Introduction
Glioma, originated from the glial cell, is the most
common intracranial primary tumors, accounting for 80% of malignant
brain tumors (1). Overall
age-adjusted incidence rate for all gliomas ranges from 4.67 to
5.73 per 100 thousand persons with a median survival 2 to 5 years.
However, the median survival is only one year for patients with
glioblastoma multiforme (GBM) (2,3).
Currently, invasive therapy achieves little benefit in glioma owing
to their less effective penetrance to brain tissue. Adjuvant
chemotherapy also has minimal efficacy. Tumor heterogeneity, low
membrane permeability, pre systemic metabolism, and blood brain
barrier (BBB) contribute to therapeutic resistance and tumor
recurrence (4). Therefore, novel
approaches for the treatment of glioma are critically required.
Ginseng, one of the most commonly used Chinese
herbal medicines in the United States, contains multiple effective
components which have been reported to have a wide variety of
biological activities including immunomodulatory, neuroprotection,
anti-aging and anti-tumor effects (5–8).
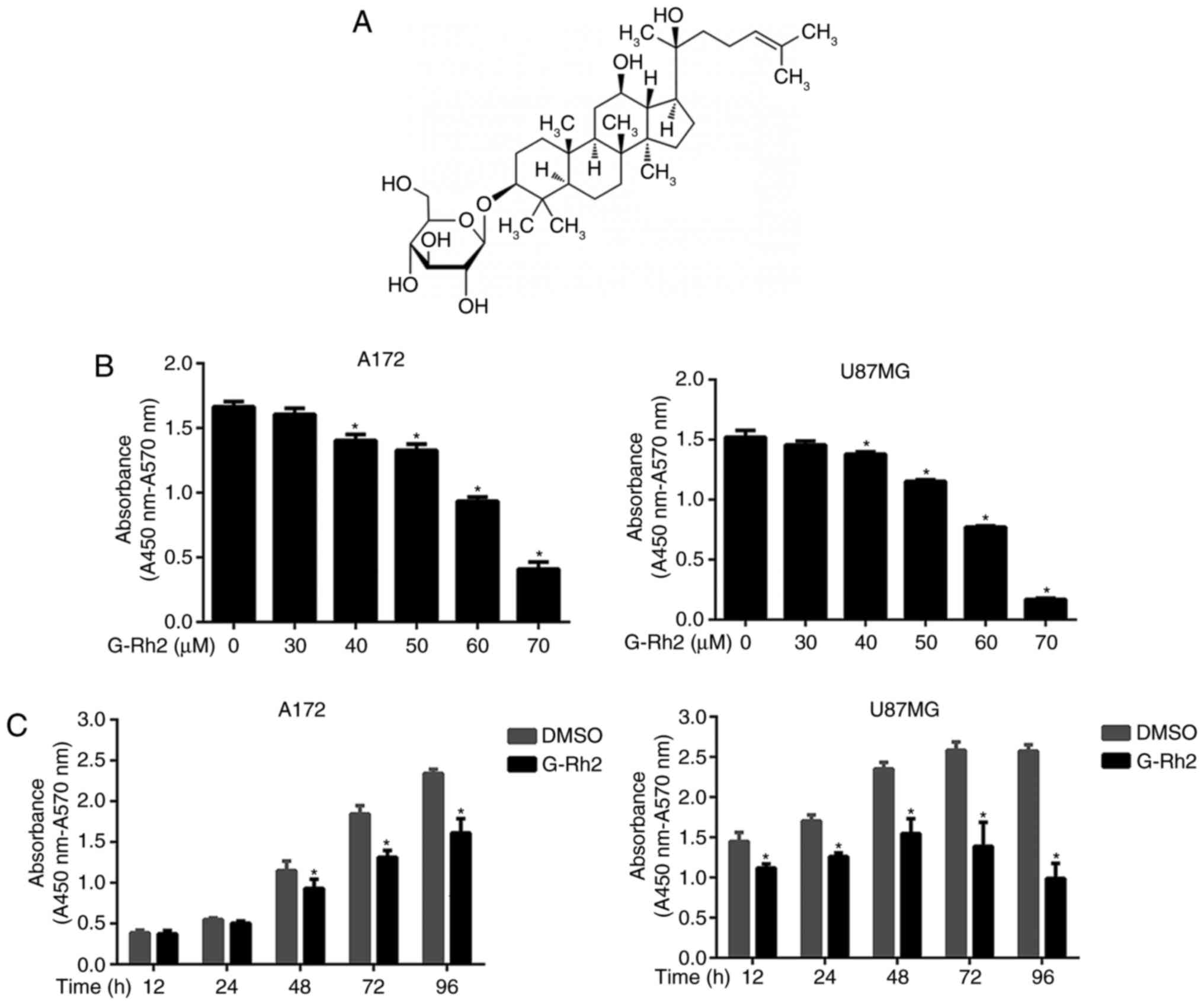
Ginsenoside Rh2 (G-Rh2), a dammarane-type glycoside compounds
(Fig. 1A) separated from Ginseng
has been reported to induce apoptosis by reactive oxygen species or
activating the p53 pathway in several tumors (9–11).
In recent years, increasing insights into the mechanism of G-Rh2
actions have been elucidated. G-Rh2 induced internalization of
rafts and caveolae, have reported to inactivate Akt and induce cell
apoptosis (12). In addition,
G-Rh2 has been shown to affect cell viability by modulating
autophagy (13). These studies
suggest that anti-tumor effect mediated by G-Rh2 might involve
multiply mechanisms.
Cell cycle deregulation is one of the most frequent
alterations associated with tumor development. Thus, the blockade
of cell cycle is regarded as a feasible strategy for targeting
tumor growth. In the process of cell proliferation, the conversion
of G1-S phase is considered to be the key step (14). Upstream signaling molecules such as
hormones or cytokines causes cell proliferation or inhibition,
which ultimately affects retinoblastoma protein (pRb). When
phosphorylated by the CDKs, pRb separate from the E2F factors to
relieve its inhibitory effect. Therefore, E2F factors promote the
transcriptions of genes causing cells to enter the S phase from the
G1 phase (15,16). CDKs activity is regulated by exact
regulatory networks, including the activated factor: Mainly CyclinD
and CyclinE, and the inhibitory factor: INK4 and CIP/KIP families.
Intensive research about the characteristics of tumor cell cycle
progression is helpful to select the optimal target drugs (17).
Previous evidence demonstrates that G-Rh2 blocks the
tumor cell proliferation via multiple mechanisms (9–13).
G-Rh2 induced the cell differentiation and cycle arrest in human
leukemia by up-regulating TGF-β expression (18). G-Rh2 decreased the cells
proliferation and induced the cell apoptosis through inhibiting
EGFR expression in gliomas (19).
However, the underlying molecular mechanistic details of G-Rh2
effects on glioma malignant cells have not been completely
elucidated. In this study, we have investigated the effects of
G-Rh2 on proliferation, cell cycle regulation of human glioma cells
and the underlying molecular mechanisms involved.
Materials and methods
Cell culture
The glioma cell lines A172 and U87MG were purchased
from the American Type Culture Collection (ATCC, Manassas, VA,
USA). Cells were cultured in Dulbecco's modified Eagle's medium
(DMEM; Gibco, Thermo Fisher Scientific Inc., Grand Island, NY, USA)
supplemented with 10% fetal bovine serum (FBS; Biowest, Nuaillé,
France), 100 U/ml penicillin and 0.1 mg/ml streptomycin and
maintained in a humidified incubator at 37°C with 5%
CO2.
Reagents
G-Rh2 (purity ≥98%) used for the present study was
purchased from Aladdin (Shanghai, China). The identity was
confirmed by liquid chromatography-mass spectrometry. G-Rh2 was
prepared in DMSO (Sigma-Aldrich, St. Louis, MO, USA) at a stock
concentration of 10 mM and diluted with fresh complete medium
immediately before use. Cell Counting Kit-8 (CCK-8; Dojindo
Laboratories, Tokyo, Japan), Cell apoptosis kit and Cell Cycle kit
(KeyGEN BioTECH, Jiangsu, China) were obtained from commercial
sources. Antibodies: CDK4 (cat. no. A0336), CyclinD (cat. no.
A1301), CDK2 (cat. no. A2254), CyclinE (cat. no. A0112), p27 (cat.
no. A0026) and β-actin (cat. no. AC006) were purchased from
ABclonal (Boston, USA). Antibodies to detect total-Akt (cat. no.
4691T) and phosphorylated-Akt (p-Akt, Ser473, cat. no. 4060T) were
purchased from Cell Signaling Technology (CST, Danvers, MA, USA).
All of the antibodies mentioned above are rabbit anti-human
antibodies and used in 1:1,000 dilutions except β-actin and p-Akt
(1:2,000 dilution).
Cell viability and cell proliferation
assay
Cytotoxicity was assessed by CCK-8 assay. Briefly,
the indicated number of cells was seeded in 96-well plate and
cultured for 24 h. A series of concentrations of G-Rh2 (30, 40, 50,
60, 70 µM) were used to treat glioma cells and an equal volume of
DMSO (final concentration, <0.1%) was used as a control. After
culturing for specified times, the cytotoxicity of G-Rh2 was
measured using a CCK-8 according to the manufacturer's
instructions. The absorbance was measured by Microplate reader
(Thermo Fisher Scientific Inc.) at 450 nm wavelength.
For measuring cellular proliferation, clone
formation assay was performed. Briefly, 150 A172 cells were
cultured in 6-well plates for 24 h and followed by the treatment
with different concentrations of G-Rh2 (0, 30, 40, 50, 60 µM) for
another 7 days. Cell clones were immobilized by methanol and
stained with crystal violet Solution before photographed and
counted. More than 50 cells were defined as a positive colony and
the number of clones was counted with Image Pro Plus 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA). Colony formation
rate=(number of clones/number of cells inoculated) ×100%.
Flow cytometric analysis
Briefly, G-Rh2-treated cells were digested by
EDTA-free Trypsin and collected by centrifugation at 400 g for 5
min. Collected cells were washed once with Phosphate buffered
saline (PBS). For cell cycle analysis, cells were fixed in 70%
ethanol overnight at 4°C and treated with RNaseA for 30 min in
water bath, and resuspended in 400 µl propidium iodide (PI) for 30
min in dark at 4°C. For cell apoptosis analysis, collected cells
were resuspended in 500 µl binding buffer with 5 µl AnnexinV-FITC
and 5 µl PI for 15 min in dark at room temperature. Cell cycle
distribution and apoptosis proportion was measured by flow
cytometry (BD LSRFortessa™; BD Biosciences, San Diego, CA, USA). A
total of 1.0×104 events were acquired for analysis using
Cell Quest software.
Western blot assay
Proteins were extracted with Cell Total Protein
Extraction Kit (KeyGEN BioTECH, Jiangsu, China). Briefly, G-Rh2
treated cells were lysed in RIPA lysis buffer (Contains Protease
inhibitor, phosphatase inhibitors and PMSF) on ice for 30 min. The
supernatants were collected after centrifugation at
18.8×103 g at 4°C for 30 min. Protein concentration was
determined by Bradford method (KeyGEN BioTECH), and whole
supernatants were mixed with 5xSDS loading buffer (contain 0.25M
Tris-HCl, 1.6M MEDTA (pH=8.5), DTT, LDS, glycerol, bromophenol
blue, pyronin Y) at a ratio of 4:1. Protein samples were heated at
100°C for 10 min and separated on SDS polyacrylamide gels. The
Separating gel concentration was determined by the size of the
target protein molecular weight. The separated proteins were then
transferred to a PVDF membrane (Merck KGaA, Darmstadt, Germany),
blocked with 5% evaporated milk and probed with a primary antibody
for 24 h. After incubation with horseradish peroxidase
(HRP)-conjugated second antibody (cat. no. bs-0295G; Bioss,
Beijing, China), membrane blots were stained with enhanced
chemiluminescent system (Fdbio Science, Hangzhou, China) to
visualize the protein bands. Densitometry of western blots was
performed with Quantity One v4.62 (Bio-Rad Laboratories, Richmond,
CA, USA). The protein levels were first normalized to loading
controls and then normalized to experimental controls. Images shown
in the figures were representative from all individuals.
Statistical analysis
Data were analyzed by one-way analysis of variance
(ANOVA) in SPSS19.0, followed by Dunnett-test when treated groups
were compared only to the control group. For plotting the analysis
findings software GraphPad Prism 6.0 was used and P<0.05 was
considered to indicate a statistically significant difference.
Results
G-Rh2 treatment decreases the
viability of human glioma cells
Initially, for determining the effect of G-Rh2 on
cell viability and Cytotoxicity, U87MG and A172 glioma cell lines
were used. With the increased concentration of G-Rh2 and the
prolongation of treatment period, the cell viability was decreased
gradually (Fig. 1B and C). The
IC50 of G-Rh2 exposure on A172 and U87MG cells for 24 h was 60.0
and 57.2 µM, respectively. Our findings demonstrated that G-Rh2
inhibits the viability of U87MG and A172 glioma cells in a
dose/time-dependent manner. Especially, at 40 µM and above
concentrations or 12–48 h of incubation time, the treatment groups
were statistically significant compared with the control group.
G-Rh2 treatment decreases the clone
formation of human A172 glioma cells
We noticed that with G-Rh2 treatment glioma cells
proliferation was decreased and the dead cells were increased in a
dose-dependent manner (Fig. 2A).
This demonstrates that G-Rh2 may have an impact on cell apoptosis
and proliferation. We further performed clone formation assay to
evaluate two important characters: Cell population dependence and
proliferation ability. It can thus discriminate between cytotoxic
(cell death) and cytostatic (decreased growth rate) effects
(20). Our findings demonstrated
that treatment with G-Rh2 significantly inhibited the clone
formation of A172 cells in a dose-dependent manner (Fig. 2B and C).
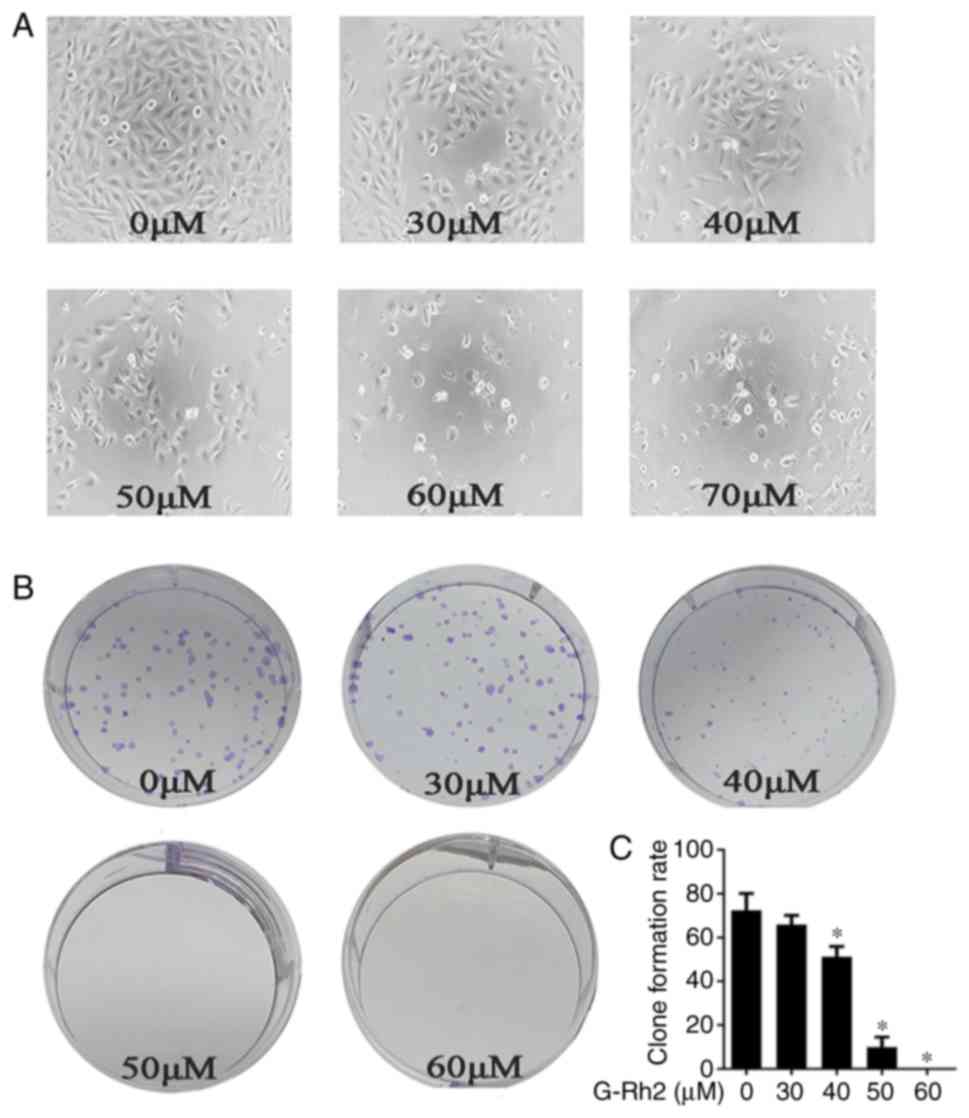 | Figure 2.Effects of G-Rh2 on the proliferation
in A172 glioma cells. (A) The A172 cells were seeded in 96-well
plates and were then treated with G-Rh2 at a series of
concentration (0, 30, 40, 50, 60, 70 µΜ) for 36 h. Morphological
changes: Scattered and fragmented shapes, reduced cell density and
increased cell gap were observed under Inverted optical microscope
(magnification, ×200); (B) The A172 cells were seeded at a density
of 150 cells in 6-well plates and cultured for 48 h before treated
with G-Rh2 at concentration (0, 30, 40, 50, 60 µΜ), and then
cultured for 7 days. With the G-Rh2 concentration increased, the
number of clones decreased gradually. 0 µM (DMSO) was used as a
control and clone formation number were quantified. (C) The data
shown represent mean ± SD of three independent experiments.
*P<0.05 vs. corresponding control group. |
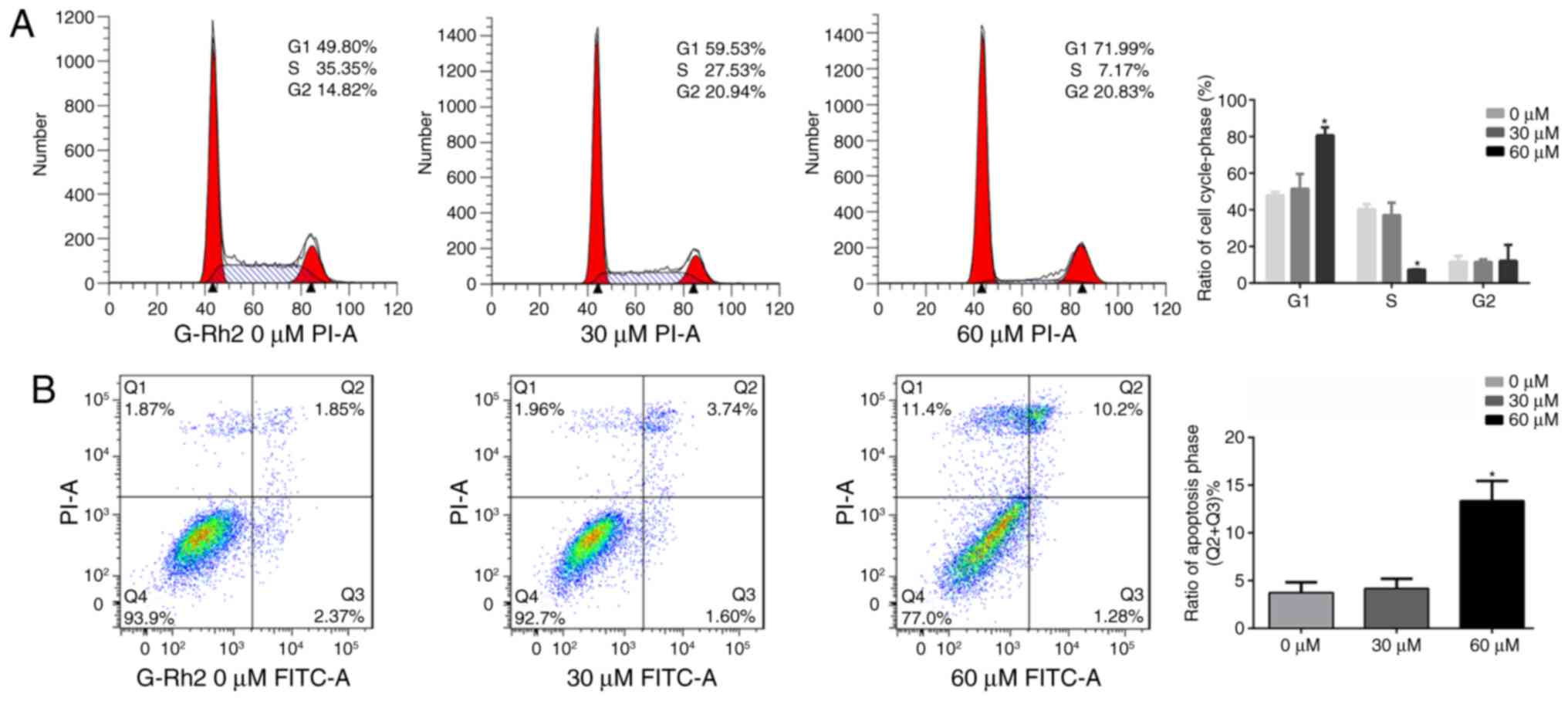
G-Rh2 treatment induces the cell cycle
arrest apoptosis in glioma cells
According clone formation assay, we speculated that
G-Rh2 may exert an anti-tumor effect in A172 cells by inhibiting
proliferation and inducing apoptosis. Therefore, we further
analyzed by flow cytometry. We found that compared to untreated
control group (47.94±1.63%), the proportion of cells in G1 phase
was significantly increased in the G-Rh2 treatment group
(51.48±7.05% at 30 µM and 80.66±7.50% at 60 µM). Correspondingly,
proportion of S-phase was markedly decreased (37.07±8.50% at 30 µM
and 7.29±0.20% at 60 µM) compared with control group (40.40±4.35%).
Therefore, these results indicated that G-Rh2 was able to block the
cell cycle progression and induce G1 phase arrest in A172 glioma
cells (Fig. 3A). Moreover, our
apoptotic analysis of A172 cells indicated that with G-Rh2
treatment apoptotic cells were increased significantly at 60 µM
(13.37%) compared to untreated control group (3.73%) as showed in
Fig. 3B.
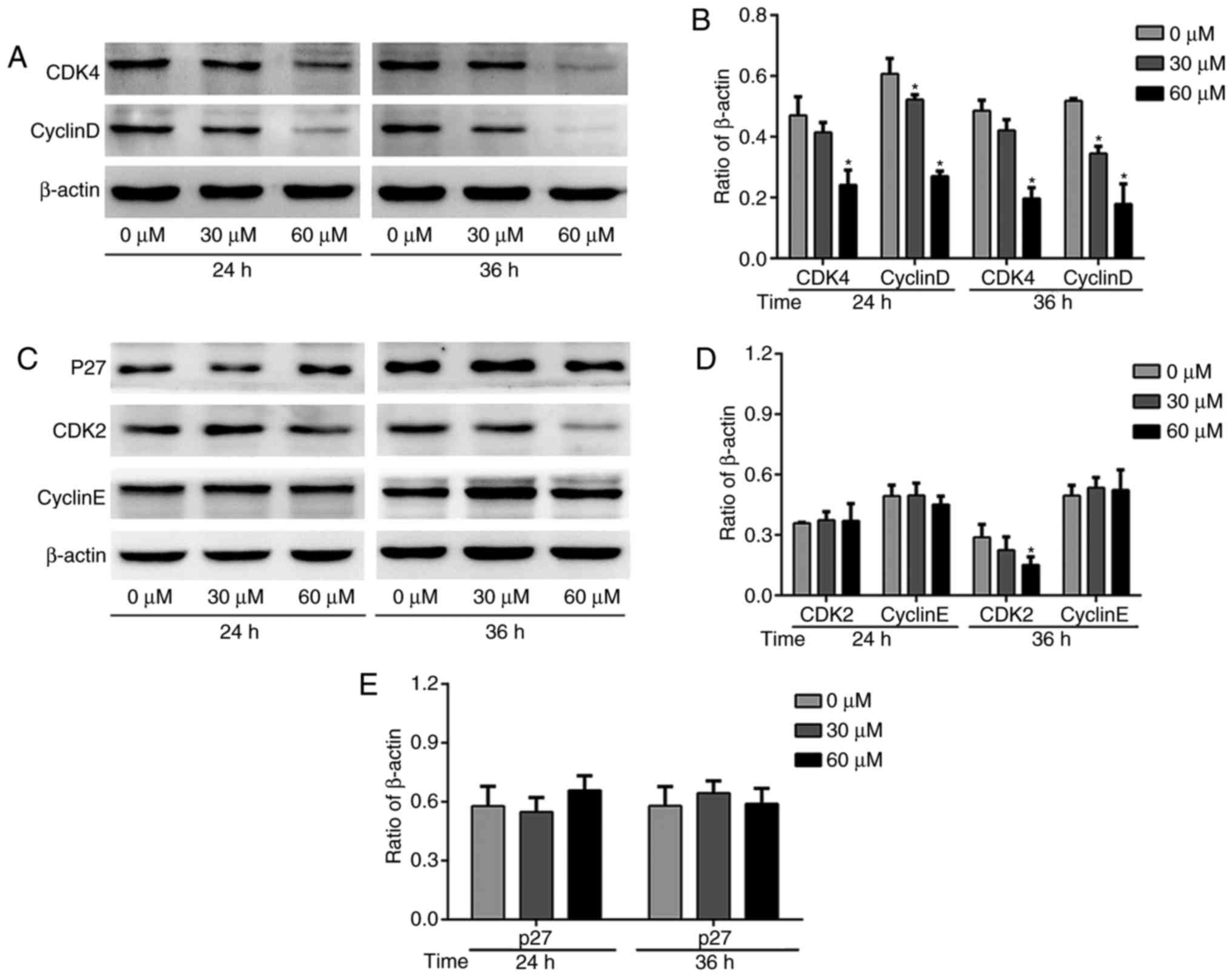
G-Rh2 induces G1 phase arrest by
inhibiting CDK4/CyclinD in A172 cells
Cell cycle related proteins are a class of proteins
with specific periodicity, which can reflect the process of cell
cycle (21). We found that upon
treatment with a series of concentrations of G-Rh2, the CDK4 was
decreased gradually (Fig. 4A and
B). CyclinD, the periodic protein regulating CDK4 activity, was
also decreased (Fig. 4A and B). We
observed modestly decreased CDK2 protein expression and no obvious
on CyclinE protein expression level with G-Rh2 treatment (Fig. 4C and D). We further investigated
the p27 protein, one of the CDKIs family members, which play an
important role in the regulation of G1-S transition to inhibit the
activation of CDK2/CyclinE complex. Consistent with the CyclinE,
there was no significant change after treatment with G-Rh2 in A172
cell line (Fig. 4C and E).
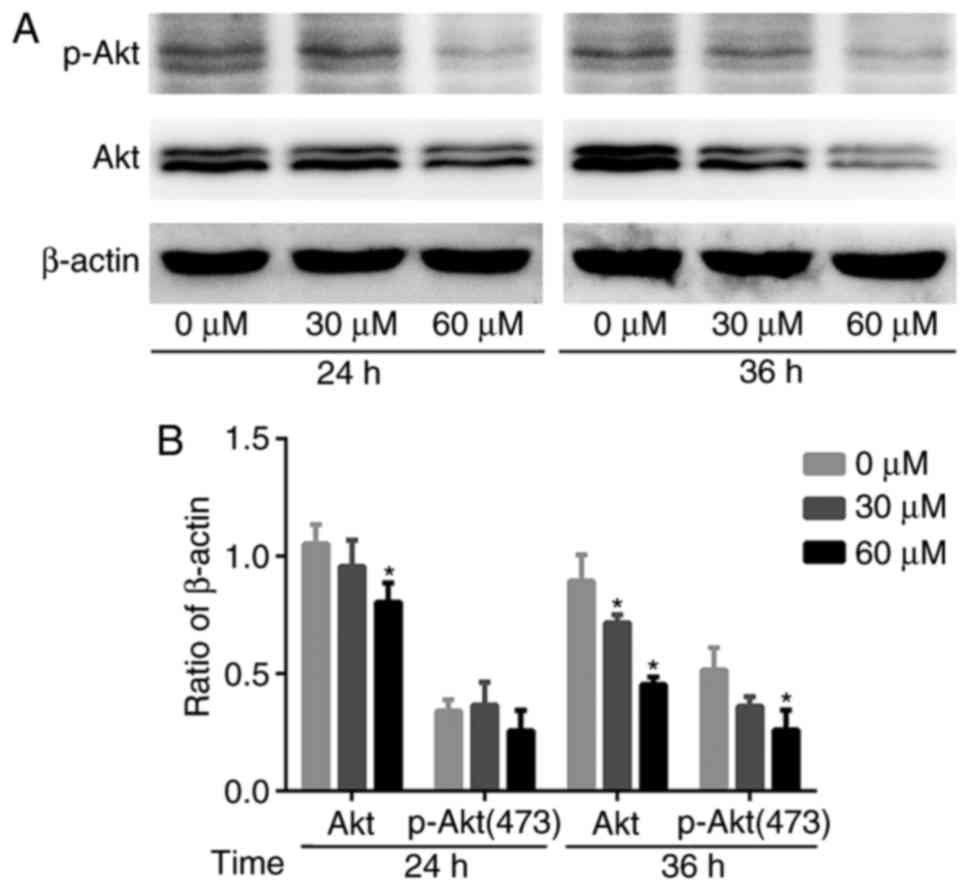
G-Rh2 reduces Akt expression and
phosphorylation
Previous studies have shown that the
phosphatidylinositol 3 kinase (PI3K)/Akt signaling pathway mediates
cell proliferation, survival and motility. Akt is implicated in the
oncogenesis of many cancers (22).
Therefore, we examined the effect of G-Rh2 treatment on Akt
expression and phosphorylation. The expression of total-Akt protein
was decreased gradually and p-Akt (Ser473) also affected by the
treatment with G-Rh2. Therefore, the results suggested that G-Rh2
might be exerting its anti-proliferative effects by inhibiting Akt
expression and phosphorylation, at least partially (Fig. 5A and B).
Discussion
Glioma patients benefit little from standard therapy
(Surgical resection combined with radiotherapy and chemotherapy).
How to improve bioavailability and penetrate the BBB have been
obstacles in chemotherapy drugs. Animal experiments show that the
bioavailability of G-Rh2 is about 16% and it distributed mainly to
the liver and gastrointestinal tissues. After oral administration
at 3 mg/kg, the concentration in plasma was 0.104 µM in dogs
(4). Our findings demonstrated
that G-Rh2 inhibits the viability of U87MG and A172 glioma cells in
a dose/time-dependent manner. The IC50 of G-Rh2 exposure on A172
and U87MG for 24 h was 60.0 and 57.2 µM, respectively. It is
necessary to develop new methods of administration or to change the
physical and chemical properties so that it can pass through BBB.
Recent studies have provided us some new ideas. For example,
liposomes, the small spherical vesicle with single or multiple
lipid bilayers, have been widely exploited due to their unique
characteristics. Animal experiments showed that the concentration
of docetaxel from docetaxel-loaded nanoliposomes in the brain was
significantly increased (23).
Esterification of G-Rh2 enhanced its cellular uptake and antitumor
activity in Human HepG2 Cells (24). The combination of G-Rh2 with
magnetic iron oxide nanoparticles increased the cell uptake
efficiency and convection-enhanced delivery (CED) treatment may
break through the BBB and greatly increased the concentration of
drugs in the brain. But their safety needs further evaluation
(25).
G-Rh2 is classified as a new anticancer drug owing
to its capability of inducing apoptosis in multiple tumor cells
(9–11). In recent years, another utility of
G-Rh2 in inhibiting tumor cell proliferation has been noticed.
Previous studies suggests that Rh2-mediated cell cycle arrest in
MCF-7 cells was accompanied by the down-regulation of CDK and
Cyclins, reduced interaction between CyclinD and CDK4/CDK6 and
increased recruitment of p15INK4B and p27KIP1 to CDK4/CyclinD and
CDK6/CyclinD complexes (26). In
addition, the Rh2-mediated G0/G1 phase cell cycle arrest in A549
lung adenocarcinoma cells has been reported to be correlated with
reduced expression of CDK6, CyclinD1 and CyclinE, but levels of
CDK4, CDK2 and CyclinA were unaltered (27). These researches indicated the
anti-tumor mechanisms involved may be cell line specific.
A172 and U87MG are classic glioma cell lines and are
derived from glioblastoma. Considering the heterogeneity of cell
response to G-Rh2, the cytotoxicity of G-Rh2 was tested at the two
cell lines. The results showed the reactivity to G-Rh2 was
consistent on both cell lines. Previous studies had shown that
G-Rh2 inhibited the expression of EGFR in A172 (19), thus we continued to explore the
following mechanism in the A172. Our further investigation revealed
that cells were arrested at G1 phase of cell cycle and the
expression of cell cycle related proteins CDK4/CyclinD was
decreased gradually with G-Rh2 treatment, which suggest that
CDK4/CyclinD is an essential regulatory target of G-Rh2 for
blocking cell cycle progression. As cell cycle progresses, CyclinE
expression is gradually increased in early G1 phase and in G1 to S
transition CDK2/CyclinE complex exerts a dominant kinase activity.
We found that with G-Rh2 treatment CyclinE level was unaltered,
whereas CDK2 expression is modestly reduced. Further investigation
revealed that p27, the main inhibitor of CDK2/CyclinE, was
unaltered as well. These results further suggested that G-Rh2
blocked cells cycle in the G1 phase. Previous studies indicated
that G-Rh2 only inhibited the kinase activity of CDKs rather than
protein expression (18). Whether
G-Rh2 exerts inhibition of CDKs activity in such manner in glioma
cells needed to be explored.
EGFR mutations are detected in half of
gliomas, which may contribute to tumorgenesis (28). The research indicated G-Rh2 may
have its anti-tumor effect through inhibiting EGFR expression in
A172 glioma cells (19). But the
downstream molecules have not yet been elucidated. The
serine/threonine kinase Akt, also known as protein kinase
B (PKB), is one of the downstream signaling molecules
regulated by EGFR. Since its discovery as a proto-oncogene,
Akt has become a major focus of attention because of its
critical regulatory role in diverse cellular processes, including
cancer progression (29,30). So we decided to investigate the
impact of G-Rh2 on Akt. Our results showed that with G-Rh2
treatment, total-Akt levels were decreased, and the Akt
phosphorylation was inhibited in a dose-dependent manner. This
finding indicates that G-Rh2 could exert anti-tumor activity
through inhibiting Akt pathway in glioma cells. Abnormal Akt
activation was observed in the majority of GBM (31). Akt phosphorylation inhibitors were
able to block effectively the proliferation, migration and invasion
in glioma cells (32). These
studies suggest that inhibition of Akt pathway is an effective
strategy to target glioma. Dozens of Akt inhibitors being in the
Phase II/III clinical trials, but so far they have not really been
used in clinical patients. Some key points such as differences in
pharmacokinetic properties and high toxicity have not been fully
resolved (33). Drug combination
therapy is well worth promoting for which contributes to enhance
the curative effect and reduce drug resistance. PI3K/Akt inhibitors
have been evaluated for their function to resensitize glioma cells
to TMZ (22). Our findings suggest
that G-Rh2 may serve as a potential Akt inhibitor and provide a new
alternative for the development of glioma treatment. Furthermore,
G-Rh2 is reported to suppress the growth of H22 hepatomas without
causing severe side effects in mouse model, which might be a
reference for safe drug use (34).
But the superior advantages compared to Akt inhibitor need further
study.
In summary, our study demonstrates that G-Rh2 exerts
an anti-tumor effect on glioma cells via mechanisms that modulate
the expression of CDK4/CyclinD complex and Akt, might have
potential applications as an effective anti-cancer drug for
glioma.
Acknowledgements
The present study was supported by the Guangdong
Natural Science Foundation (2016A030313525); the Science and
Technology Program of Guangzhou (201607010015); the Guangdong
Province Medical Research Foundation (A2015381) and the President
Foundation of Nanfang Hospital, Southern Medical University
(2014C016). We would like to thank Professor Qinghe Meng for the
suggestion and guidance.
References
|
1
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ostrom QT, Bauchet L, Davis FG, Deltour I,
Fisher JL, Langer CE, Pekmezci M, Schwartzbaum JA, Turner MC, Walsh
KM, et al: The epidemiology of glioma in adults: A ‘state of the
science’ review. Neuro-Oncol. 16:896–913. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang H, Xu T, Jiang Y, Xu H, Yan Y, Fu D
and Chen J: The challenges and the promise of molecular targeted
therapy in malignant gliomas. Neoplasia. 17:239–255. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gu Y, Wang GJ, Sun JG, Jia YW, Wang W, Xu
MJ, Lv T, Zheng YT and Sai Y: Pharmacokinetic characterization of
ginsenoside Rh2, an anticancer nutrient from ginseng, in rats and
dogs. Food Chem Toxicol. 47:2257–2268. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bahrke MS and Morgan WR: Evaluation of the
ergogenic properties of ginseng: An update. Sports Med. 29:113–133.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jin Y, Kotakadi VS, Ying L, Hofseth AB,
Cui X, Wood PA, Windust A, Matesic LE, Pena EA, Chiuzan C, et al:
American ginseng suppresses inflammation and DNA damage associated
with mouse colitis. Carcinogenesis. 29:2351–2359. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shen CY, Jiang JG, Yang L, Wang DW and Zhu
W: Anti-ageing active ingredients from herbs and nutraceuticals
used in traditional Chinese medicine: Pharmacological mechanisms
and implications for drug discovery. Br J Pharmacol. 174:1395–1425.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cui X, Jin Y, Poudyal D, Chumanevich AA,
Davis T, Windust A, Hofseth A, Wu W, Habiger J, Pena E, et al:
Mechanistic insight into the ability of American ginseng to
suppress colon cancer associated with colitis. Carcinogenesis.
31:1734–1741. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li B, Zhao J, Wang CZ, Searle J, He TC,
Yuan CS and Du W: Ginsenoside Rh2 induces apoptosis and
paraptosis-like cell death in colorectal cancer cells through
activation of p53. Cancer Lett. 301:185–192. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park HM, Kim SJ, Kim JS and Kang HS:
Reactive oxygen species mediated ginsenoside Rg3- and Rh2-induced
apoptosis in hepatoma cells through mitochondrial signaling
pathways. Food Chem Toxicol. 50:2736–2741. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guo XX, Li Y, Sun C, Jiang D, Lin YJ, Jin
FX, Lee SK and Jin YH: p53-dependent Fas expression is critical for
Ginsenoside Rh2 triggered caspase-8 activation in HeLa cells.
Protein Cell. 5:224–234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park EK, Lee EJ, Lee SH, Koo KH, Sung JY,
Hwang EH, Park JH, Kim CW, Jeong KC, Park BK and Kim YN: Induction
of apoptosis by the ginsenoside Rh2 by internalization of lipid
rafts and caveolae and inactivation of Akt. Br J Pharmacol.
160:1212–1223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu S, Chen M, Li P, Wu Y, Chang C, Qiu Y,
Cao L, Liu Z and Jia C: Ginsenoside rh2 inhibits cancer stem-like
cells in skin squamous cell carcinoma. Cell Physiol Biochem.
36:499–508. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hengstschläger M, Braun K, Soucek T,
Miloloza A and Hengstschläger-Ottnad E: Cyclin-dependent kinases at
the G1-S transition of the mammalian cell cycle. Mutat Res.
436:1–9. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giacinti C and Giordano A: RB and cell
cycle progression. Oncogene. 25:5220–5227. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Munro S, Carr SM and La Thangue NB:
Diversity within the pRb pathway: Is there a code of conduct.
Oncogene. 31:4343–4352. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lapenna S and Giordano A: Cell cycle
kinases as therapeutic targets for cancer. Nat Rev Drug Discov.
8:547–566. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chung KS, Cho SH, Shin JS, Kim DH, Choi
JH, Choi SY, Rhee YK, Hong HD and Lee KT: Ginsenoside Rh2 induces
cell cycle arrest and differentiation in human leukemia cells by
upregulating TGF-β expression. Carcinogenesis. 34:331–340. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li S, Gao Y, Ma W, Guo W, Zhou G, Cheng T
and Liu Y: EGFR signaling-dependent inhibition of glioblastoma
growth by ginsenoside Rh2. Tumor Biol. 35:5593–5598. 2014.
View Article : Google Scholar
|
|
20
|
Plumb JA: Cell sensitivity assays:
Clonogenic assay. Methods Mol Med. 88:159–164. 2004.PubMed/NCBI
|
|
21
|
Diaz-Moralli S, Tarrado-Castellarnau M,
Miranda A and Cascante M: Targeting cell cycle regulation in cancer
therapy. Pharmacol Ther. 138:255–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Burris HA III: Overcoming acquired
resistance to anticancer therapy: Focus on the PI3K/AKT/mTOR
pathway. Cancer Chemother Pharmacol. 71:829–842. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shaw TK, Mandal D, Dey G, Pal MM, Paul P,
Chakraborty S, Ali KA, Mukherjee B, Bandyopadhyay AK and Mandal M:
Successful delivery of docetaxel to rat brain using experimentally
developed nanoliposome: A treatment strategy for brain tumor. Drug
Deliv. 24:346–357. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen F, Deng ZY, Zhang B, Xiong ZX, Zheng
SL, Tan CL and Hu JN: Esterification of Ginsenoside Rh2 enhanced
its cellular uptake and antitumor activity in human HepG2 cells. J
Agric Food Chem. 64:253–261. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaluzova M, Bouras A, Machaidze R and
Hadjipanayis CG: Targeted therapy of glioblastoma stem-like cells
and tumor non-stem cells using cetuximab-conjugated iron-oxide
nanoparticles. Oncotarget. 6:8788–8806. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Choi S, Kim TW and Singh SV: Ginsenoside
Rh2-mediated G1 phase cell cycle arrest in human breast cancer
cells is caused by p15Ink4B and p27Kip1-dependent inhibition of
cyclin-dependent kinases. Pharm Res. 26:2280–2288. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cheng CC, Yang SM, Huang CY, Chen JC,
Chang WM and Hsu SL: Molecular mechanisms of ginsenoside
Rh2-mediated G1 growth arrest and apoptosis in human lung
adenocarcinoma A549 cells. Cancer Chemother Pharmacol. 55:531–540.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Viana-Pereira M, Lopes JM, Little S,
Milanezi F, Basto D, Pardal F, Jones C and Reis RM: Analysis of
EGFR overexpression, EGFR gene amplification and the EGFRvIII
mutation in portuguese high-grade gliomas. Anticancer Res.
28:913–920. 2008.PubMed/NCBI
|
|
29
|
Hill MM and Hemmings BA: Inhibition of
protein kinase B/Akt: Implications for cancer therapy. Pharmacol
Ther. 93:243–251. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yoeli-Lerner M and Toker A: Akt/PKB
signaling in cancer: A function in cell motility and invasion. Cell
Cycle. 5:603–605. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gallia GL, Tyler BM, Hann CL, Siu IM,
Giranda VL, Vescovi AL, Brem H and Riggins GJ: Inhibition of Akt
inhibits growth of glioblastoma and glioblastoma stem-like cells.
Mol Cancer Ther. 8:386–393. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chautard E, Ouédraogo ZG, Biau J and
Verrelle P: Role of Akt in human malignant glioma: From oncogenesis
to tumor aggressiveness. J Neurooncol. 117:205–215. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mayer IA and Arteaga CL: The PI3K/AKT
pathway as a target for cancer treatment. Annu Rev Med. 67:11–28.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lv Q, Rong N, Liu LJ, Xu XL, Liu JT, Jin
FX and Wang CM: Antitumoral activity of (20R)- and
(20S)-Ginsenoside Rh2 on transplanted hepatocellular carcinoma in
mice. Planta Med. 82:705–711. 2016. View Article : Google Scholar : PubMed/NCBI
|