Introduction
Adiponectin (APN), a 30-kDa protein hormone, is
primarily secreted by adipocytes into the circulation and is
encoded by apM1. It is also secreted by the kidney (1–3) and
placenta (4). The bioactivity of
APN is mediated through APN receptors (AdipoRs), among which
AdipoR1 and AdipoR2 are well defined and are expressed at different
locations with different functions (5). AdipoR1 binds to globular APN (gAd) to
activate and phosphorylate adenosine monophosphate-activated
protein kinase (AMPK) in the kidney. AdipoR1 action is similar in
the skeletal muscle, synovial fibroblasts, atrial cells and
endothelial cells (1,5–10).
AdipoR2 is primarily expressed in the liver and weakly expressed in
the kidney, and mediates the activation of peroxisome
proliferator-activated receptor α (5,11–13).
Studies have demonstrated that AdipoR1 protects the kidney against
inflammatory, fibrotic or oxidative damage through the activation
of AMPK (14–19).
The role of circulating APN has been widely
investigated in different nephropathies. In patients with type 2
diabetes, circulating APN is reduced in the absence of diabetic
nephropathy or during early nephropathy (20,21),
but is increased when advanced albuminuria develops (21). In adenine-induced chronic kidney
disease models, APN levels in the serum and urine are increased
(22). In the offspring of rats
receiving a high-fat and high-fructose diet, hypoadiponectinemia
was observed, which was accompanied by increased urinary albumin
excretion (UAE), glomerulosclerosis and renal transforming growth
factor-β1 expression, and reduced podocin (19). In addition, serum APN levels are
reported to be inversely correlated with UAE (19). These results indicate that
circulating APN may protect against renal injury.
Hypoadiponectinemia is associated with a lower degree of
renoprotection and is therefore associated with albuminuria during
early diabetes. Higher serum APN levels during established diabetic
nephropathy may be a protective response to mitigate renal
lesions.
However, blood may not be an optimal choice for
measuring APN levels, as serum APN is influenced by a low
glomerular filtration rate or by medication (14). There is limited evidence concerning
the intrarenal distribution of APN in kidney diseases. The
expression of APN in glomerular endothelia is dramatically reduced
in patients with diabetic nephropathy (2) or systemic lupus erythematosus with
glomerular hypercellularity, sclerosis and interstitial
inflammation (3). In an
adenine-induced chronic kidney disease model, renal AdipoR1 and
AdipoR2 mRNA was elevated and positively correlated with serum and
urinary APN levels (22). These
findings indicate that the local APN pathway may also be protective
against renal injury. However, Perri et al (11) reported an increase in APN protein,
AdipoR1 mRNA and the downstream phosphorylated
(p)-AMPK/p-extracellular signal-regulated kinase/p-c-Jun N-terminal
kinase (JNK) activities in lipopolysaccharide (LPS)-treated HK-2
cells. Notably, APN mediates the nuclear translocation of nuclear
factor-κB (NF-κB) and p-c-Fos/p-c-Jun (activator protein 1), which
are both induced by pJNK and promote the transactivation of tumor
necrosis factor (TNF)-α, thereby worsening renal inflammation in an
autocrine-dependent manner instead of exerting an anti-inflammatory
effect (11). This controversial
finding challenges the physiological effect of APN and merits
further investigation.
Uric acid is an independent predictor of the
development, progression and prognosis of chronic kidney disease
(23–27). It causes renal inflammation in a
crystal-dependent and independent manner (28). Soluble uric acid (SUA) has been
reported to induce inflammation through neutrophil and monocyte
chemotaxis (29,30) or through the activation of an NLR
family pyrin domain-containing (NLRP) 3 inflammasome and the
secretion of TNF-α (31). Our
previous study demonstrated that SUA activated NLRP3 and increased
interleukin (IL) 1β production in cultured primary human renal
proximal tubule epithelial cells (PTECs) (32). Based on the association between
inflammation and APN signaling, APN may have a critical role in
SUA-induced inflammation. However, to the best of our knowledge, no
data currently exists to support or refute this hypothesis.
In the current study, we evaluated the in
vivo intrarenal expression of APN and AdipoR1 by
immunohistochemistry and the renal pathology in a hyperuricemic
(HUA) model. We further investigated the protein expressions of APN
pathway and that of NLRP3 by immunoblotting and immunofluorescence
in cultured PTECs exposed to SUA.
Materials and methods
Materials
The primary human renal PTEC cell line (cat. no.
4100) and epithelial cell culture medium (EpiCM; cat. no. 4101)
were provided by ScienCell Research Laboratories, Inc. (San Diego,
CA, USA). BioXtra uric acid and oxonic acid potassium salt were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Primary antibodies against APN and AdipoR1 were from Abcam
(Cambridge, UK), and anti-NLRP3 was purchased from Novus
Biologicals, LLC (Littleton, CO, USA). GAPDH primary antibody and
goat anti-mouse and goat anti-rabbit IgG horseradish peroxidase
(HRP)-conjugated secondary antibodies for immunoblotting were
obtained from Sino Biological, Inc. (Beijing, China). For
immunohistochemistry, the goat anti-mouse and goat anti-rabbit
fluorescein isothiocyanate (FITC)-conjugated secondary antibodies
were from Abcam (Cambridge, UK). The goat anti-mouse and goat
anti-rabbit IgG HRP-conjugated secondary antibodies were purchased
from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Hydrogen
peroxide (cat. no. ZLI-9312), ethylenediaminetetraacetic acid
(EDTA; cat. no. ZLI-9068) solution for antigen retrieval and bovine
serum albumin (BSA; cat. no. ZLI-9027) were purchased from OriGene
Technologies, Inc. (Beijing, China). 3,3′-diaminobenzidine (DAB)
and DAPI were provided by OriGene Technologies, Inc. Instant
hematoxylin and eosin (H&E; cat. nos. ar1180-1 and ar1180-2)
solution were purchased from Boster Biological Technology
(Pleasanton, CA, USA).
SUA preparation
SUA was freshly prepared prior to each experiment as
previously described (33).
Briefly, the BioXtra uric acid was dissolved in 1 M NaOH at a final
concentration of 50 mg/ml. The solution was filtered (22 µm pore
size) and tested for mycoplasma and endotoxin (31). There were no detectable crystals
(polarizing microscopy) in the solution neither at the onset nor
during the incubation period.
Cell culture
PTECs were incubated at 37°C in a humidified 5%
CO2 and 95% air atmosphere. Cells of less than four
passages were used in the following experiments. In all
experiments, PTECs were cultured in epithelial cell medium, which
comprised 500 ml basal medium, 50 ml fetal bovine serum, 5 ml
epithelial cell growth supplement and 5 ml penicillin/streptomycin
solution (32). The medium was
changed every other day.
Cell viability
Growth-arrested PTECs were seeded onto 96-well
plates (0.25×105 cells per well) and exposed to SUA at
increasing concentrations (0, 12.5, 25, 50, 100, 150 and 200 µg/ml)
for different durations (24, 48 and 72 h). Control cells were
treated with blank medium. A commercial MTT assay kit (Amresco,
LLC, Solon, OH, USA) was used to measure the cell viability of
PTECs. Cells were incubated with MTT solution for 4 h and crystals
were dissolved in dimethyl sulfoxide. Cell viability was recorded
as a percentage change in the absorbance (570 nm) of treated cells
compared with the control cells.
HUA rat models
Male, 8-weeks-old specific pathogen-free
Sprague-Dawley rats (n=12; 200-250 g) were bred at the Animal
Center in the School of Pharmacy, Fudan University (Shanghai,
China). Rats were kept in groups in a 12 h light/dark cycle at 25°C
with free access to food and water. Rats were acclimatized for 1
week prior to being divided into the following three groups (n=4
per group): Control group, where mice received routine chow; HUA
group, which received oral administration of a uricase inhibitor,
oxonic acid (750 mg/kg), daily for 10 weeks; and the febuxostat
(FEB) group, which received 10 weeks oral gavage of oxonic acid
(750 mg/kg) daily, with 4 weeks gastric FEB (5 mg/kg) daily after 6
weeks of oxonic acid treatment. After 10 weeks, rats were
intraperitoneally anesthetized with 2% sodium pentobarbital (40
mg/kg). Blood was collected into non-heparinized tubes by cardiac
puncture after 8 h fasting until rats died due to exsanguination.
Blood was centrifuged at 2,000 × g for 10 min at 4°C to harvest
sera samples. Serum creatinine (SCr), uric acid, blood urea
nitrogen (BUN) and glucose levels were determined using a P800
automatic biochemical analyzer (Roche Diagnostics Beijing, China).
Fasting insulin (FINS) was measured with a i2000 chemiluminescence
analyzer (Abbott Pharmaceutical Co., Ltd., Lake Bluff, IL, USA).
Homeostasis model assessment-insulin resistance (HOMA-IR) was also
deduced, as described previously (34). Urine samples were collected 24 h
prior to sacrifice. The 24 h urinary protein (UP) was detected with
the sulfosalicylic acid method using a BN ProSpec®
protein analyzer (Siemens Healthineers, Erlangen, Germany). Kidneys
were collected and weighed, and the renal cortex was fixed with 4%
paraformaldehyde for 24 h at room temperature, embedded in paraffin
and submitted for H&E and immunohistochemical staining. The
remaining tissues were stored at −80°C for western blotting assays.
All animal procedures were in accordance with National Institute of
Health guidelines (35) and were
approved by the Animal Care and Use Committee of Fudan University
(Shanghai, China).
Immunofluorescence, histological and
immunohistochemical staining
Following treatment with control or 100 µg/ml SUA
for 48 h, PTECs were seeded at a density of 4×105
cells/well and were fixed with 4% paraformaldehyde pre-cooled at
4°C for 15 min, permeabilized with 0.2% Triton-X-100 for 15 min and
blocked with 10% BSA (OriGene Technologies, Inc.) for 1 h at room
temperature, prior to incubation with antibodies against APN (cat.
no. ab22554; 1:100) and AdipoR1 (cat. no. ab126611; 1:200) for 1 h
at room temperature. All dilutions were made in 1% BSA in PBS.
Subsequently, cells were washed three times with PBS prior to
incubation with goat anti-mouse (cat. no. ab6785; 1:2,000) and
anti-rabbit (cat. no. ab6717; 1:500) FITC-conjugated secondary
antibodies for 50 min at 37°C, followed by PBS washing and staining
with DAPI for 5 min at room temperature. Images were acquired using
a confocal fluorescence microscope (Leica TCS-SP5; Leica
Microsystems GmbH, Wetzlar, Germany). Two pathologists evaluated
the cellular localization of each protein in a blinded manner.
In rats, renal tissue samples were fixed in 4%
paraformaldehyde at room temperature for 24 h, dehydrated in a
graded alcohol series, washed in xylene and paraffin-embedded prior
to being cut into 3 µm sections. Instant H&E solution was
subsequently used to stain renal slides for 6 min at room
temperature. Intrarenal APN, AdipoR1 and NLRP3 expression was
determined by immunohistochemistry and semi-quantification on 3 µm
paraffin-embedded sections of renal cortices (2). Sections were deparaffinized,
endogenous peroxidase activity was blocked with 3% hydrogen
peroxide (OriGene Technologies, Inc.) and antigens were retrieved
by EDTA (cat. no. ZLI-9068; 1:50) in a microwave oven for 20 min.
Slides were blocked in 5% BSA (OriGene Technologies, Inc.) at 37°C
for 30 min. Primary antibodies against APN (cat. no. Ab22554,
1:1,000), AdipoR1 (cat. no. Ab 126611, 1:2,000) and NLRP3 (cat. no.
NBP2-12446, 1:1,000) were subsequently incubated at 4°C overnight,
followed by incubation with horseradish peroxidase-conjugated
secondary antibodies (cat. no. sc-2012; 1:200 and cat. no. sc-2005;
1:200) purchased from Santa Cruz Biotechnology, Inc. at 37°C for 1
h. The color reaction was induced by DAB, followed by
counterstaining with 10% Mayer's hematoxylin at room temperature
for 1 min. PBS was used as the negative control. Two pathologists
blinded to the nature of samples reviewed histological sections
under light microscopy. Positive signals were indicated as brown.
Quantification was performed by examining 10 (magnification, ×400)
fields per sample. The area percentage for the stained tubules was
evaluated by eye and the proportion score (PS) was graded as 0 (no
positive staining), 1 (≤5%), 2 (6–10%), 3 (11–20%) or 4 (>20%).
The intensity of staining was assessed as intensity score (IS) and
was scaled as 0 (no staining), 1 (weak staining or light yellow), 2
(moderate staining or yellowish brown) or 3 (strong staining or
brown). The final histological score was expressed as a product of
PS and IS, as described by Yu et al (36).
Western blot analysis
Upon confluence, PTECs were exposed to freshly
prepared SUA solution (0, 50, 100 and 200 µg/ml) in basal medium
for 48 h. Cells were subsequently harvested in cold PBS and
suspended in total radioimmunoprecipitation assay buffer (RIPA;
Beyotime Institute of Biotechnology, Haimen, China). Rats renal
cortex tissue was collected and lysed in RIPA buffer
(Sigma-Aldrich; Merck KGaA). The concentrations of total protein in
cell lysates were determined by a Bio-Rad Protein assay (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) using the Bradford method.
Lysates (40 µg) were resolved on 11% SDS-PAGE gels, transferred
onto a nitrocellulose membrane and probed with antibodies against
APN (cat. no. ab22554; 1:1,000), AdipoR1 (cat. no. ab126611,
1:2,000) and NLRP3 (cat. no. NBP2-12446, 1:1,000) at 4°C overnight
after 30 min blocking with 5% BSA at room temperature. Membranes
were incubated with secondary antibodies, including horseradish
peroxidase-conjugated goat anti-mouse (SSA007, 1:1,000) and goat
anti-rabbit IgG (SSA004, 1:1,000) antibodies for 1 h at room
temperature. Protein immunoblots were developed using
RapidStep™ ECL Reagent (cat. no. 345818; Merck KGaA) and
analyzed through the ImageQuant LAS 4000 software (GE Healthcare,
Chicago, IL, USA). A GAPDH antibody (cat. no. 10094-T52; 1:10,000;
Sino Biological, Inc.) was used as an internal control.
Statistical analysis
All statistical analyses were performed using SPSS
21.0 software (IBM Corp., Armonk, NY, USA). Normally distributed
data, including kidney/body mass ratio, BUN, SCr, serum uric acid,
HOMA-IR and 24 h urinary protein (UP) for rats, the cell viability
of PTECs, the expression levels of APN, AdipoR1 and NLRP3 protein
in cultured PTECs or renal tissues from rats were expressed as the
mean ± standard deviation. Discrepancies between groups were
analyzed by one-way analysis of variance and Least Significant
Difference test. Non-normally distributed data such as the renal
histological scores of rats in different groups were analyzed by
Kruskal-Wallis and Nemenyi test. P<0.05 was considered to
indicate a statistically significant difference.
Results
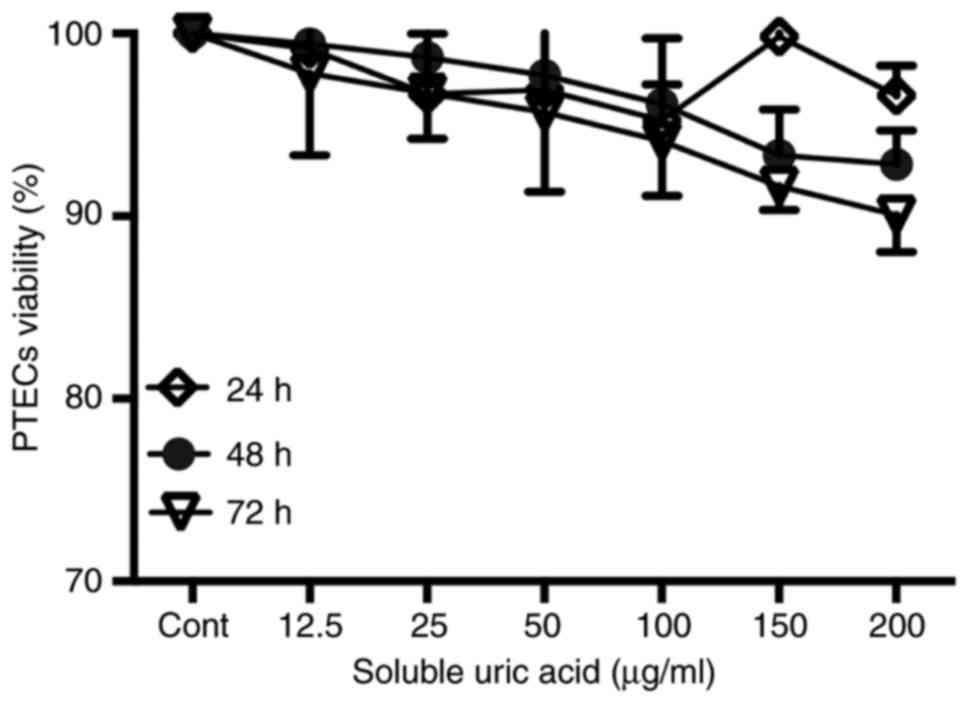
Viability of PTECs exposed to SUA
PTECs seeded in 96-well plates were incubated with
different concentrations of SUA (12.5–200 µg/ml) for predefined
periods. Cell viabilities were assessed by a commercial MTT assay.
As demonstrated in Fig. 1,
increasing concentrations of SUA had no significant effect on the
cell viability of PTECs compared with the blank medium treatment.
As 100 µg/ml uric acid is a common hyperuricemia level in human
serum, this dose was employed in the following experiments.
Biochemical alterations in HUA rat
models
All rats survived the treatment with no alterations
in their eating and drinking behavior. The behavior of rats was
evaluated through observing the appetite of each animal. Following
sacrifice, only serum uric acid levels exhibited significant
differences between the groups, and no obvious differences were
observed for kidney/body mass ratio, BUN, SCr, 24 h UP or the
HOMA-IR (Table I).
 | Table I.Serum and urine analysis of rats in
different treatment groups. |
Table I.
Serum and urine analysis of rats in
different treatment groups.
| Parameter | Control (n=4) | HUA (n=4) | FEB (n=4) |
|---|
| KI | 0.62±0.04 | 0.66±0.04 | 0.64±0.07 |
| BUN, mmol/l | 7.08±0.74 | 5.95±0.32 | 6.40±1.16 |
| SCr, µmol/l | 40.78±2.21 | 41.22±5.13 | 41.90±4.74 |
| Serum uric acid,
µmol/; | 83.24±35.23 |
155±36.18a |
16.50±1.32a,b |
| HOMA-IR | 7.09±1.74 | 6.29±1.83 | 7.23±1.14 |
| 24 h UP, g | 12.42±5.82 | 11.11±3.90 | 12.58±2.89 |
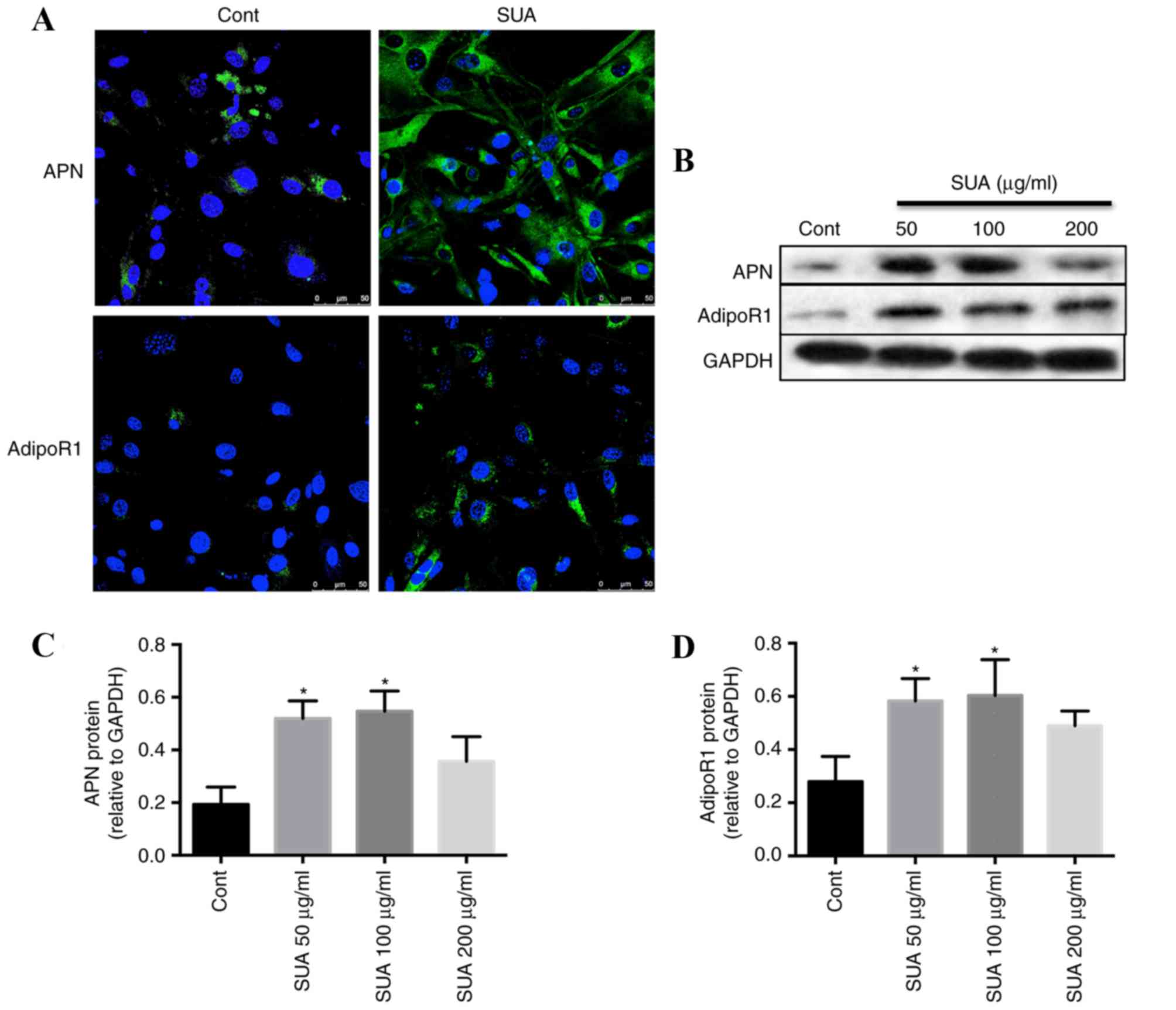
Immunofluorescence analysis of the
expression of APN and AdipoR1 in control and SUA-treated PTECs
The control PTECs cultured in basal medium exhibited
positive staining for APN and AdipoR1 within the cytoplasm
(Fig. 2A). Compared with the basal
medium control cells, SUA (100 µg/ml) incubation for 48 h markedly
increased the expression of cytoplasmic APN and transmembrane
AdipoR1 in cultured PTECs (Fig.
2A).
SUA promotes the expression of APN
signaling in PTECs and HUA rats
Western blotting demonstrated that APN (Fig. 2B and C) and AdipoR1 (Fig. 2B and D) protein levels in PTEC cell
lysates were significantly increased dose-dependently compared with
control cells following treatment with 50 and 100 µg/ml SUA, with
APN and AdiopoR1 levels peaking at 100 µg/ml SUA. However,
expression levels declined following treatment with 200 µg/ml SUA
(Fig. 2B-D).
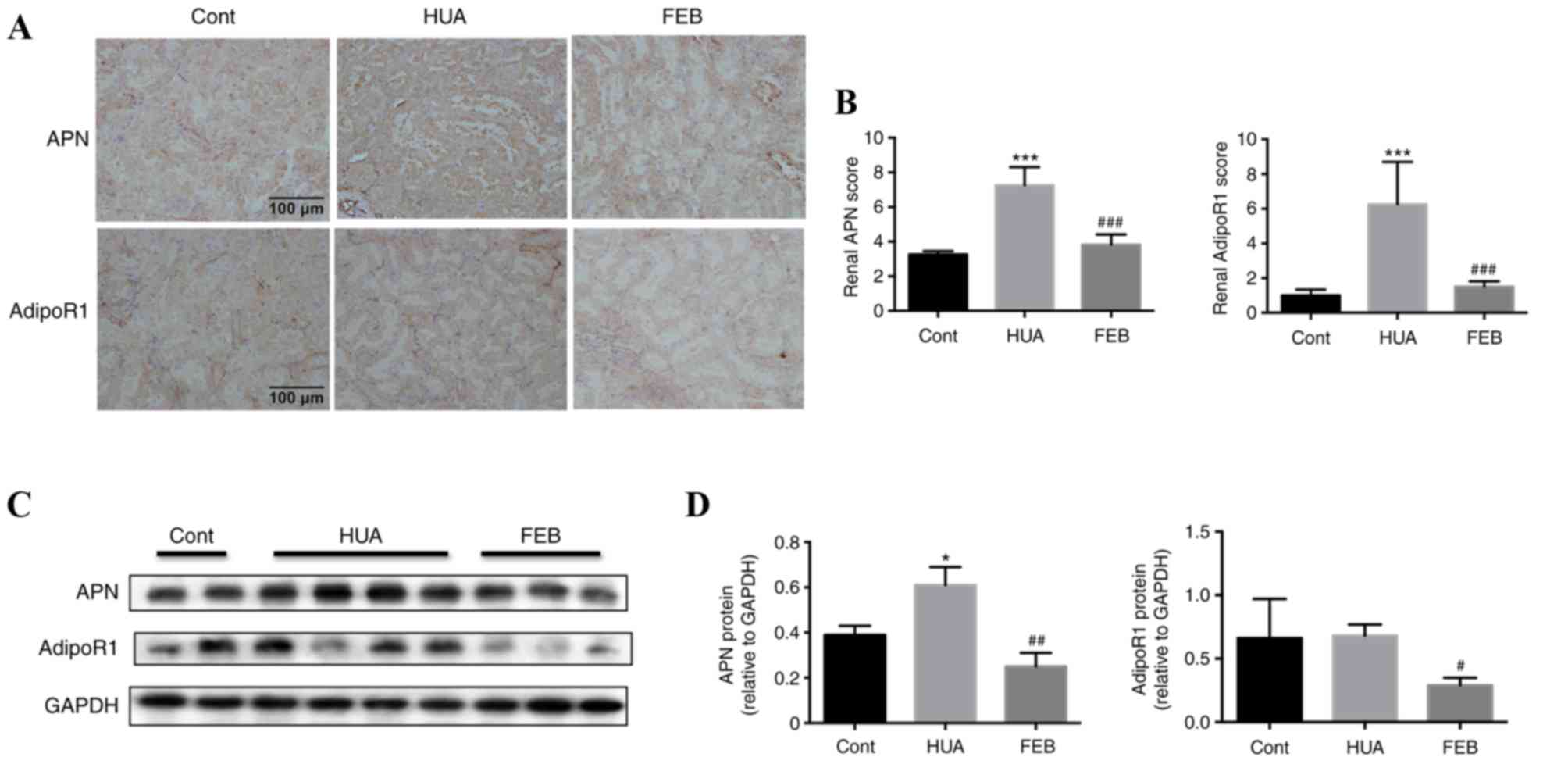
There was visible in vivo APN and AdipoR1
immunohistochemical staining within proximal renal tubules of
control rats. APN localized predominantly in the cytoplasm of the
renal tubular epithelia, whereas AdipoR1 was detectable on the
membrane and within the cytoplasm (Fig. 3A). Immunohistochemistry staining
revealed intense staining of APN and AdipoR1 within the proximal
renal tubules in HUA rats compared with the control group, however,
staining was weaker in the FEB group (Fig. 3A). Tubular APN and AdipoR1 were
quantitatively higher in HUA rats compared with the control group,
while FEB treatment significantly reduced the expression of APN and
AdipoR1 in HUA rats (Fig. 3B).
Furthermore, western blot analysis also demonstrated that APN
levels were markedly higher in HUA rats compared with the control
group, while APN levels in the FEB group were significantly lower
compared with the HUA group (Fig. 3C
and D). No statistically significant difference was observed in
AdipoR1 expression between the HUA and control groups, however,
AdipoR1 levels were significantly lower in the FEB group compared
with the HUA group (Fig. 3C and
D).
SUA induces renal inflammation
HUA rats exhibited a prominent loss of brush
borders, detachment of renal tubular epithelial cells and
interstitial mononuclear cell infiltration, compared with the
control and FEB groups (Fig. 4A).
In addition, intense NLRP3 staining within the tubules of HUA rats,
and not control or FEB groups, was observed (Fig. 4B). In addition, protein levels of
NLRP3 in the renal cortices were significantly higher in HUA rats
compared with the control and FEB groups (Fig. 4C). Furthermore, PTECs exposed to
SUA for 48 h exhibited increased NLRP3 expression in a
dose-dependent manner (Fig. 4D).
NLRP3 levels declined following treatment with an SUA concentration
of 200 µg/ml (Fig. 4D).
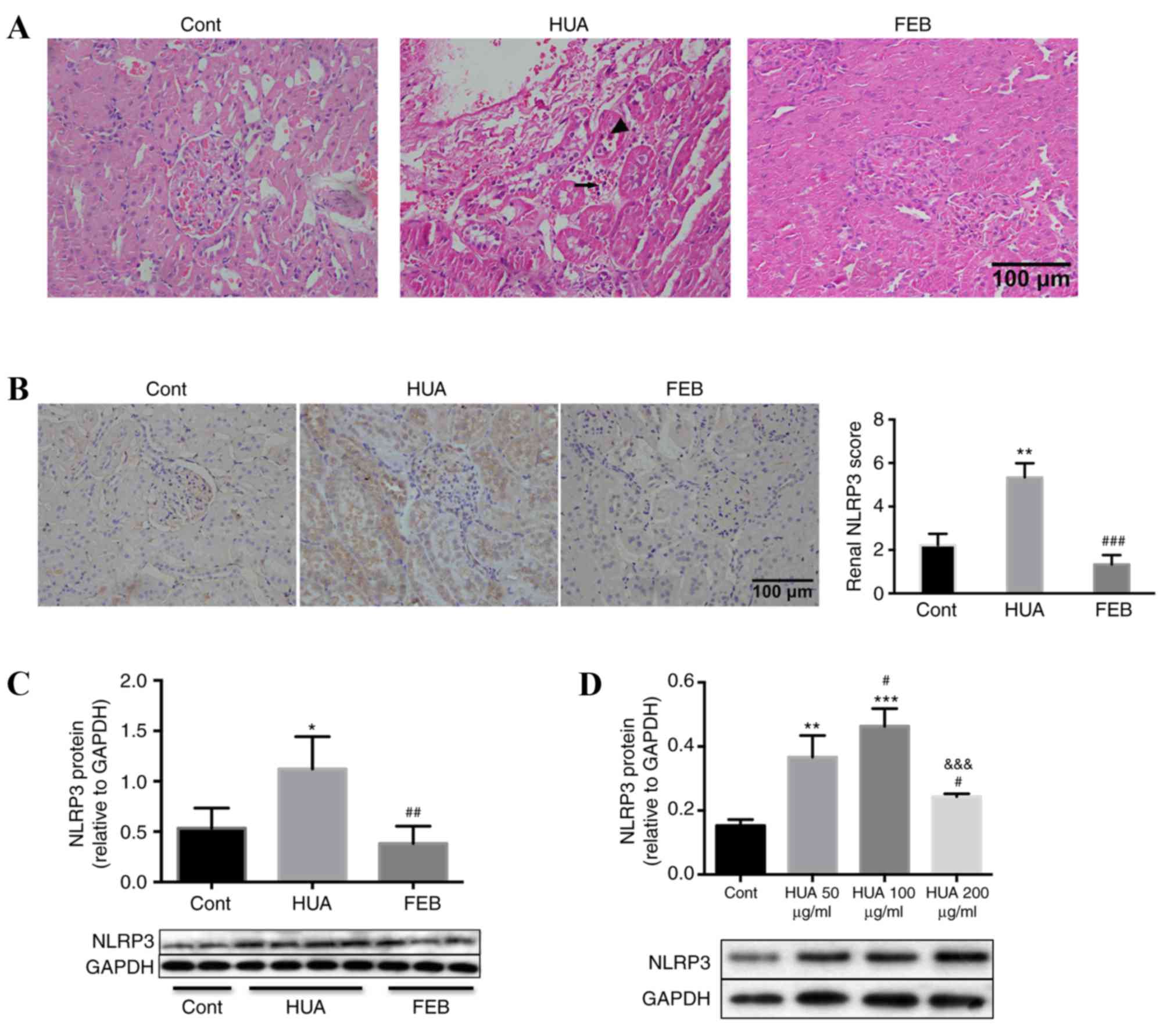 | Figure 4.Uric acid induced renal proximal
tubule inflammation in vivo and in vitro. (A)
Hematoxylin and eosin staining demonstrated that HUA rats exhibited
increased tubular epithelial cell detachment (indicated by
arrowhead) and interstitial mononuclear cell infiltration
(indicated by arrow) compared with the control and FEB groups.
Magnification, ×400. (B) Immunohistochemical staining in rat renal
tissues also indicated an increased intensity of NLRP3 staining
within the tubular cells of HUA rats compared with control and FEB
groups, which was evaluated in a blinded manner. Magnification,
×400. (C) Western blot analysis demonstrated that NLRP3 protein
levels in the renal cortices of HUA rats were significantly higher
compared with the control and FEB groups, n=4 per group. (D)
Western blot analysis demonstrated that cultured PTECs exhibited a
dose-dependent increase in NLRP3 protein levels following treatment
with SUA, compared with the control group, although levels were
reduced at 200 µg/ml SUA. Bars represent data from four independent
experiments. For parts B-D, *P<0.05, **P<0.01 and
***P<0.001 vs. control; for parts B and C,
##P<0.01 and ###P<0.001 vs. HUA group;
for part D, #P<0.05 vs. 50 µg/ml SUA group and
&&&P<0.001 vs. 100 µg/ml SUA group. HUA,
hyperuricemia; FEB, febuxostat; NLRP3, NLR family pyrin
domain-containing 3; PTECs, proximal tubule epithelial cells; SUA,
soluble uric acid; Cont, control. |
Discussion
Our previous study demonstrated that SUA is a
newly-recognized danger signal and induces the activation of the
NLRP3 inflammasome, maturation of caspase-1, cleavage of IL-1β and
the overexpression of intercellular cell adhesion molecule (ICAM)-1
through toll-like receptor (TLR)4 in human PTECs (32). However, it is unclear whether SUA,
as a trigger of inflammation, stimulates the expression of APN and
its receptor. In the present study, the results demonstrated that
APN and its receptor AdipoR1 were constantly expressed in human
PTECs, and were simultaneously upregulated upon exposure to SUA,
which was also associated with increased NLRP3 expression. These
findings indicate that the APN-AdipoR1 axis may participate in the
modulation of SUA-induced inflammatory reactions in human
PTECs.
It is established that APN is an adipocyte-derived
cytokine (37) that exhibits
extensive effects, including insulin-sensitization,
anti-inflammatory, anti-fibrotic and anti-atherosclerosis effects,
and lowers the risk of metabolic disorders (6,12,38–40).
Certain studies have focused on APN signaling in the kidneys
(1,3,11)
and have demonstrated that APN was detectable on the endothelial
surfaces of glomerular capillaries, podocytes, intrarenal
arterioles, peritubular capillaries, smooth muscle cells and
tubular epithelial cells in healthy and injured kidneys (1–3), in
addition to cultured PTECs (11).
APN is reported to protect podocytes against oxidative stress
(16,17), inhibit glycogen synthase in distal
tubules (13), mitigate
interlobular arteriosclerosis (41) and prevent renal diseases in the
offspring of maternal rats that were exposed to a high-fat and
high-fructose diet (19). APN
expression was also modulated by metabolic dysfunction in the
presence of hypertension or hyperuricemia in chronic kidney disease
(42,43). Baldwin et al (44) demonstrated that the levels of APN
mRNA in fat tissues and circulating APN were reduced in obese mice
with metabolic syndrome and hyperuricemia, while levels were
increased upon lowering uric acid with allopurinol. In patients
with hyperuricemia, circulating APN levels were also reduced
(45), and increased serum uric
acid levels were associated with hypoadiponectinemia in patients
with essential hypertension (46).
Therefore, circulating APN is associated with hyperuricemia.
Furthermore, serum APN levels may be used to predict the risk of
mortality and cardiovascular disease in patients with renal disease
in a biphasic manner; a high APN serum level (>20 mg/l) is
associated with higher cardiovascular risk, while lower serum APN
levels (<15 mg/l) also predict a higher cardiovascular risk
(14). However, the effects of
systemic APN are not identical to local renal APN, and limited
information is available concerning the biological importance of
APN in renal cells following SUA administration. Therefore, the
present study focused on the role of local renal APN in HUA-induced
kidney injury. To the best of our knowledge, the present study is
the first to demonstrate that SUA induced the expression of APN in
PTECs in vitro and that APN was overexpressed even in rats
with mild HUA. Furthermore, this increase was reversible at low
uric acid concentrations, indicating that APN may be involved in
the initiation and the development of SUA-associated tubular
injury. Notably, in the current study, the results for intrarenal
expression of APN were consistent with previous reports (11,47).
However, contradictory findings in diabetic nephropathy are also
present (2). As uric acid is a
proinflammatory factor and also a component of metabolic syndrome,
it is difficult to determine whether the uric acid-induced APN
upregulation is mediated though inflammatory signaling or metabolic
pathways. Therefore, the biological effect of APN and its mechanism
requires further investigation. As APN was upregulated upon SUA
stimulation, future studies should aim to clarify the regulatory
function of APN on SUA-induced inflammation. To elucidate the
potential mechanism, in vitro or in vivo APN knockout
models may improve the understanding of how APN modulates the
tubular inflammation induced by SUA.
Previous studies have revealed that APN receptors,
including AdipoR1 and AdipoR2, are expressed in human renal
proximal tubule cells, and the expression of AdipoR1 mRNA was
higher compared with AdipoR2 expression. Although AdipoR1 consists
of 7 transmembrane domains, it is structurally and functionally
different from G-protein-coupled receptors. AdipoR1 is the most
abundant APN receptor in the kidneys, and expressed by PTECs and
various glomerular cells (1,12,13,22).
AdipoR1 has a high affinity for the globular domain of APN
(5) and transmits its downstream
signaling through AMPK phosphorylation (48). In the adenine-induced chronic
kidney disease model, intrarenal AdipoR1 expression in the
glomerular endothelium and tubular epithelial cells was
upregulated, while AdipoR1 mRNA was positively correlated with
serum and urine APN, and 24 h UP (22). In patients with uremia, the
expression of AdipoR1 in the muscle and circulatory levels were
increased, which was also associated with elevated catalytic AMPK,
lower acetyl-CoA carboxylase phosphorylation and decreased
carnitine palmitoyl transferase levels. These findings indicate
that uremia leads to the upregulation of AdipoR1 accompanied and
APN resistance (49).
The expression of AdipoR1 in different animal models
has been previously investigated. Recently, Akita diabetic mice
with no increases in SCr were reported to exhibit increased
glomerular expression of AdipoR1 (50). Inversely, streptozocin
(STZ)-induced diabetic rodents (51–53)
and mesangial cells (HBYZ-1) exposed to high glucose (53) exhibited AdipoR1 downregulation.
However, to the best of our knowledge, the mechanism by which
AdipoR1 expression may be regulated in uric acid-treated renal
tubule cells has not been previously investigated. In the current
study, AdipoR1 was predominantly expressed in renal tubule
epithelial cells of rats, with increased levels in rats with oxonic
acid-induced hyperuricemia and in cultured human PTECs, compared
with the respective control groups. The protein levels of AdipoR1
during SUA exposure were modulated in a similar manner to
intrarenal APN levels, which indicates that APN may participate in
SUA-induced renal injury through AdipoR1. APN was reported to
downregulate the inflammatory cytokine monocyte chemotactic protein
(MCP)-1 (12) and attenuate
angiotensin II-induced NF-κB activation and fibronectin expression
in proximal tubular cells (15),
which was dependent on AdipoR1 (12,15).
Furthermore, it has been demonstrated that APN exerts
anti-inflammatory effects and is therefore renoprotective in
various renal diseases. Delaigle et al (54) demonstrated that, in human and
rodent myotubes, APN was increased in vivo and in
vitro in response to inflammatory stimuli. The upregulation of
the APN pathway in renal diseases has also been considered as
compensatory feedback to mitigate renal injuries. Our previous
study revealed that SUA activated the NLRP3-caspase-1-IL-1β pathway
in cultured PTECs through a TLR4-dependent mechanism (32). In the present study, SUA may have
initially activated the NLRP3 inflammasome pathway and subsequently
triggered APN-AdipoR1 signaling to mitigate local inflammation. As
AdipoR1 is proven to be the most crucial functional receptor for
APN, it is reasonable to hypothesize that inflammation and renal
damage would be exaggerated if the function of AdipoR1 was
inhibited by silencing AdipoR1 in future experiments. Upon
silencing of AdipoR1, SUA-induced inflammatory damage would fail to
effectively activate the APN-AdipoR1 pathway and subsequently
weaken the anti-inflammatory function of APN. Therefore, AdipoR1
knockout studies may clarify the involvement of AdipoR1-mediated
APN signaling in the modulation of SUA-induced NLRP3 expression in
PTECs.
Previous studies have investigated the role of the
APN-AdipoR1 pathway in renal diseases. Evidence regarding the role
of APN in renal tissues is controversial. Certain reports indicate
that APN is proinflammatory in the kidneys, and the pathophysiology
may involve the activation of NF-κB, the recruitment of
inflammatory cells, the promotion of macrophage migration and the
induction of various cytokines and chemokines, including IL-6,
TNF-α, MCP-1 and macrophage inflammatory protein-2 (11,47).
In cultured human synovial fibroblasts, recombinant full-length APN
was demonstrated to enhance IL-6 expression via the
AdipoR1/AMPK/p38/IκB kinaseα/β and NF-κB pathways (7). However, other reports, using
overexpression and/or deletion of APN-associated genes and the
addition of recombinant APN, have demonstrated that the APN-AdipoR1
pathway possesses anti-inflammatory effects. These effects include
the mitigation of macrophage infiltration in the remnant kidney
following partial nephrectomy (18), reduced endothelin-1 and
plasminogen-1 production in the renal cortex of STZ-induced
diabetic rats (55) and the
reduction of NF-κB activity in angiotensin II-stimulated human
renal tubular cells (15) and
high-glucose-treated mesangial cells (50). The upstream mediators of the
inflammatory response were further identified through the use of
experimental autoimmune myocarditis models in mice, which indicated
that TLR4 may have a critical role in mediating the anti-cardiac
inflammation effect of adenovirus-mediated overexpression of APN
(56). The results of the current
study, in addition to those of previous studies (32,57),
indicates potential cross-talk between the NLRP3-IL-1β pathway and
APN signaling, in a TLR4-dependent manner. Consistently, in the
present study, the renal tissues of HUA rats exhibited marked NLRP3
immunostaining, in addition to structural and inflammatory injuries
within the tubulointerstitium. However, no marked damage was
observed in the glomerular layer of hyperuricemic rats under light
microscopy. These results are consistent with data obtained by
Sanchez-Lozada et al (58);
in oxonic acid-induced rats with mild hyperuricemia, the average
serum uric acid concentration was 3.07±0.20 mg/dl and no marked
glomerular structural changes were observed. Mazzali et al
(59) also demonstrated preserved
renal architecture in mild hyperuricemia induced by 2% oxonic acid.
In the present study, the mean level of serum uric acid in HUA rats
was (155.00±36.18) µmol/l, which was lower compared with levels in
the study by Sanchez-Lozada et al (58). Tubulointerstitial damage was
reversed by interventions to reduce uric acid levels in the present
study and a previous report (60).
The alterations in the tubular APN-AdipoR1 axis upon SUA-induced
inflammation indicates that APN-AdipoR1 axis activation may be an
autocrine-associated positive feedback mechanism for alleviating
renal injuries. Further investigation is required to elucidate the
mechanism underlying NLRP3 modulation. To improve the understanding
of the SUA-associated alterations in NLRP3 signaling and its
association with the APN pathway, the evaluation of serum and renal
levels of cytokines downstream of NLRP3, such as IL-18 and IL-1β,
may be useful.
APN is a multimeric complex with various molecular
weights and isoforms (61), and
the phenotype of each isoform may differ (62). Furthermore, recombinant gAd is
derived from a wide variety of sources, which may partially explain
the contradictory findings concerning APN function in different
reports. Standardized procedures regarding the isoform, molecular
weight and origin of APN may provide increased information in the
future. In addition, the anti-inflammatory properties of APN are
beneficial for renal injury and may act against the proinflammatory
effects of SUA. To clarify the protective role of APN on
SUA-induced inflammation, in vivo and in vitro APN
knockout models are required. To elucidate the mechanism by which
the NLRP3 inflammasome interacts with the APN-AdipoR1 signaling
pathway, further studies should include AdipoR1 knockout
experiments, in addition to AMPK activation or inactivation
experiments, to determine whether, and how, the APN-AdipoR1-AMPK
pathway regulates the NLRP3 inflammasome in SUA-treated PTECs.
Certain limitations were associated with the present
study. For example, the food intake and other behavior indices of
rats were not accurately quantified. In addition, the APN and
AdipoR1 levels in 200 µg/ml SUA-treated PTECs, unlike those treated
with 50 and 100 µg/ml SUA, did not exhibit significant differences
compared with the control group. This may be partially attributed
to the fact that the effect of SUA on APN, AdipoR1 and NLRP3
expression may not always be linear, and may also be due to
apoptosis occurring in 200 µg/ml SUA-treated PTECs (63). Further study should investigate
cells damage prior to changes in cell viability, including
necrotic, apoptotic and fibrotic processes of PTECs. Furthermore,
an in vivo model of renal specific APN-knockout in
hyperuricemic animals would improve the understanding of the role
of APN in the process of renal tubular inflammation. In summary,
the current study demonstrated that SUA regulated the expression of
APN and AdipoR1 in PTECs. The APN-AdipoR1 pathway in turn may exert
a protective role during SUA-induced renal proximal tubule
inflammatory injury and has potential as a viable therapeutic
target in urate nephropathy.
Acknowledgements
The present study was supported by Shanghai
Municipal Commission of Health and Family Planning, Key Developing
Disciplines (grant no. 2015ZB0501) and Jinshan Science and
Technology Committee of Shanghai (grant no. 2016-3-02). Certain
results in this paper were presented as a poster in the 6th
Oriental Congress of Nephrology 2016 (Shanghai, China).
References
|
1
|
Perri A, Vizza D, Lofaro D, Gigliotti P,
Leone F, Brunelli E, Malivindi R, De Amicis F, Romeo F, De Stefano
R, et al: Adiponectin is expressed and secreted by renal tubular
epithelial cells. J Nephrol. 26:1049–1054. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
von Eynatten M, Liu D, Hock C, Oikonomou
D, Baumann M, Allolio B, Korosoglou G, Morcos M, Campean V, Amann
K, et al: Urinary adiponectin excretion: A novel marker for
vascular damage in type 2 diabetes. Diabetes. 58:2093–2099. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rovin BH, Song H, Hebert LA, Nadasdy T,
Nadasdy G, Birmingham DJ, Yung Yu C and Nagaraja HN: Plasma, urine,
and renal expression of adiponectin in human systemic lupus
erythematosus. Kidney Int. 68:1825–1833. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen J, Tan B, Karteris E, Zervou S, Digby
J, Hillhouse EW, Vatish M and Randeva HS: Secretion of adiponectin
by human placenta: Differential modulation of adiponectin and its
receptors by cytokines. Diabetologia. 49:1292–1302. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yamauchi T, Kamon J, Ito Y, Tsuchida A,
Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et
al: Cloning of adiponectin receptors that mediate antidiabetic
metabolic effects. Nature. 423:762–769. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iwabu M, Yamauchi T, Okada-Iwabu M, Sato
K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata
M, et al: Adiponectin and AdipoR1 regulate PGC-1alpha and
mitochondria by Ca(2+) and AMPK/SIRT1. Nature. 464:1313–1319. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tang CH, Chiu YC, Tan TW, Yang RS and Fu
WM: Adiponectin enhances IL-6 production in human synovial
fibroblast via an AdipoR1 receptor, AMPK, p38, and NF-kappa B
pathway. J Immunol. 179:5483–5492. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cao T, Gao Z, Gu L, Chen M, Yang B, Cao K,
Huang H and Li M: AdipoR1/APPL1 potentiates the protective effects
of globular adiponectin on angiotensin II-induced cardiac
hypertrophy and fibrosis in neonatal rat atrial myocytes and
fibroblasts. PLoS One. 9:e1037932014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Farias JM, Maggi RM, Tromm CB, Silva LA,
Luciano TF, Marques SO, Lira FS, de Souza CT and Pinho RA: Exercise
training performed simultaneously to a high-fat diet reduces the
degree of insulin resistance and improves adipoR1-2/APPL1 protein
levels in mice. Lipids Health Dis. 11:1342012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu GZ, Liang B, Lau WB, Wang Y, Zhao J,
Li R, Wang X, Yuan Y, Lopez BL, Christopher TA, et al: High
glucose/High Lipids impair vascular adiponectin function via
inhibition of caveolin-1/AdipoR1 signalsome formation. Free Radic
Biol Med. 89:473–485. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Perri A, Vizza D, Lupinacci S, Toteda G,
De Amicis F, Leone F, Gigliotti P, Lofaro D, La Russa A and
Bonofiglio R: Adiponectin secreted by tubular renal cells during
LPS exposure worsens the cellular inflammatory damage. J Nephrol.
29:185–194. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shen YY, Hughes JT, Charlesworth JA, Kelly
JJ and Peake PW: Adiponectin is present in the urine in its native
conformation, and specifically reduces the secretion of MCP-1 by
proximal tubular cells. Nephrology (Carlton). 13:405–410. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cammisotto PG, Londono I, Gingras D and
Bendayan M: Control of glycogen synthase through ADIPOR1-AMPK
pathway in renal distal tubules of normal and diabetic rats. Am J
Physiol Renal Physiol. 294:F881–F889. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Christou GA and Kiortsis DN: The role of
adiponectin in renal physiology and development of albuminuria. J
Endocrinol. 221:R49–R61. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fang F, Liu GC, Kim C, Yassa R, Zhou J and
Scholey JW: Adiponectin attenuates angiotensin II-induced oxidative
stress in renal tubular cells through AMPK and cAMP-Epac signal
transduction pathways. Am J Physiol Renal Physiol. 304:F1366–F1374.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sharma K, Ramachandra Rao S, Qiu G, Usui
HK, Zhu YQ, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, et al:
Adiponectin regulates albuminuria and podocyte function in mice. J
Clin Invest. 118:1645–1656. 2008.PubMed/NCBI
|
|
17
|
Rutkowski JM, Wang ZV, Park ASD, Zhang J,
Zhang D, Hu MC, Moe OW, Susztak K and Scherer PE: Adiponectin
promotes functional recovery after podocyte ablation. J Am Soc
Nephrol. 24:268–282. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ohashi K, Iwatani H, Kihara S, Nakagawa Y,
Komura N, Fujita K, Maeda N, Nishida M, Katsube F, Shimomura I, et
al: Exacerbation of albuminuria and renal fibrosis in subtotal
renal ablation model of adiponectin-knockout mice. Arterioscler
Thromb Vasc Biol. 27:1910–1917. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yamada-Obara N, Yamagishi SI, Taguchi K,
Kaida Y, Yokoro M, Nakayama Y, Ando R, Asanuma K, Matsui T, Ueda S,
et al: Maternal exposure to high-fat and high-fructose diet evokes
hypoadiponectinemia and kidney injury in rat offspring. Clin Exp
Nephrol. 20:853–861. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yilmaz MI, Saglam M, Qureshi AR, Carrero
JJ, Caglar K, Eyileten T, Sonmez A, Cakir E, Oguz Y, Vural A, et
al: Endothelial dysfunction in type-2 diabetics with early diabetic
nephropathy is associated with low circulating adiponectin. Nephrol
Dial Transplant. 23:1621–1627. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fujita H, Morii T, Koshimura J, Ishikawa
M, Kato M, Miura T, Sasaki H, Narita T, Ito S and Kakei M: Possible
relationship between adiponectin and renal tubular injury in
diabetic nephropathy. Endocr J. 53:745–752. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu Y, Bao BJ, Fan YP, Shi L and Li SQ:
Changes of adiponectin and its receptors in rats following chronic
renal failure. Ren Fail. 36:92–97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kamei K, Konta T, Hirayama A, Suzuki K,
Ichikawa K, Fujimoto S, Iseki K, Moriyama T, Yamagata K, Tsuruya K,
et al: A slight increase within the normal range of serum uric acid
and the decline in renal function: Associations in a
community-based population. Nephrol Dial Transplant. 29:2286–2292.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li L, Chen Y, Zhao Y, Zeng X, Liu F and Fu
P: Is hyperuricemia an independent risk factor for new-onset
chronic kidney disease? A systematic review and meta-analysis based
on observational cohort studies. BMC Nephrol. 15:1–12. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chou YC, Kuan JC, Yang T, Chou WY, Hsieh
PC, Bai CH, You SL, Chen CH, Wei CY and Sun CA: Elevated uric acid
level as a significant predictor of chronic kidney disease: A
cohort study with repeated measurements. J Nephrol. 28:457–462.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hovind P, Rossing P, Tarnow L, Johnson RJ
and Parving HH: Serum uric acid as a predictor for development of
diabetic nephropathy in type 1 diabetes: An inception cohort study.
Diabetes. 58:1668–1671. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Madero M, Sarnak MJ, Wang X, Greene T,
Beck GJ, Kusek JW, Collins AJ, Levey AS and Menon V: Uric acid and
long-term outcomes in CKD. Am J Kidney Dis. 53:796–803. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shimada M, Johnson RJ, May WS Jr,
Lingegowda V, Sood P, Nakagawa T, Van QC, Dass B and Ejaz AA: A
novel role for uric acid in acute kidney injury associated with
tumour lysis syndrome. Nephrol Dial Transplant. 24:2960–2964. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cirillo P, Gersch MS, Mu W, Scherer PM,
Kim KM, Gesualdo L, Henderson GN, Johnson RJ and Sautin YY:
Ketohexokinase-dependent metabolism of fructose induces
proinflammatory mediators in proximal tubular cells. J Am Soc
Nephrol. 20:545–553. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han HJ, Lim MJ, Lee YJ, Lee JH, Yang IS
and Taub M: Uric acid inhibits renal proximal tubule cell
proliferation via at least two signaling pathways involving PKC,
MAPK, cPLA2, and NF-kappaB. Am J Physiol Renal Physiol.
292:F373–F381. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xiao J, Fu C, Zhang X, Zhu D, Chen W, Lu Y
and Ye Z: Soluble monosodium urate, but not its crystal, induces
toll like receptor 4-dependent immune activation in renal mesangial
cells. Mol Immunol. 66:310–318. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xiao J, Zhang XL, Fu CS, Han R, Chen WJ,
Lu YJ and Ye ZB: Soluble uric acid increases NALP3 inflammasome and
interleukin-1β expression in human primary renal proximal tubule
epithelial cells through the Toll-like receptor 4-mediated pathway.
Int J Mol Med. 35:1347–1354. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Eisenbacher JL, Schrezenmeier H,
Jahrsdörfer B, Kaltenmeier C, Rojewski MT, Yildiz T, Beyer T, Erle
A, Wiegmann DS, Grassl S, et al: S100A4 and uric acid promote
mesenchymal stromal cell induction of IL-10+/IDO+ lymphocytes. J
Immunol. 192:6102–6110. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Matthews DR, Hosker JP, Rudenski AS,
Naylor BA, Treacher DF and Turner RC: Homeostasis model assessment:
Insulin resistance and beta-cell function from fasting plasma
glucose and insulin concentrations in man. Diabetologia.
28:412–419. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
National Research Council, . Guide for the
Care and Use of Laboratory Animals. The National Academies Press;
Washington, DC: 1996, https://www.nap.edu/read/5140/chapter/1
|
|
36
|
Yu P, Bu H, Wang H, Zhao G, Zhang J and
Zhou Q: Comparative study on image analysis and manual counting of
immunohistichemistry. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi.
20:288–290. 2003.(In Chinese). PubMed/NCBI
|
|
37
|
Scherer PE, Williams S, Fogliano M,
Baldini G and Lodish HF: A novel serum protein similar to C1q,
produced exclusively in adipocytes. J Biol Chem. 270:26746–26749.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hata A, Yonemoto K, Shikama Y, Aki N,
Kosugi C, Tamura A, Ichihara T, Minagawa T, Kuwamura Y, Miyoshi M,
et al: Cut-off value of total adiponectin for managing risk of
developing metabolic syndrome in male japanese workers. Plos One.
10:e01183732015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hayashi M, Shibata R, Takahashi H, Ishii
H, Aoyama T, Kasuga H, Yamada S, Ohashi K, Maruyama S, Matsuo S, et
al: Association of adiponectin with carotid arteriosclerosis in
predialysis chronic kidney disease. Am J Nephrol. 34:249–255. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zoico E, Garbin U, Olioso D, Mazzali G,
Fratta Pasini AM, Di Francesco V, Sepe A, Cominacini L and Zamboni
M: The effects of adiponectin on interleukin-6 and MCP-1 secretion
in lipopolysaccharide-treated 3T3-L1 adipocytes: Role of the
NF-kappaB pathway. Int J Mol Med. 24:847–851. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Iwasa Y, Otsubo S, Ishizuka T, Uchida K
and Nitta K: Influence of serum high-molecular-weight and total
adiponectin on arteriosclerosis in IgA nephropathy patients.
Nephron Clin Pract. 108:c226–c232. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Park CS, Ihm SH, Park HJ, Shin WS, Kim PJ,
Chang K, Kim HY, Youn HJ, Chung WS, Seung KB and Kim JH:
Relationship between plasma adiponectin, retinol-binding protein 4
and uric Acid in hypertensive patients with metabolic syndrome.
Korean Circ J. 41:198–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hosoya T, Ohno I, Nomura S, Hisatome I,
Uchida S, Fujimori S, Yamamoto T and Hara S: Effects of
topiroxostat on the serum urate levels and urinary albumin
excretion in hyperuricemic stage 3 chronic kidney disease patients
with or without gout. Clin Exp Nephrol. 18:876–884. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Baldwin W, McRae S, Marek G, Wymer D,
Pannu V, Baylis C, Johnson RJ and Sautin YY: Hyperuricemia as a
mediator of the proinflammatory endocrine imbalance in the adipose
tissue in a murine model of the metabolic syndrome. Diabetes.
60:1258–1269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tsioufis C, Kyvelou S, Dimitriadis K,
Syrseloudis D, Sideris S, Skiadas I, Katsi V, Stefanadi E, Lalos S,
Mihas C, et al: The diverse associations of uric acid with
low-grade inflammation, adiponectin and arterial stiffness in
never-treated hypertensives. J Hum Hypertens. 25:554–559. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nishizawa T, Taniura T and Nomura S:
Effects of febuxostat on platelet-derived microparticles and
adiponectin in patients with hyperuricemia. Blood Coagul
Fibrinolysis. 26:887–892. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jin X, Chen J, Hu Z, Chan L and Wang Y:
Genetic deficiency of adiponectin protects against acute kidney
injury. Kidney Int. 83:604–614. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yamauchi T, Kamon J, Minokoshi Y, Ito Y,
Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, et al:
Adiponectin stimulates glucose utilization and fatty-acid oxidation
by activating AMP-activated protein kinase. Nat Med. 8:1288–1295.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Martinez Cantarin MP, Keith SW, Waldman SA
and Falkner B: Adiponectin receptor and adiponectin signaling in
human tissue among patients with end-stage renal disease. Nephrol
Dial Transplant. 29:2268–2277. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fang F, Bae EH, Hu A, Liu GC, Zhou X,
Williams V, Maksimowski N, Lu C, Konvalinka A, John R and Scholey
JW: Deletion of the gene for adiponectin accelerates diabetic
nephropathy in the Ins2 (+/C96Y) mouse. Diabetologia. 58:1668–1678.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tamura Y, Murayama T, Minami M, Matsubara
T, Yokode M and Arai H: Ezetimibe ameliorates early diabetic
nephropathy in db/db mice. J Atheroscler Thromb. 19:608–618. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Guo Z and Zhao Z: Effect of
N-acetylcysteine on plasma adiponectin and renal adiponectin
receptors in streptozotocin-induced diabetic rats. Eur J Pharmacol.
558:208–213. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ji H, Wu L, Ma X, Ma X and Qin G: The
effect of resveratrol on the expression of AdipoR1 in kidneys of
diabetic nephropathy. Mol Biol Rep. 41:2151–2159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Delaigle AM, Jonas JC, Bauche IB, Cornu O
and Brichard SM: Induction of adiponectin in skeletal muscle by
inflammatory cytokines: In vivo and in vitro studies.
Endocrinology. 145:5589–5597. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nakamaki S, Satoh H, Kudoh A, Hayashi Y,
Hirai H and Watanabe T: Adiponectin reduces proteinuria in
streptozotocin-induced diabetic Wistar rats. Exp Biol Med
(Maywood). 236:614–620. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jenke A, Wilk S, Löbel M, Stehr J, Poller
W, Eriksson U, Schultheiss HP, Scheibenbogen C and Skurk C:
Adiponectin inhibits toll-like receptor 4 mediated cardiac
inflammation and injury. J Am Coll Cardiol. 59:E15502012.
View Article : Google Scholar
|
|
57
|
Martinon F, Pétrilli V, Mayor A, Tardivel
A and Tschopp J: Gout-associated uric acid crystals activate the
NALP3 inflammasome. Nature. 440:237–241. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sánchez-Lozada LG, Tapia E, Avila-Casado
C, Soto V, Franco M, Santamaria J, Nakagawa T, Rodriguez-Iturbe B,
Johnson RJ and Herrera-Acosta J: Mild hyperuricemia induces
glomerular hypertension in normal rats. Am J Physiol Renal Physiol.
283:F1105–F1110. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mazzali M, Hughes J, Kim YG, Jefferson JA,
Kang DH, Gordon KL, Lan HY, Kivlighn S and Johnson RJ: Elevated
uric acid increases blood pressure in the rat by a novel
crystal-independent mechanism. Hypertension. 38:1101–1106. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wang C, Pan Y, Zhang QY, Wang FM and Kong
LD: Quercetin and allopurinol ameliorate kidney injury in
STZ-treated rats with regulation of renal NLRP3 inflammasome
activation and lipid accumulation. PLoS One. 7:e382852012.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang ZV, Schraw TD, Kim JY, Khan T, Rajala
MW, Follenzi A and Scherer PE: Secretion of the adipocyte-specific
secretory protein adiponectin critically depends on thiol-mediated
protein retention. Mol Cell Biol. 27:3716–3731. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Marek G, Pannu V, Shanmugham P, Pancione
B, Mascia D, Crosson S, Ishimoto T and Sautin YY: Adiponectin
resistance and proinflammatory changes in the visceral adipose
tissue induced by fructose consumption via ketohexokinase-dependent
pathway. Diabetes. 64:508–518. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Verzola D, Ratto E, Villaggio B, Parodi
EL, Pontremoli R, Garibotto G and Viazzi F: Uric acid promotes
apoptosis in human proximal tubule cells by oxidative stress and
the activation of NADPH oxidase NOX 4. Plos One. 9:e1152102014.
View Article : Google Scholar : PubMed/NCBI
|