Introduction
Apoptosis, also known as programmed cell death,
plays important roles in a variety of cellular events in both
physiological and pathological settings and disease development
(1,2), including the pathogenesis of heart
diseases such as myocardial infarction (MI) and heart failure (HF)
(3,4), as evidenced by the findings that
abnormal apoptosis of myocytes promotes the development of HF,
arrhythmogenic right ventricular dysplasia, and dilated
cardiomyopathy (2). Moreover, very
low levels of cardiac myocyte apoptosis were found to be sufficient
to cause cardiomyopathy and HF, and correspondingly, the inhibition
of myocyte apoptosis prevented the development of cardiomyopathy
(5). Therefore, targeting
apoptosis is a potential avenue for the clinical treatment of
certain cardiac diseases such as HF.
It has been well documented that the
renin-angiotensin-aldosterone system (RAAS) is activated in and
contributes to the pathogenesis of a number of cardiovascular
diseases, including HF and MI (6).
Angiotensin II (AngII), as an important component of the RAAS, is
also increased in these cardiac disease settings and induces
cardiomyocyte apoptosis (7).
Therefore, angiotensin-converting enzyme inhibitors (ACEIs) have
been used to treat patients with HF and MI in the clinic (8–11).
Although it is well known that AngII induces cardiomyocyte
apoptosis via at least two mechanisms: Oxidative stress and DNA
damage (7,12), the exact mechanisms linking AngII
stimulation and pathological changes remain uncertain, and the
complete network that mediates the cellular responses to AngII
treatment remains to be deciphered.
MicroRNAs (miRNAs) are a class of short, noncoding
RNAs that downregulate the expression of target genes by binding to
the 3′-untranslated region (3′UTR). Numerous studies have reported
dysregulation and involvement of miRNA expression in pathological
states including cardiovascular disorders (13). For instance, miR-21 protects
myocytes against H2O2-induced injury by
targeting PDCD4 (14),
and miR-214 promotes cell survival and suppresses cell apoptosis
(15,16). In addition, miR-31 inhibits tumor
metastasis though blocking the cell cycle and inducing apoptosis,
and it is regarded as a tumor suppressor-related miRNA (17). The role of miRNAs in the RAAS
system is just emerging (18). For
example, miR-155, which plays an important role in the pathogenesis
of cardiac diseases (19), serves
as a negative regulator of AngII-promoted cell proliferation via
targeting AT1R (20), while
overexpression of miR-132 and miR-212 potentiates AngII activity
and mediates the expression of a number of genes involved in AngII
signaling (21,22). However, whether other miRNAs and
novel mechanisms are involved in AngII-mediated apoptosis remains
to be elucidated.
MiR-31a-5p is the leading member of miR-31 family,
but limited studies have been performed to investigate its roles in
physiological and pathophysiological events. A previous study has
shown that MiR-31a-5p is linked to colorectal cancer (23). Our more recent work revealed that
the miR-31a-5p level was significantly higher in the hearts of rats
with post-infarction HF than that in the sham group (24). However, the exact role of
miR-31a-5p in the pathogenesis of HF remains uncertain. Given the
increased expression of miR-31a-5p in failing hearts, we
hypothesized that miR-31a-5p may play a potential role in the
development of HF. In the present study, we investigated whether
miR-31a-5p mediates myocardial apoptosis in H9C2 cells, a cardiac
cell line, in response to AngII stimulation.
Materials and methods
Cell culture
H9C2 cells were obtained from the Center Laboratory
of China-Japan Union Hospital. Cells were cultured in Dulbecco's
modified Eagle medium with Nutrient Mixture F-12 (DMEM/F12)
containing 10% fetal bovine serum and 1% antibiotics in a modular
incubator with an atmosphere of 5% CO2/95% air at 37°C.
When cells reached 70–80% confluence, they were seeded in 6-well
and 96-well plates.
AngII stimulation and
transfection
H9C2 cells in 6-well plates were transfected with
FAM fluorescently labeled miRNA mimics, inhibitor, and negative
control (NC; GenePharma, Shanghai, China), respectively, with
Fugene HD (Promega Corp., Madison, WI, USA). The transfection was
performed in two groups: Vehicle and AngII groups. Experiments were
executed in three independent assays, with each carried out in
duplicate. Transfection efficiency was evaluated by a fluorescence
microscope at 6 to 24 h after transfection. AngII (10−9
M) was added to cells of the AngII group; this concentration has
been reported to present a maximal apoptotic rate in myocytes
(25).
Flow cytometry
After transfection, H9C2 cells in different groups
were digested by EDTA-free trypsin, washed twice with
phosphate-buffered saline (PBS), and then resuspended in 500 µl of
1× binding buffer. Harvested cells were incubated with 5 µl of
Annexin V fluorescein isothiocyanate and 5 µl of propidium iodide
(Annexin V:FITC Apoptosis Detection kit I; BD Biosciences, San
Diego, CA, USA) for 20 min at room temperature. Thereafter, the
cell suspensions were analyzed by flow cytometry (BD LSRFortessa
X-20; BD Biosciences). The data were analyzed using FlowJo 7.61
software (Treestar Inc., Ashland, OR, USA).
Total RNA extraction and reverse
transcription (RT)
Total RNA of transfected cells was extracted using
TRIzol reagent (Takara Bio, Inc., Otsu, Japan), and the RNA
concentration and integrity were evaluated by a NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE,
USA) and agarose gel electrophoresis. A total of 1 µg of purified
RNA was used for reverse transcription using an RT kit (Takara Bio,
Inc.), according to the manufacturer's protocol. RT primers for
miR-31a-5p were designed using the stem ring method. cDNA was
stored at −80°C.
Prediction of target genes and primer
design
The target genes for miR-31a-5p were predicted using
bioinformatics tools. The specific primers for these genes were
designed by Primer 5.0. The selected genes were further verified by
real-time PCR. A 20-µl system consisting of 2 µl of cDNA, 0.4 µl of
forward/reverse primers, 10 µl of SYBR-Green qPCR Master Mix
(Takara Bio, Inc.), and 7.2 µl of RNase-free water was run on an
Mx3005P (Agilent Technologies, Inc., Santa Clara, CA, USA). The
results were analyzed using the 2−ΔΔCT method. The data
were analyzed using SPSS Statistics 19 software (IBM Corp., Armonk,
NY, USA).
Total protein extraction and western
blot
Total protein was extracted from H9C2 cells using
RIPA protein lysis buffer (Beyotime Institute of Biotechnology,
Haimen, China) with 1% protease inhibitors (Bestbio, Shanghai,
China). Briefly, cells in different groups were harvested using
trypsin, washed with cold PBS twice, incubated with 100 µl of lysis
buffer on ice for 30 min, and centrifuged at 12,000 rpm for 18–30
min. The concentration of protein products was monitored using a
BCA kit (KenGen BioTECH, Nanjing, China), according to the
manufacturer's protocol. Supernatant protein products were stored
at −80°C.
For western blot, briefly, 25 µg of protein samples
from different transfection groups were resolved by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred to
polyvinylidenefluoride membranes. Membranes were probed with rabbit
polyclonal anti-rat tumor protein p53 (Tp53) and rabbit monoclonal
anti-β-actin primary antibodies (Abcam, USA) at 4°C overnight.
Then, the membranes were washed with tris-buffered saline
containing Tween-20 and further incubated with the appropriate
horseradish peroxidase-conjugated secondary antibody (Abcam,
Cambridge, MA, USA) at room temperature for 1.5 h. The protein
bands were detected by an ImageQuant LAS 4000 Mini instrument (GE
Healthcare Bio-Sciences AB, Uppsala, Sweden) and a SuperSignal West
Pico Chemiluminescence kit (Thermo Fisher Scientific Inc., Waltham,
MA, USA). The protein bands were quantified by ImageJ software
(National Institutes of Health, Bethesda, MD, USA).
Caspase-3 activity determination
Caspase-3 activity was determined using a Caspase-3
Activity Assay kit (Bestbio), according to the manufacturer's
protocol. Briefly, 50 µg of protein sample was added to 90 µl of
detection buffer and 10 µl of Ac-DEVD-pNA, and the mixture was
incubated at 37°C for 1 h. Next, the absorbance at 405 nm was
measured. The ratio of the reading from the experimental group to
that of the control group was the relative caspase-3 activity.
Rat apoptosis RT2
Profiler™ PCR Array analysis
The Rat Apoptosis RT2
Profiler™ PCR Array (Qiagen, GmbH, Hilden, Germany)
contains 84 apoptotic pathway-related genes. Total RNA was
extracted using an RNA extraction kit (Qiagen GmbH, Hilden,
Germany), according to the manufacturer's protocol. RT reactions
were performed using 1 ng of RNA and an RT kit (Qiagen).
Housekeeping genes such as β-actin and β-2 microglobulin were used
for normalization. The values of controls in the plate were used to
determine the expression of each target gene present in the plate.
Equal aliquots of cDNA and RT2 SYBR Green qPCR Master
Mix (Qiagen) were added to each well of the same PCR Array plate
containing the gene-specific primer sets. The data with the
threshold values were exported and analyzed using the
RT2 Profiler PCR Array Data Analysis template v. 3.3
(SuperArray Biosciences; Qiagen).
Luciferase reporter assay
To verify that Tp53 is a direct target for
miR-31a-5p, we constructed a target reporter using the
pmiR-RB-REPORT™ carrier (Ribobio, Guangzhou, China). An
approximately 200-bp fragment of the 3′UTR of Tp53 mRNA was
synthesized (Genewiz, Inc., South Plainfield, NJ, USA), which
carried two restriction sites, NotI (GC^GGCCGC) and
XhoI (C^TCGAG). This fragment was then inserted into the
vector pmiR-RB-REPORT™ (designed as
pmiR-RB-REPORT™-WT) on these two sites. Targeted
mutagenesis was performed to generate the desired mutations on the
potential miR-31a-5p target site (from WT, TCCTTTCTTGCCATTTTA, to mutant
TCCTTCATGTAGCATTTTA), designed as
pmiR-RB-REPORT™-mut. H9C2 cells seeded in 24-well plates
were cotransfected with pmiR-RB-REPORT™-WT or
pmiR-RB-REPORT™-mut together with miR-31a-5p mimics or
NC duplex (GenePharma) using a FuGene HD transfection reagent.
pmiR-RB-REPORT™ was transfected as a control. After 48 h, the cells
were harvested, and the luciferase activity was measured using a
dual-luciferase reporter assay kit (Promega Corp.) and recorded
with a multi-plate reader (Synergy 2; BioTek Instruments, Inc.,
Winooski, VT, USA).
Statistical analysis
Data are presented as the mean ± standard deviation
and were compared using a two-tailed t-test for two groups or
one-way analysis of variance for multiple groups. Correlation
analysis was performed using Spearman's correlation coefficient.
P<0.05 was considered to indicate a statistically significant
difference.
Results
MiR-31a-5p attenuated AngII-induced
apoptosis
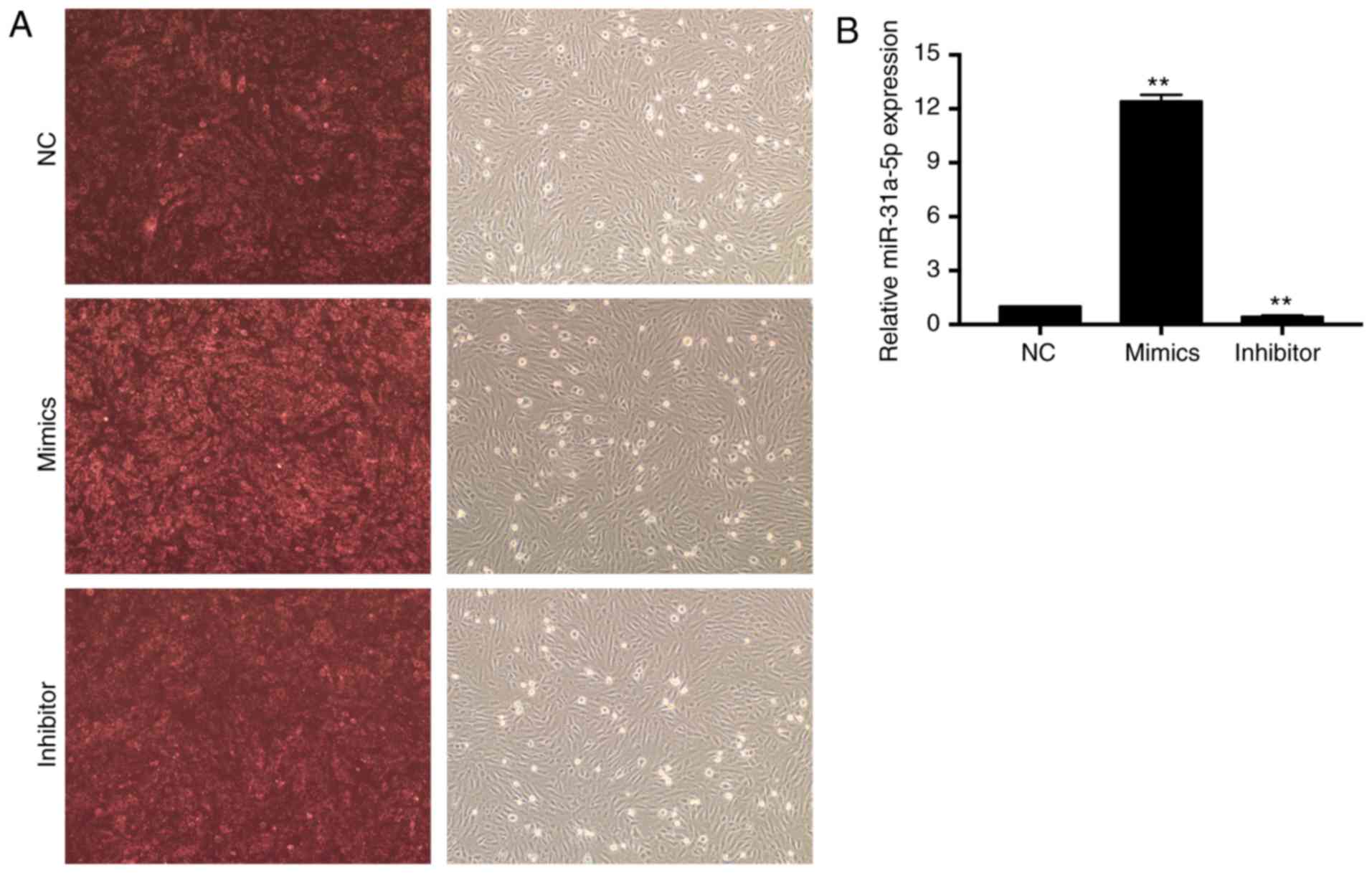
The transfection efficiency of FAM-labeled siRNAs
was first evaluated by fluorescence microscopy. As shown in
Fig. 1A, 80–90% of cells were
stained red. Next, real-time PCR was performed to examine the
expression levels of transfected miR-31a-5p. As shown in Fig. 1B, the expression level of
miR-31a-5p in the group transfected with mimics was significantly
higher than that in the control group (P<0.01), while the
miR-31a-5p level in the inhibitor group was greatly suppressed
compared with the NC group (P<0.01).
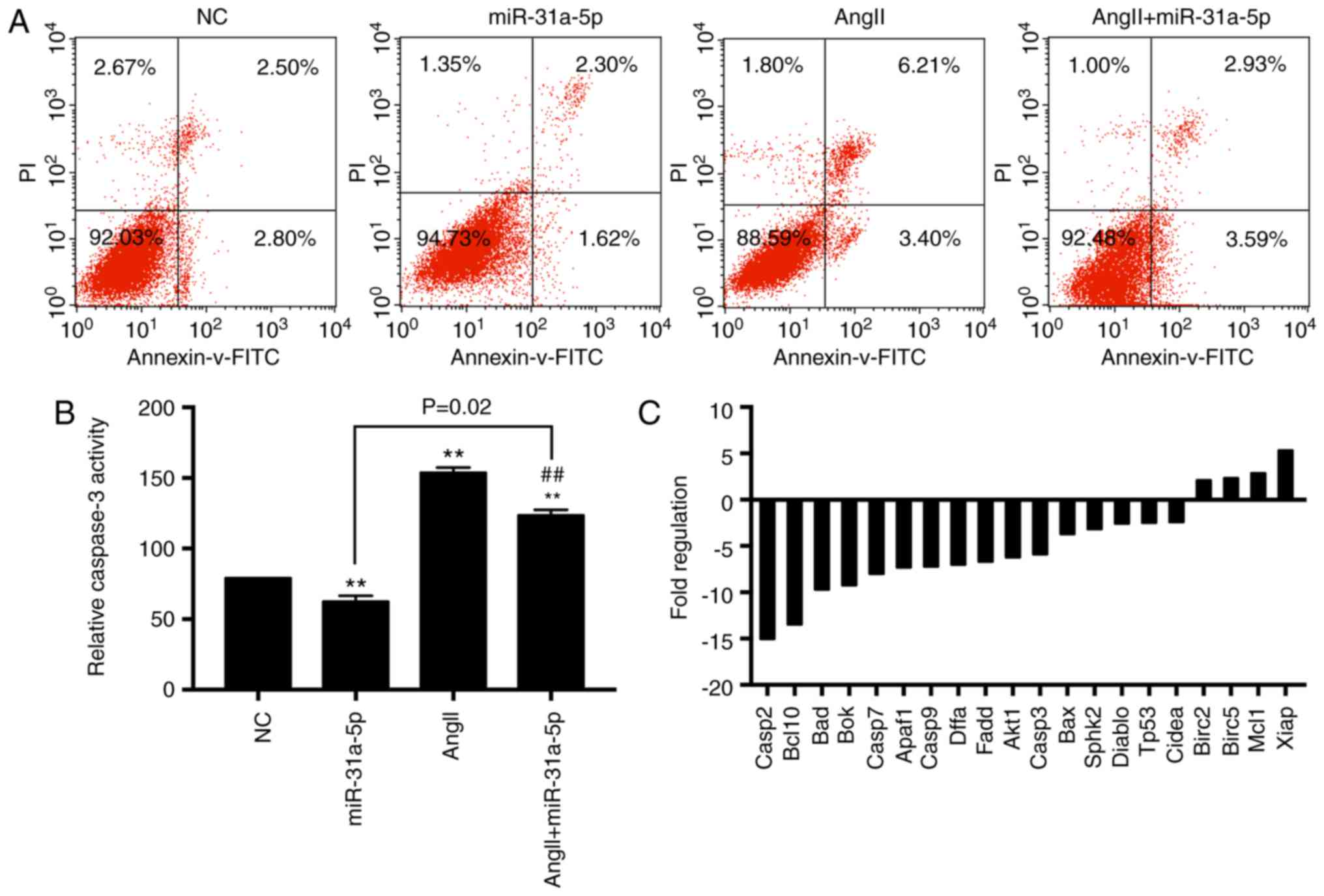
Next, we evaluated cell apoptosis in different
groups with fluorescence correlation microscopy. Overexpression of
miR-31a-5p decreased the apoptotic rate induced by AngII, while
miR-31a-5p only slightly decreased cell apoptosis in the absence of
AngII (Fig. 2A). We also measured
the caspase-3 activity, and the results were consistent with those
obtained by fluorescence correlation microscopy (Fig. 2B). Therefore, we concluded that
miR-31a-5p protects myocytes against AngII-induced apoptosis.
Next, we measured the expression of
apoptosis-related genes by RT2 Profiler™ PCR
Array analysis and found that 20 apoptosis-related genes exhibited
differential expression in the mimic-transfected group compared to
the control group, among which 16 proapoptotic genes were
downregulated and 4 antiapoptotic genes were upregulated (Fig. 2C).
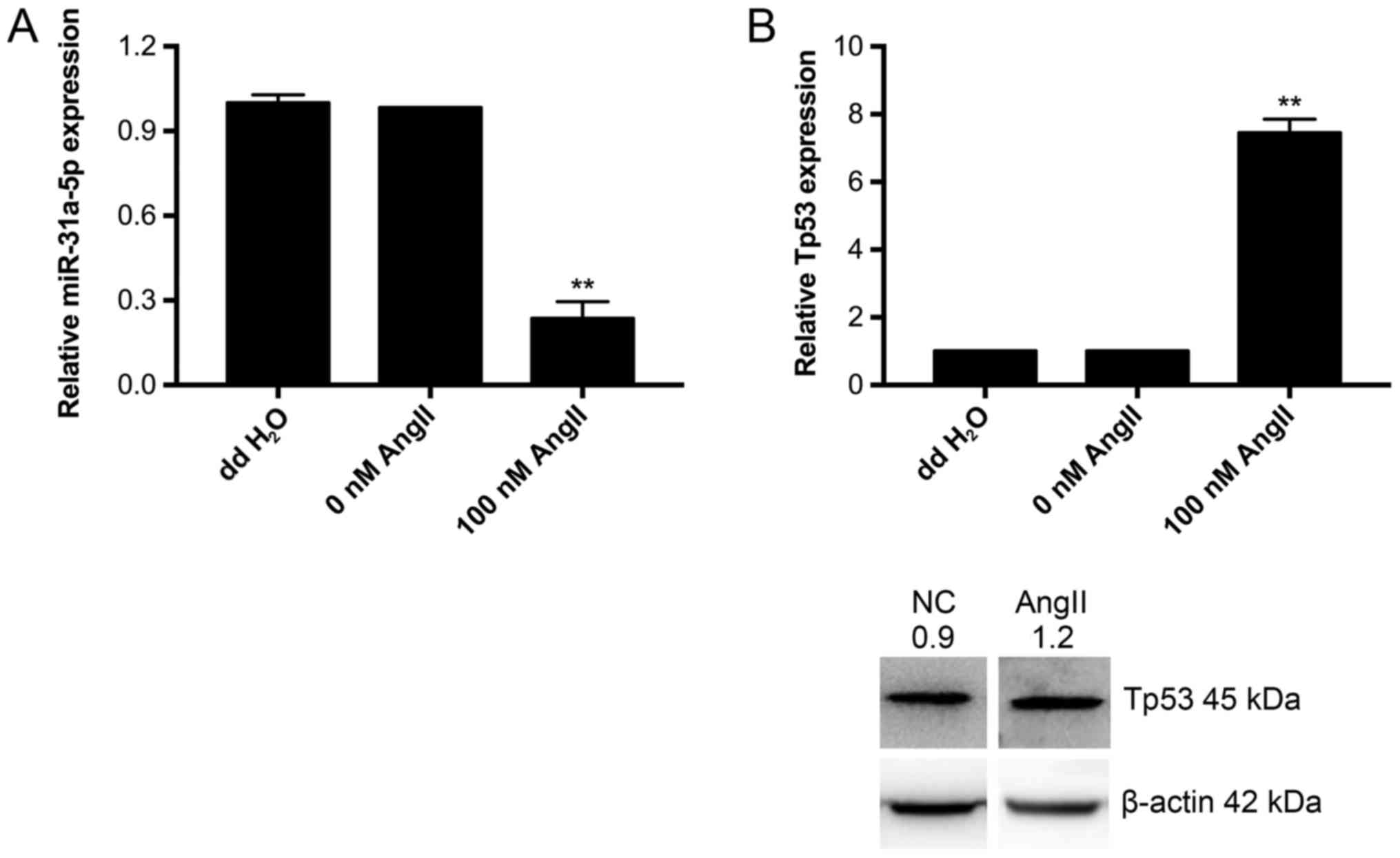
AngII decreases the expression of
miR-31a-5p and increases the expression of Tp53
After stimulation with 10−9 M AngII for
24 h, we measured the expression of miR-31a-5p and Tp53 by
real-time PCR and western blot. The results showed a significant
decrease in miR-31a-5p expression (Fig. 3A) and a substantial increase in
Tp53 expression (Fig. 3B). Hence,
we conclude that miR-31a-5p and Tp53 participate in AngII-induced
apoptosis.
Tp53 is a direct target of
miR-31a-5p
We screened several genes involved in cell cycle
progression and apoptosis. tumor protein 53 (Tp53) was one of them.
We also found that miR-31a-5p complementarily base paired with the
3′UTR region of Tp 53 mRNA (Fig.
4). Moreover, Tp53 was shown to be significantly downregulated
in an HF animal model in our previous study (unpublished).
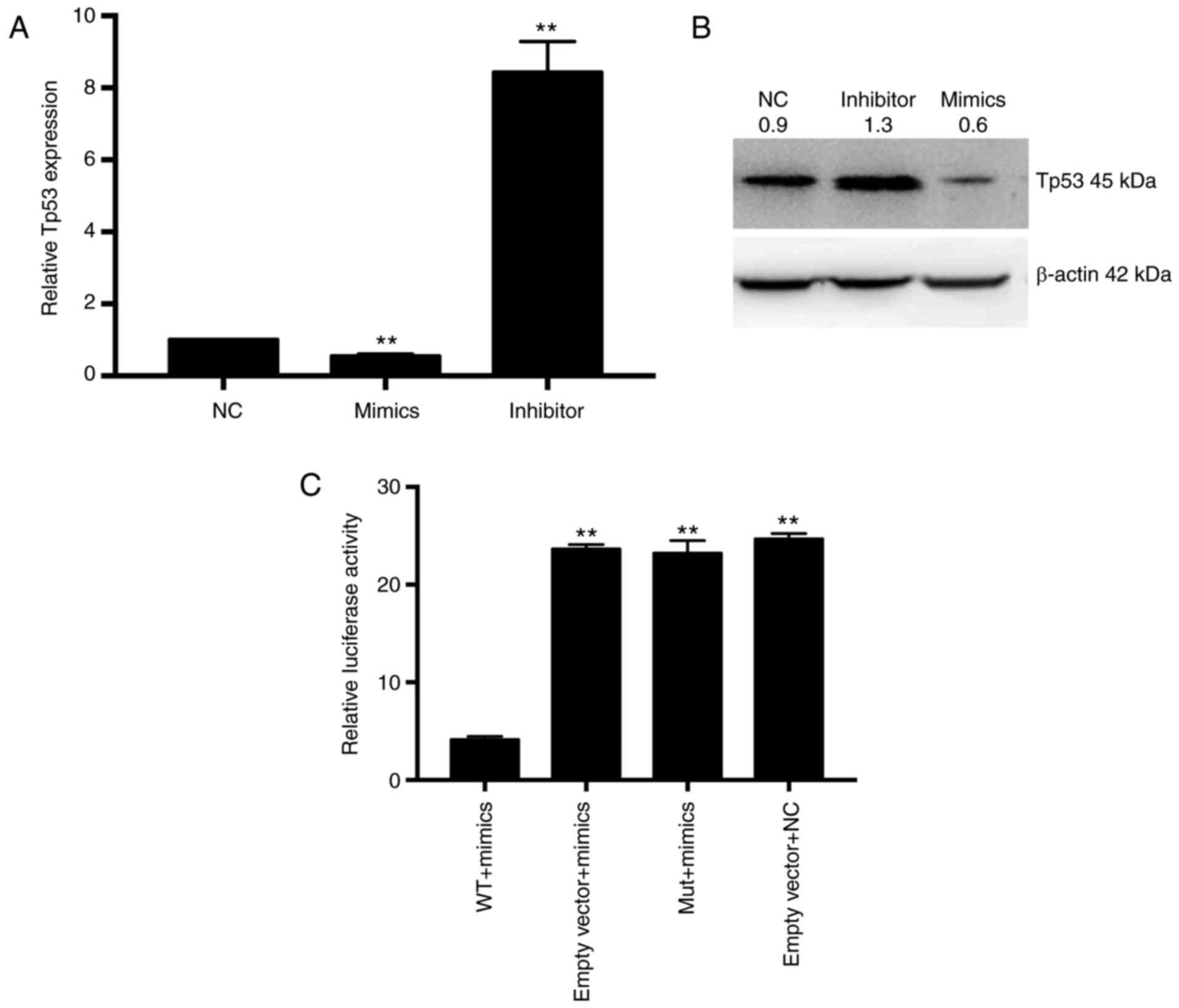
Therefore, Tp53 was chosen for further investigation. We first
examined the Tp53 expression level in different transfection groups
using real-time PCR (Fig. 5A) and
western blot (Fig. 5B).
Overexpression of miR-31a-5p inhibited the Tp53 expression level,
while the miR-31a-5p inhibitor increased the Tp53 level. Since
miRNAs bind to the seed sequence in the 3′UTR of target genes to
cause degradation or decrease the expression level of target genes,
we subcloned the wild-type and mutated target sequence of
miR-31a-5p located on the 3′UTR of Tp53 into pmiR-RB-REPORT™ to
explore whether miR-31a-5p could mediate their activity. As shown
in Fig. 5C, the activity of
pmiR-RB-REPORT™-WT but not pmiR-RB-REPORT™-mut was significantly
inhibited by miR-31a-5p mimics. Collectively, we argue that Tp53 is
a direct target of miR-31a-5p.
Discussion
In the present study, we investigated the role of
miR-31a-5p in apoptosis in AngII-stimulated H9C2 cells, a cardiac
cell line. We found that i) the miR-31a-5p level was decreased by
AngII stimulation, ii) the Tp53 level was increased by AngII
stimulation, and iii) Tp53 was a direct target of miR-31a-5p.
Previously, we showed that miR-31a-5p was
significantly upregulated in the hearts of rats with end-stage HF
compared to that in the sham-operation group. It is widely
recognized that multi-factorial mechanisms are involved in the
pathogenesis of HF, including increased activity of the RAAS and
apoptosis. It is well documented that AngII is increased in
pressure overload-induced cardiac hypertrophy (26), which contributes to cell
hypertrophy, apoptosis, and fibroblast proliferation in vivo
(27,28) as well as myocardial cell apoptosis
in vitro (25). Indeed,
ACEI has been used to treat HF effectively in the clinic, and the
inhibition of ACE reduces cardiomyocyte apoptosis (29,30),
suggesting the implication of AngII signaling in cardiomyocyte
apoptosis in vivo. Also, AngII is recognized as one of the
triggers of G-protein-coupled receptor (GPCR) signaling pathways
(31). In our study, miR-31a-5p
negatively influenced AngII-induced cell apoptosis, suggesting that
miR-31a-5p is involved in mediating GPCR signaling. In addition,
since increased apoptosis of myocardial cells induces a loss of
normal heart tissues and contributes to the pathogenesis and
progression of cardiomyopathy (32), suppression of apoptosis is a
potentially effective treatment for a number of cardiovascular
diseases including HF (33).
Consistent with the previous findings, in the present study, we
found that AngII stimulation increased apoptosis and decreased
miR-31a-5p expression in H9C2 cells. Correspondingly, the increased
expression of miR-31a-5p attenuated AngII-linked cell apoptosis, as
evidenced by a decreased cell apoptotic rate and caspase-3 activity
in AngII-treated cells with overexpressed miR-31a-5p, although
miR-31a-5p only slightly reduced cell apoptosis in the absence of
AngII. In contrast, the decreased expression of miR-31a-5p
heightened AngII-induced cell apoptosis. Thus, it appears that
miR-31a-5p is more involved in AngII-induced cardiomyocyte
apoptosis than in the basal level of apoptosis. It will be
interesting to explore whether miR-31a-5p is involved in other
stress-induced myocardial cell apoptosis.
To further understand the molecular basis underlying
miR-31a-5p mediation of cell apoptosis, we analyzed the expression
profile of the apoptosis-related genes after miR-31a-5p
overexpression in cells using a Rat Apoptosis RT2
Profiler™ PCR Array. We found that 16 proapoptotic genes
were downregulated and 4 were upregulated, including the members of
the caspase family and the Bcl2 family. The caspase family
proteases are well known to serve important roles in initiation and
development of apoptosis, which are influenced by either the
extrinsic or intrinsic cues. Among these caspase family members
detected, caspases 8 and 9 are regarded as initiators to receive
signals from upstream, and caspases 3, 6, and 7 are death executors
(1,34,35).
The Bcl2 family including anti/proapoptotic genes is involved in
the intrinsic apoptosis pathway; they are localized in the outer
membrane of mitochondria and govern mitochondrial perforation or
caspase recruitment (36). Thus,
it appears that miR-31a-5p mediates AngII-induced apoptosis through
affecting the expression of a number of apoptosis-related
genes.
In the present study, we also identified a novel
target of miR-31a-5p, Tp53. Both Tp53, the most frequently mutated
gene in cancers, and miRNAs are implicated in a variety of cellular
events and disease development, and the functional interaction
between Tp53 signaling and miRNAs has also been explored previously
(37,38). For instance, Tp53 has been shown to
govern the expression of miR-34 family members, which in turn
potentiate Tp53-regulated apoptosis (39,40).
Also, Tp53 and its signaling are mediated by miRNAs. For example,
miR-504 has been reported to suppress Tp53 expression once
ectopically expressed, thus impairing Tp53-mediated apoptosis
(41). In addition, miR-125b, a
brain-enriched miRNA, negatively regulates the expression of Tp53
(42). Moreover, overexpression of
miR-125b has been shown to diminish the expression level of Tp53
and promote cell apoptosis, while a decreased expression of
miR-125b did the opposite (42).
In the present study, we found that both miR-31a-5p and Tp53 were
mediated by AngII stimulation. Furthermore, we observed an
increased expression of Tp53, which coincided with a decreased
expression of miR-31a-5p. Further examination using bioinformatics
tools revealed that the 3′UTR of Tp53 mRNA contains the specific
(i.e., seed) sequence for potential binding of miR-31a-5p. We
further confirmed that Tp53 was a novel direct target of
miR-31a-5p, as revealed by the findings that miR-31a-5p suppressed
the activity of pmiR-RB-REPORT™-WT but not
pmiR-RB-REPORT™-mut. Together, these results suggested
that pmiR-RB-REPORT™-WT bound to the seed sequence of
the 3′UTR of TP53 mRNA and downregulated its expression.
In conclusion, our research demonstrated that the
miR-31a-5p level is suppressed under AngII stimulation and that
overexpression of miR-31a-5p reduces AngII-induced apoptosis in
H9C2 cells. We further revealed that miR-31a-5p mediates
AngII-triggered apoptosis at least in part through directly
targeting Tp53 to its 3′UTR of mRNA. Therefore, our findings add
another layer of complexity to the functional interaction between
Tp53 and miRNAs. Further work should be directed at examining
whether miR-31a-5p is a general regulator of apoptosis in response
to different external stressors in the cardiovascular setting.
Acknowledgements
The present study was supported by National Natural
Science Foundation of China (grant no. 81570360).
References
|
1
|
Salvesen GS and Dixit VM: Caspase
activation: The induced-proximity model. Proc Natl Acad Sci USA.
96:pp. 10964–10967. 1999; View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Narula J, Haider N, Virmani R, DiSalvo TG,
Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW and Khaw BA:
Apoptosis in myocytes in end-stage heart failure. N Engl J Med.
335:1182–1189. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: A basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Colucci WS: Apoptosis in the heart. N Engl
J Med. 335:1224–1226. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wencker D, Chandra M, Nguyen K, Miao W,
Garantziotis S, Factor SM, Shirani J, Armstrong RC and Kitsis RN: A
mechanistic role for cardiac myocyte apoptosis in heart failure. J
Clin Invest. 111:1497–1504. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ma TK, Kam KK, Yan BP and Lam YY:
Renin-angiotensin-aldosterone system blockade for cardiovascular
diseases: Current status. Br J Pharmacol. 160:1273–1292. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Suzuki J, Iwai M, Nakagami H, Wu L, Chen
R, Sugaya T, Hamada M, Hiwada K and Horiuchi M: Role of angiotensin
II-regulated apoptosis through distinct AT1 and AT2 receptors in
neointimal formation. Circulation. 106:847–853. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Demers C, Mody A, Teo KK and McKelvie RS:
ACE inhibitors in heart failure: What more do we need to know? Am J
Cardiovasc Drugs. 5:351–359. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smith WH and Ball SG: ACE inhibitors in
heart failure: An update. Basic Res Cardiol. 95 Suppl 1:I8–I14.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao H, Qi G, Han Y, Shen X, Yao F, Xuan
C, Gu Y, Qian SY, Zeng Q, O'Rourke ST, et al:
20-hydroxyeicosatetraenoic acid is a key mediator of angiotensin
II-induced apoptosis in cardiac myocytes. J Cardiovasc Pharmacol.
66:86–95. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schröder D, Heger J, Piper HM and Euler G:
Angiotensin II stimulates apoptosis via TGF-beta1 signaling in
ventricular cardiomyocytes of rat. J Mol Med (Berl). 84:975–983.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Grishko V, Pastukh V, Solodushko V,
Gillespie M, Azuma J and Schaffer S: Apoptotic cascade initiated by
angiotensin II in neonatal cardiomyocytes: Role of DNA damage. Am J
Physiol Heart Circ Physiol. 285:H2364–H2372. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cakmak HA, Coskunpinar E, Ikitimur B,
Barman HA, Karadag B, Tiryakioglu NO, Kahraman K and Vural VA: The
prognostic value of circulating microRNAs in heart failure:
Preliminary results from a genome-wide expression study. J
Cardiovasc Med (Hagerstown). 16:431–437. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cheng Y, Liu X, Zhang S, Lin Y, Yang J and
Zhang C: MicroRNA-21 protects against the H(2)O(2)-induced injury
on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol.
47:5–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Flynt AS, Li N, Thatcher EJ,
Solnica-Krezel L and Patton JG: Zebrafish miR-214 modulates
Hedgehog signaling to specify muscle cell fate. Nat Genet.
39:259–263. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lv G, Shao S, Dong H, Bian X, Yang X and
Dong S: MicroRNA-214 protects cardiac myocytes against H2O2-induced
injury. J Cell Biochem. 115:93–101. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Valastyan S, Chang A, Benaich N, Reinhardt
F and Weinberg RA: Activation of miR-31 function in
already-established metastases elicits metastatic regression. Genes
Dev. 25:646–659. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pacurari M and Tchounwou PB: Role of
MicroRNAs in renin-angiotensin-aldosterone system-mediated
cardiovascular inflammation and remodeling. Int J Inflam.
2015:1015272015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rayner KJ: MicroRNA-155 in the heart: The
right time at the right place in the right cell. Circulation.
131:1533–1535. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Elton TS, Selemon H, Elton SM and
Parinandi NL: Regulation of the MIR155 host gene in physiological
and pathological processes. Gene. 532:1–12. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jeppesen PL, Christensen GL, Schneider M,
Nossent AY, Jensen HB, Andersen DC, Eskildsen T, Gammeltoft S,
Hansen JL and Sheikh SP: Angiotensin II type 1 receptor signalling
regulates microRNA differentially in cardiac fibroblasts and
myocytes. Br J Pharmacol. 164:394–404. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Eskildsen TV, Schneider M, Sandberg MB,
Skov V, Brønnum H, Thomassen M, Kruse TA, Andersen DC and Sheikh
SP: The microRNA-132/212 family fine-tunes multiple targets in
Angiotensin II signalling in cardiac fibroblasts. J Renin
Angiotensin Aldosterone Syst. 16:1288–1297. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mlcochova J, Faltejskova-Vychytilova P,
Ferracin M, Zagatti B, Radova L, Svoboda M, Nemecek R, John S, Kiss
I, Vyzula R, et al: MicroRNA expression profiling identifies
miR-31-5p/3p as associated with time to progression in wild-type
RAS metastatic colorectal cancer treated with cetuximab.
Oncotarget. 6:38695–38704. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu X, Meng H, Jiang C, Yang S, Cui F and
Yang P: Differential microRNA expression and regulation in the rat
model of post-infarction heart failure. PLoS One. 11:e01609202016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cigola E, Kajstura J, Li B, Meggs LG and
Anversa P: Angiotensin II activates programmed myocyte cell death
in vitro. Exp Cell Res. 231:363–371. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kajstura J, Mansukhani M, Cheng W, Reiss
K, Krajewski S, Reed JC, Quaini F, Sonnenblick EH and Anversa P:
Programmed cell death and expression of the protooncogene bcl-2 in
myocytes during postnatal maturation of the heart. Exp Cell Res.
219:110–121. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sadoshima J and Izumo S: Molecular
characterization of angiotensin II-induced hypertrophy of cardiac
myocytes and hyperplasia of cardiac fibroblasts. Critical role of
the AT1 receptor subtype. Circ Res. 73:413–423. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kajstura J, Cigola E, Malhotra A, Li P,
Cheng W, Meggs LG and Anversa P: Angiotensin II induces apoptosis
of adult ventricular myocytes in vitro. J Mol Cell Cardiol.
29:859–870. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Goussev A, Sharov VG, Shimoyama H,
Tanimura M, Lesch M, Goldstein S and Sabbah HN: Effects of ACE
inhibition on cardiomyocyte apoptosis in dogs with heart failure.
Am J Physiol. 275:H626–H631. 1998.PubMed/NCBI
|
|
30
|
Bäcklund T, Palojoki E, Saraste A,
Grönholm T, Eriksson A, Lakkisto P, Vuolteenaho O, Nieminen MS,
Voipio-Pulkki LM, Laine M and Tikkanen I: Effect of vasopeptidase
inhibitor omapatrilat on cardiomyocyte apoptosis and ventricular
remodeling in rat myocardial infarction. Cardiovasc Res.
57:727–737. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu CH, Gong Z, Liang ZL, Liu ZX, Yang F,
Sun YJ, Ma ML, Wang YJ, Ji CR, Wang YH, et al: Arrestin-biased AT1R
agonism induces acute catecholamine secretion through TRPC3
coupling. Nat Commun. 8:143352017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Prech M, Marszałek A, Schröder J, Filas V,
Lesiak M, Jemielity M, Araszkiewicz A and Grajek S: Apoptosis as a
mechanism for the elimination of cardiomyocytes after acute
myocardial infarction. Am J Cardiol. 105:1240–1245. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim NH and Kang PM: Apoptosis in
cardiovascular diseases: Mechanism and clinical implications.
Korean Circ J. 40:299–305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Thornberry NA and Lazebnik Y: Caspases:
Enemies within. Science. 281:1312–1316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sadowski-Debbing K, Coy JF, Mier W, Hug H
and Los M: Caspases-their role in apoptosis and other physiological
processes as revealed by knock-out studies. Arch Immunol Ther Exp
(Warsz). 50:19–34. 2002.PubMed/NCBI
|
|
36
|
Crow MT, Mani K, Nam YJ and Kitsis RN: The
mitochondrial death pathway and cardiac myocyte apoptosis. Circ
Res. 95:957–970. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hünten S, Siemens H, Kaller M and
Hermeking H: The p53/microRNA network in cancer: Experimental and
bioinformatics approaches. Adv Exp Med Biol. 774:77–101. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Feng Z, Zhang C, Wu R and Hu W: Tumor
suppressor p53 meets microRNAs. J Mol Cell Biol. 3:44–50. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tazawa H, Tsuchiya N, Izumiya M and
Nakagama H: Tumor-suppressive miR-34a induces senescence-like
growth arrest through modulation of the E2F pathway in human colon
cancer cells. Proc Natl Acad Sci USA. 104:pp. 15472–15477. 2007;
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Raver-Shapira N, Marciano E, Meiri E,
Spector Y, Rosenfeld N, Moskovits N, Bentwich Z and Oren M:
Transcriptional activation of miR-34a contributes to p53-mediated
apoptosis. Mol Cell. 26:731–743. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hu W, Chan CS, Wu R, Zhang C, Sun Y, Song
JS, Tang LH, Levine AJ and Feng Z: Negative regulation of tumor
suppressor p53 by microRNA miR-504. Mol Cell. 38:689–699. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Le MT, Teh C, Shyh-Chang N, Xie H, Zhou B,
Korzh V, Lodish HF and Lim B: MicroRNA-125b is a novel negative
regulator of p53. Genes Dev. 23:862–876. 2009. View Article : Google Scholar : PubMed/NCBI
|