLR4, Toll-like receptor 4; MβCD, methyl-β-cyclodextrin; PDTC,
ammonium pyrrolidinedithiocarbamate; ROS, reactive oxygen
species. |
Fluorescence microscopy detection of
the co-localization of TLR4 and HIF-1α
TLR4 and HIF-1α were detected by immunofluoresce
following treatment as described in the Treatment groups for
24 h. Localization of TLR4 and HIF-1α in cells was investigated
using fluorescent microscopy, and TLR4 and HIF-1α were labeled
using immunofluorescent staining (Fig.
6). HIF-1α expression was labeled in green and TLR4 expression
was labeled in red. TLR4 localized to the cell surface but HIF-1α
primarily localized to the cytoplasm. TLR4 was co-localized with
HIF-1α in cervical cancer cells.
Discussion
The TLR4 receptor is expressed on the surface of a
number of immune cells and serves a role in innate immunity against
bacterial infection. The activation of TLR4 signaling leads to
secretion of cytokines and regulate adaptive immune responses. TLR4
is expressed on multiple tumor cell surfaces, including in lung,
breast and cervical cancer (11,30,31).
Activation of TLR4 signaling on tumor surfaces may promote
proliferation and inhibit apoptosis in tumor cells and, unlike
activation on immune cell surfaces, this may promote tumor growth
(32–34). A previous study indicated that the
elevated expression of TLR4 in cervical cancer cells was positively
associated with increased HIF-1α activity and this is implicated in
cervical cancer growth (23).
HIF-1α is a marker of malignant prognosis and
excessive activation of HIF-1α promotes the growth, invasion and
metastasis of tumor cells, and induces tumor cell resistance to
radiotherapy and chemotherapy (35). Excessive activation of HIF-1α is
associated with inhibition of prolyl hydroxylase enzyme (PHD)
activity, which indicates that ROS may inhibit PHDs, and prevent
the degradation and enhance the stability of HIF-1α (36). A previous study demonstrated that
increased levels of ROS maintain elevated HIF-1α activity in
cervical cancer cells during hypoxia (26). However, the role of elevated
expression of HIF-1α in non-hypoxic areas of cervical cancer
remains to be elucidated.
TLR4 signaling balances the activation of NF-κB
signaling, which inhibits HIF-1α degradation to maintain its
activity (37–39). The present study and previous
studies demonstrated that activation of the TLR4 signaling pathway
is associated with lipid rafts (14,26),
which are microdomains located within the cell membrane that are
rich in cholesterol, sphingolipids and the liquid ordered phase of
receptors. Lipid rafts are dynamic functional platforms through
which external signals are transmitted via receptors to the cells
(40). Lipid rafts provide
activation and transduction platform for the initiation of the TLR4
signaling pathway, which may not be activated when expression of
proteins such as (such as CD14) that form the lipid structure or
fluidity is inhibited. Lipid rafts are not only enriched with
sphingolipids and cholesterol that float within the phospholipid
bilayer cell membrane but also contain many signaling proteins.
These rafts can serve as platforms for the formation of
multicomponent complexes. Many transmembrane receptors are
recruited into lipid raft upon stimulation and resulting in
downstream signaling (41).
Therefore, a TLR4 signaling effect is more apparent following
stimulation of lipid raft function (42). LPS binds to the complex of CD14 and
lipopolysaccharide binding protein (LBP), forming a LPS-LBP-mCD14
complex that can activate the TLR4-MD2-MyD88 recognition complex or
the TLR4-TRAM-TRIF complex on target cells. Then the complexes can
activate NF-κB or the interferon regulatory factor 3 pathway to
cause secretion of tumor necrosis factor-α, interleukin-2 (IL-2),
IL-6, and interferon-γ (23). In
addition, lipid rafts aid in promoting and maintaining ROS levels
derived from NADPH oxidase in cells (43). ROS levels generated by NADPH
oxidase decrease following lipid raft damage, and aggregation of
lipid rafts on cell membranes may modify NADPH oxidase subunits,
causing activation of NADPH oxidase and an increase in
intracellular ROS levels (44–46).
However, the mechanism underlying the lipid raft-mediated
contribution to cervical cancer development, by maintaining
increased HIF-1α activity mediated by TLR4, remains to be
elucidated.
Therefore, the authors of the present study
hypothesized that TLR4 combined with its ligands may trigger the
molecular flow of lipid rafts and induce alterations in their
spatial configuration. Conformational alterations could provide
conditions for the aggregation of NADPH oxidase subunits to the
lipid rafts and then activating redox signals, inhibiting HIF-1α
degradation and increasing its expression.
The present study investigated the proliferation and
cloning of SiHa cells, and alterations in ROS levels, NADPH oxidase
activity and HIF-1α expression following inhibition or activation
of TLR4, MyD88 and NF-κB signaling, and inhibition of lipid raft
functionality. Compared with the control group, LPS stimulated TLR4
signaling which promoted cervical cancer cell growth and increased
HIF-1α expression, NADPH oxidase activity and ROS content.
Silencing of TLR4 expression using siTLR4 inhibited the growth of
cervical cancer cells and reduced HIF-1α expression, NADPH oxidase
activity and ROS content. Therefore, TLR4 may be hypothesized to be
associated with cervical cancer initiation, and the development and
maintenance of elevated HIF-1α expression in cervical cancer cells.
The underlying mechanism may be associated with ROS produced by
NADPH oxidase. Compared with TLR4 signal activation alone,
inhibition of lipid raft functionality following TLR4 activation
significantly inhibited cervical cancer growth and HIF-1α
expression levels, NADPH oxidase activity and ROS levels.
Therefore, lipid rafts serve a role in the activation of TLR4
signaling and ROS content by altering NADPH oxidase activity.
Compared with TLR4 signal activation alone (LPS group), inhibition
of MyD88 and NF-κB signaling following TLR4 activation inhibited
SiHa cell growth and reduced HIF-1α activity. Inhibition of NF-κB
signaling did not exhibit the same effect as TLR4 signaling with
respect to inhibition of SiHa cell growth and the reduction of
HIF-1α activity. NADPH oxidase activity and ROS content decreased
following inhibition of TLR4 signaling, but NADPH oxidase activity
and ROS content were not significantly altered following inhibition
of NF-κB signaling, compared with the LPS group. TLR4 binds LPS via
auxiliary factors lymphocyte antigen 69 and CD14, and
intracellularly transduces signals by activating downstream MyD88
signal transduction molecules, eventually leading to the activation
of NF-κB signaling (47). MyD88 is
an upstream signal of NF-κB (48–50)
which may be associated with the activation of NADPH oxidase
activity independently of NF-κB signaling and may be used to
measure the activity of TLR4. From the present results, it was
demonstrated that NF-κB signaling may be involved in cervical
cancer growth and may increase HIF-1α activity via other
mechanisms, independently of NADPH oxidase.
Compared with inhibition of NF-κB signaling,
inhibition of lipid raft functionality following TLR4 signaling
activation by LPS significantly inhibited cervical cancer cell
growth and reduced intracellular HIF-1α expression, NADPH oxidase
activity and ROS levels. Therefore, based on the results of the
present study, it may be hypothesized that TLR4 primarily increases
NADPH oxidase activity via the lipid raft-associated pathway rather
than the NF-κB signaling pathway and, subsequently, TLR4 is
involved in the maintenance of the elevated expression of HIF-1α,
which promotes cervical cancer. TLR4 and HIF-1α were
immunofluorescently labeled in the present study and their cellular
locations were identified. TLR4 was located on the cell surface,
and HIF-1α was localized to the cytoplasm. TLR4 content was
proportional the levels of HIF-1α. Inhibition of TLR4 signaling and
lipid raft functionality inhibited HIF-1α expression, suggesting
that TLR4 and lipid rafts aid in maintaining elevated HIF-1α
activity in cervical cancer cells.
In conclusion, TLR4 signaling, lipid rafts and NF-κB
signaling contribute to cervical cancer. TLR4 signaling may trigger
ROS production by lipid rafts/NADPH oxidase-dependent mechanisms to
maintain elevated HIF-1α expression. However, the results of the
present study suggested that this may be a separate process from
the activation of NF-κB, which is downstream of TLR4 signaling.
Stimulation of TLR4 may increase HIF-1α expression, although this
effect was more evidently promoted by the TLR4/lipid rafts/NADPH
oxidase pathway compared with the TLR4-NF-κB signaling pathway.
NF-κB signaling may promote cervical cancer through other
mechanisms. The present study suggested a novel mechanism
underlying elevated HIF-1α expression and tumor formation. In
addition, TLR4 signaling pathway intervention may regulate the
lipid raft/NADPH oxidase/ROS/HIF-1α signaling pathway and affect
cancer. TLR4 signal transduction may be suppressed following
inhibition of lipid raft function, and a TLR4 signaling pathway
inhibitor or lipid raft interference may inhibit cancer growth and
aid in the development of novel therapies for the treatment of
cervical cancer.
Acknowledgements
The present study was supported by the Natural
Science Foundation of China (grant no. 81302273) and the Science
and Technology Department Support Project of Hubei Province, China
(grant no. 2015BCA313).
References
|
1
|
Wei KR, Chen WQ, Zhang SW, Zheng RS, Wang
YN and Liang ZH: Epidemiology of uterine corpus cancer in some
cancer registering areas of China from 2003–2007. Zhonghua Fu Chan
Ke Za Zhi. 47:445–451. 2012.(In Chinese). PubMed/NCBI
|
|
2
|
Sharma N, Akhade AS and Qadri A: Src
kinases central to T-cell receptor signaling regulate TLR-activated
innate immune responses from human T cells. Innate Immun.
22:238–244. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Galli R, Starace D, Busà R, Angelini DF,
Paone A, De Cesaris P, Filippini A, Sette C, Battistini L, Ziparo E
and Riccioli A: TLR stimulation of prostate tumor cells induces
chemokine-mediated recruitment of specific immune cell types. J
Immunol. 184:6658–6669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Molteni M, Marabella D, Orlandi C and
Rossetti C: Melanoma cell lines are responsive in vitro to
lipopolysaccharide and express TLR-4. Cancer Lett. 235:75–83. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
MacRedmond R, Greene C, Taggart CC,
McElvaney N and O'Neill S: Respiratory epithelial cells require
Toll-like receptor 4 for induction of human beta-defensin 2 by
lipopolysaccharide. Respir Res. 6:1162005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oblak A and Jerala R: Toll-like receptor 4
activation in cancer progression and therapy. Clin Dev Immunol.
2011:6095792011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roxburgh CS and McMillan DC: The role of
the in situ local inflammatory response in predicting recurrence
and survival in patients with primary operable colorectal cancer.
Cancer Treat Rev. 38:451–466. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Woo JK, Kang JH, Jang YS, Ro S, Cho JM,
Kim HM, Lee SJ and Oh SH: Evaluation of preventive and therapeutic
activity of novel non-steroidal anti-inflammatory drug, CG100649,
in colon cancer: Increased expression of TNF-related
apoptosis-inducing ligand receptors enhance the apoptotic response
to combination treatment with TRAIL. Oncol Rep. 33:1947–1955. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Goto Y, Arigami T, Kitago M, Nguyen SL,
Narita N, Ferrone S, Ferrone S, Morton DL, Irie RF and Hoon DS:
Activation of Toll-like receptors 2, 3, and 4 on human melanoma
cells induces inflammatory factors. Mol Cancer Ther. 7:3642–3653.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu L and Chen S: Toll-like receptors
expressed in tumor cells: Targets for therapy. Cancer Immunol
Immunother. 57:1271–1278. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nishimura M and Naito S: Tissue-specific
mRNA expression profiles of human toll-like receptors and related
genes. Biol Pharm Bull. 28:886–892. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Weng Y, Shi Y, Xia X, Wang S and
Duan H: Expression and functional analysis of Toll-like receptor 4
in human cervical carcinoma. J Membr Biol. 247:591–599. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song J, Fan HJ, Li H, Ding H, Lv Q and Hou
SK: Zingerone ameliorates lipopolysaccharide-induced acute kidney
injury by inhibiting Toll-like receptor 4 signaling pathway. Eur J
Pharmacol. 772:108–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fessler MB and Parks JS: Intracellular
lipid flux and membrane microdomains as organizing principles in
inflammatory cell signaling. J Immunol. 187:1529–1535. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nishi H, Sasaki T, Nagamitsu Y, Terauchi
F, Nagai T, Nagao T and Isaka K: Hypoxia inducible factor-1
mediates upregulation of urokinase-type plasminogen activator
receptor gene transcription during hypoxia in cervical cancer
cells. Oncol Rep. 35:992–998. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Myllyharju J and Koivunen P:
Hypoxia-inducible factor prolyl 4-hydroxylases: Common and specific
roles. Biol Chem. 394:435–448. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yatabe N, Kyo S, Maida Y, Nishi H,
Nakamura M, Kanaya T, Tanaka M, Isaka K, Ogawa S and Inoue M:
HIF-1-mediated activation of telomerase in cervical cancer cells.
Oncogene. 23:3708–3715. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jing S, Xu Q, Jing S, Zhao Z, Zhao Z, Wu
F, Liu Q, Cheng Y and Wang J: Effect of silencing HIF-1α by RNA
interference on adhesion and invasion of the human nasopharyngeal
carcinoma cell line CNE-1. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke
Za Zhi. 50:929–933. 2015.(In Chinese). PubMed/NCBI
|
|
20
|
Tong Y, Yang H, Xu X, Ruan J, Liang M, Wu
J and Luo B: Effect of a hypoxic microenvironment after
radiofrequency ablation on residual hepatocellular cell migration
and invasion. Cancer Sci. 108:753–762. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jiang JH, Wang ZR, Jiang L, Bao Y and
Cheng YX: Mechanism of anti-tumor effect of HIF-1alpha silencing on
cervical cancer in nude mice. Zhonghua Zhong Liu Za Zhi.
31:820–825. 2009.(In Chinese). PubMed/NCBI
|
|
22
|
Peyssonnaux C, Cejudo-Martin P, Doedens A,
Zinkernagel AS, Johnson RS and Nizet V: Cutting edge: Essential
role of hypoxia inducible factor-1alpha in development of
lipopolysaccharide-induced sepsis. J Immunol. 178:7516–7519. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cheng YX, Hu M, Chen L, Huang JL, Xia LB,
Li BS, Zhou LM and Hong L: The mechanism of lipid raft mediating
chemoresistance of cervical cancer. Saudi Med J. 33:508–514.
2012.PubMed/NCBI
|
|
24
|
Niecknig H, Tug S, Reyes BD, Kirsch M,
Fandrey J and Berchner-Pfannschmidt U: Role of reactive oxygen
species in the regulation of HIF-1 by prolyl hydroxylase 2 under
mild hypoxia. Free Radic Res. 46:705–717. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cheng Y, Chen G, Hong L, Zhou L, Hu M, Li
B, Huang J, Xia L and Li C: How does hypoxia inducible factor-1α
participate in enhancing the glycolysis activity in cervical
cancer? Ann Diagn Pathol. 17:305–311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cheng YX, Qi XY, Huang JL, Hu M, Zhou LM,
Li BS and Xu XX: Toll-like receptor 4 signaling promotes the
immunosuppressive cytokine production of human cervical cancer. Eur
J Gynaecol Oncol. 33:291–294. 2012.PubMed/NCBI
|
|
27
|
Feng Z, Wang Z, Yang M, Zhou L and Bao Y:
Polysaccharopeptide exerts immunoregulatory effects via
MyD88-dependent signaling pathway. Int J Biol Macromol. 82:201–207.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carlson CG, Stein L, Dole E, Potter RM and
Bayless D: Agents which inhibit NF-κB signaling block spontaneous
contractile activity and negatively influence survival of
developing myotubes. J Cell Physiol. 231:788–797. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sághy É, Szőke É, Payrits M, Helyes Z,
Börzsei R, Erostyák J, Jánosi TZ, Sétáló G Jr and Szolcsányi J:
Evidence for the role of lipid rafts and sphingomyelin in
Ca2+-gating of Transient Receptor Potential channels in
trigeminal sensory neurons and peripheral nerve terminals.
Pharmacol Res. 100:101–106. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhan Z, Xie X, Cao H, Zhou X, Zhang XD,
Fan H and Liu Z: Autophagy facilitates TLR4- and TLR3-triggered
migration and invasion of lung cancer cells through the promotion
of TRAF6 ubiquitination. Autophagy. 10:257–268. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu L, Wang L, Li M, Zhong J, Wang Z and
Chen S: Expression of toll-like receptor 4 is downregulated during
progression of cervical neoplasia. Cancer Immunol Immunother.
59:1021–1028. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Szczepanski MJ, Czystowska M, Szajnik M,
Harasymczuk M, Boyiadzis M, Kruk-Zagajewska A, Szyfter W, Zeromski
J and Whiteside TL: Triggering of Toll-like receptor 4 expressed on
human head and neck squamous cell carcinoma promotes tumor
development and protects the tumor from immune attack. Cancer Res.
69:3105–3113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang B, Zhao J, Li H, He KL, Chen Y, Chen
SH, Mayer L, Unkeless JC and Xiong H: Toll-like receptors on tumor
cells facilitate evasion of immune surveillance. Cancer Res.
65:5009–5014. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang EL, Qian ZR, Nakasono M, Tanahashi T,
Yoshimoto K, Bando Y, Kudo E, Shimada M and Sano T: High expression
of Toll-like receptor 4/myeloid differentiation factor 88 signals
correlates with poor prognosis in colorectal cancer. Br J Cancer.
102:908–915. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Iovine B, Guardia F, Irace C and
Bevilacqua MA: l-carnosine dipeptide overcomes acquired resistance
to 5-fluorouracil in HT29 human colon cancer cells via
downregulation of HIF1-alpha and induction of apoptosis. Biochimie.
127:196–204. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Acker T, Fandrey J and Acker H: The good,
the bad and the ugly in oxygen-sensing: ROS, cytochromes and
prolyl-hydroxylases. Cardiovasc Res. 71:195–207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nishi K, Oda T, Takabuchi S, Oda S, Fukuda
K, Adachi T, Semenza GL, Shingu K and Hirota K: LPS induces
hypoxia-inducible factor 1 activation in macrophage-differentiated
cells in a reactive oxygen species-dependent manner. Antioxid Redox
Signal. 10:983–995. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tewari R, Choudhury SR, Ghosh S, Mehta VS
and Sen E: Involvement of TNFα-induced TLR4-NF-κB and TLR4-HIF-1α
feed-forward loops in the regulation of inflammatory responses in
glioma. Journal of Molecular Medicine. 2012.90(1): 67–80.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lai FB, Liu WT, Jing YY, Yu GF, Han ZP,
Yang X, Zeng JX, Zhang HJ, Shi RY, Li XY, et al: Lipopolysaccharide
supports maintaining the stemness of CD133(+) hepatoma cells
through activation of the NF-κB/HIF-1α pathway. Cancer Lett.
378:131–141. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Patra SK: Dissecting lipid raft
facilitated cell signaling pathways in cancer. Biochim Biophys
Acta. 1785:182–206. 2008.PubMed/NCBI
|
|
41
|
Wickström SA, Alitalo K and Keski-Oja J:
Endostatin associates with lipid rafts and induces reorganization
of the actin cytoskeleton via down-regulation of RhoA activity. J
Biol Chem. 278:37895–37901. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chansrichavala P, Chantharaksri U, Sritara
P, Ngaosuwankul N and Chaiyaroj SC: Atorvastatin affects TLR4
clustering via lipid raft modulation. Int Immunopharmacol.
10:892–899. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Das S, Alhasson F, Dattaroy D, Pourhoseini
S, Seth RK, Nagarkatti M, Nagarkatti PS, Michelotti GA, Diehl AM,
Kalyanaraman B and Chatterjee S: NADPH oxidase-derived
peroxynitrite drives inflammation in mice and human nonalcoholic
steatohepatitis via TLR4-Lipid raft recruitment. Am J Pathol.
185:1944–1957. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gorudko IV, Mukhortava AV, Caraher B, Ren
M, Cherenkevich SN, Kelly GM and Timoshenko AV: Lectin-induced
activation of plasma membrane NADPH oxidase in cholesterol-depleted
human neutrophils. Arch Biochem Biophys. 516:173–181. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bieberich E: It's a lipid's world:
Bioactive lipid metabolism and signaling in neural stem cell
differentiation. Neurochem Res. 37:1208–1229. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Eum SY, Andras I, Hennig B and Toborek M:
NADPH oxidase and lipid raft-associated redox signaling are
required for PCB153-induced upregulation of cell adhesion molecules
in human brain endothelial cells. Toxicol Appl Pharmacol.
240:299–305. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gribar SC, Anand RJ, Sodhi CP and Hackam
DJ: The role of epithelial Toll-like receptor signaling in the
pathogenesis of intestinal inflammation. J Leukoc Biol. 83:493–498.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tanaka T, Legat A, Adam E, Steuve J, Gatot
JS, Vandenbranden M, Ulianov L, Lonez C, Ruysschaert JM, Muraille
E, et al: DiC14-amidine cationic liposomes stimulate myeloid
dendritic cells through Toll-like receptor 4. Eur J Immunol.
38:1351–1357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kim D and Kim JY: Anti-CD14 antibody
reduces LPS responsiveness via TLR4 internalization in human
monocytes. Mol Immunol. 57:210–215. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Campbell JS, Riehle KJ, Brooling JT, Bauer
RL, Mitchell C and Fausto N: Proinflammatory cytokine production in
liver regeneration is Myd88-dependent, but independent of Cd14,
Tlr2, and Tlr4. J Immunol. 176:2522–2528. 2006. View Article : Google Scholar : PubMed/NCBI
|