Introduction
Chronic and acute stress to the heart results in a
pathological remodeling response accompanied by cardiomyocyte
hypertrophy, fibrosis, myocyte degeneration and apoptosis, finally
contributing to heart failure (HF) (1). HF is a progressive disorder
characterized by poor quality of life and poor prognosis. In recent
years HF has reached endemic proportions in the industrialized
world, with 5.7 million patients affected by this disease in the
United States (2). Similarly
striking data are also observed in Europe and Asia, and the
incidence of critical risk factors for HF have also markedly
increased (3,4). The prevalence of HF is expected to
rise due to the aging population and improved treatment of
cardiovascular diseases that precede HF (5). Therefore, it is imperative that novel
methods to improve the treatment of HF are identified.
Cardiac remodeling is an important determinant in
the clinical outcome of HF; it is linked to disease progression and
poor prognosis (6). Cardiac
fibrosis is an important contributor to the pathophysiology of
cardiac remodeling caused by hypertension. Galectin-3 is a
β-galactoside-binding lectin that appears to be a mediator of
cardiac fibrosis in several experimental studies (7,8). The
potential HF-promoting actions of galectin-3 have been described
(9), although the precise
mechanisms still warrant further investigation. Galectin-3 is
produced by macrophages and fibroblasts, but not by cardiomyocytes,
in rat and mouse models of progressive cardiac remodeling (10,11).
The pro-fibrotic characteristics of galectin-3 have also been
demonstrated in models of pathological fibrosis in kidney and
liver, suggesting that fibrosis may be a hallmark of cardiac
remodeling and HF (12). Although
several prognostic studies of patients with HF have indicated the
independent predictive value of galectin-3 for cardiovascular
outcomes, others have questioned whether galectin-3 is truly
independently associated with adverse cardiac events (13,14).
The observations provide evidence for the hypothesis that
longstanding elevated galectin-3 may not signify disease, but
likely represents a unique phenotype at high risk for the
development and progression of heart failure.
The mechanism of galectin-3 as a promoter of the
development of HF has not been investigated. The present study
aimed to explore the mechanism by which galectin-3 may be
associated with cardiomyocyte apoptosis and survival during HF.
Materials and methods
Isolation of cardiomyocytes and cell
culture
Isolation of cardiomyocytes and cell culture was
performed as previously described (15). In brief, animals were anesthetized
by an intraperitoneal injection of chloral hydrate (300 mg/kg).
Perfusion with fixatives (4% paraformaldehyde, 2.5% glutaraldehyde)
was used as the primary method of euthanasia. The heart was excised
and perfused using the retrograde Langendorff technique.
Subsequently, the perfusion buffer was supplemented with 0.6%
collagenase II and 0.6% bovine serum albumin (both from Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), and the buffer
was recirculated for 12–15 min at 25°C. The dissociated cells,
including the majority of the cardiomyocytes, were filtered through
a 200 µm mesh. The cardiomyocytes were preplated for 1 h [2 mM
L-glutamine and 10% FBS in Dulbecco's modified Eagle's medium
(DMEM); Invitrogen; Thermo Fisher Scientific, Inc.] at 37°C in a
humidified atmosphere of 5% CO2 and 95% air to enrich
the culture, and were subsequently cultured in serum-free DMEM
(Invitrogen; Thermo Fisher Scientific, Inc.) for 24 h at 37°C in a
humidified atmosphere of 5% CO2 and 95% air.
Overexpression and construction of
stable cell lines
The galectin-3 coding sequence was purchased from
Sangon Biotech Co., Ltd. (Shanghai, China) and the sequence was
cloned into the pcDNA3.1(+) vector (Sangon Biotech Co., Ltd.). The
primers used were as follows: Galectin-3 forward,
5′-CGGGATCCTTAGATCATGGCGTGGTTAGC-3′ and reverse,
5′-CGGGATCCTTAGATCATGGCGTGGTTAGC-3′. The sequences were annealed
and digested using EcoRI and BamHI, and were ligated
into the pcDNA3.1(+) vector. An empty pCDNA3.1(+) vector served as
the negative control (NC). The pCDNA3.1(+) vector encoding
galectin-3 (1 µg) was subsequently transfected into the
cardiomyocytes using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
instructions. All assays were performed 48 h following
transfection.
Cell viability analysis
Following transfection, cardiomyocytes were plated
into 96-well plates at 1×103 cells/well in a final
volume of 100 µl, and cultured overnight. Cell viability was
determined using a Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) assay, according to the
manufacturer's instructions. In brief, at 24, 48 and 72 h following
transfection, 10 µl CCK-8 solution was added to each well and
incubated for 1 h at 37°C and 5% CO2. Following
incubation, absorbance was measured at 450 nm using a microplate
reader (SM600 Labsystem; Shanghai Utrao Medical Instrument Co.,
Ltd., Shanghai, China).
Cell cycle and apoptosis analysis
For the cell cycle assay, 3×104
transfected cells were collected and fixed in 70% ethanol at −20°C
overnight. The cells were subsequently washed in PBS and
resuspended in staining solution containing 20 µg/ml propidium
iodide (PI; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and 200
µg/ml RNase A. Cell cycle was assessed by flow cytometry, using BD
Accuri C6 software version 1.0.264.21 (BD Biosciences, Franklin
Lakes, NJ, USA). For the cell apoptosis assay, 3×104
transfected cells were collected by trypsinization (Jrdun
Biotechnology Co., Ltd., Shanghai, China) and incubated with 195 µl
Annexin V-fluorescein isothiocyanate for 15 min at 4°Cand 5 µl PI
for 5 min at 4°C, prior to analysis using a flow cytometer (BD
Biosciences, Franklin Lakes, NJ, USA). Following this, the results
were subsequently analyzed using CellQuest software (version 5.1;
BD Biosciences).
Animals and treatment
A total of 18 male Dahl salt-sensitive rats (weight,
200–250 g) purchased from Shanghai SLAC Laboratory Animal Co., Ltd.
(Shanghai, China) were fed a high-salt (HS) diet (8.0% NaCl)
starting at 8 weeks of age, which progresses to a model of
congestive HF (16). Rats (6/cage)
were housedin an animal facility at 25°C, with a relative humidity
of 60–70% and received food and water ad libitum. After 1
week on the HS diet, 25 µg/kg lentivirus containing gal-3 short
hairpin (sh)RNA sequence (pLKO-shGAL3 cat. TRC0000029305;
OpenBiosystems; Thermo Fisher Scientific, Inc.) was intravenously
injected via the tail vein (6/goup). The shRNA scramble sequence
was used as a transduction control for 3 weeks. After 4 weeks on
the HS diet, the rats were sacrificed and weighed. Cardiac tissue
samples were collected at the indicated time-points for molecular
or histological examination, which were frozen at −80°C and fixed
with 4% paraformaldehyde for 48 h at 25°C. The survival time of
rats with galectin-3 knockdown treatment was measured during the 4
weeks. The investigation conformed to the Guide for the Care and
Use of Laboratory Animals published by the U.S. National Institutes
of Health (17). The study was
approved by the ethics committee of Zhongnan Hospital of Wuhan
University (Wuhan, China).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
For in vitro and in vivo RT-qPCR
analyses, RNA was extracted from cardiomyocytes or cardiac tissue
with TRIzol (Life Technologies; Thermo Fisher Scientific, Inc.). A
total of 1 µg RNA was used to generate cDNA using a M-MLV RT
Reagent kit according to the manufacturer's instructions (Thermo
Fisher Scientific, Inc.). To detect the level of galectin-3,
proliferating cell nuclear antigen (PCNA), B-cell lymphoma 2
(Bcl-2) and Bcl-2-associated X protein (Bax), qPCR was performed
with a SYBR Green qPCR Master mix (Takara Biotechnology Co., Ltd.,
Dalian, China) and data collection was conducted using an ABI 7500
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The PCR
cycling conditions were as follows: 95°C for 10 min, followed by 40
cycles at 95°C for 15 sec and 60°C for 45 sec, and a final
extension step of 95°C for 15 sec, 60°C for 1 min, 95°C for 15 sec
and 60°C for 15 sec. GAPDH was used an endogenous control for
normalization. The gene expression was calculated using the
2−ΔΔCq method (18).
The primers (Sangon Biotech Co., Ltd.) used were as follows:
Galectin-3 forward, 5′-CATTGTGTGTAACACGAAGCAGGAC-3′ and reverse,
5′-CTGCAGTAGGTGAGCATCGTTGA-3′; GAPDH forward,
5′-GGAATCCACTGGCGTCTTCA-3′ and reverse, 5′-GGTTCACGCCCATCACAAAC-3′;
Bcl-2 forward, 5′-CCACCTGTGGTCCATCTGAC-3′ and reverse,
5′-CAATCCTCCCCCAGTTCACC-3′; Bax forward, 5′-GTCATCTCGCTCTGGTACGG-3′
and reverse, 5′-CACACACACAAAGCTGCTCC-3′; PCNA forward,
5′-CCCTGGAGCCCTTGAAGAAG-3′ and reverse,
5′-AGATGCACAACTTCTCGGCA-3′.
Western blot analysis
Mouse monoclonal antibodies against galectin-3
(1:1,000; cat. no. sc-374253) and the rabbit polyclonal antibodies
against Bax (1:500; cat. no. sc-493) and Bcl-2 (1:1,000; cat. no.
sc-492) were purchased from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA). Rabbit monoclonal antibodies against PCNA (1:1,000; cat.
no. 13110) and GAPDH (1:1,000; cat. no. 5174) were purchased from
Cell Signaling Technology, Inc. (Danvers, MA, USA). Horseradish
peroxidase-conjugated goat anti-rabbit (1:1,000; cat. no. A0208)
and anti-mouse (1:1,000; cat. no. A0216) secondary antibodies were
purchased from Beyotime Institute of Biotechnology (Haimen, China).
Proteins were detected by western blot analysis as previously
described (19). The total cell
lysates were extracted using radioimmunoprecipitation buffer
(Amyjet Scientific, Inc., Wuhan, China) according to the
manufacturer's instructions. The protein concentration was
determined using a Bicinchoninic Acid Protein Assay kit (cat. no.
PICPI23223; Thermo Fisher Scientific, Inc.). A total of 15 µl of
proteins were subjected to 10 or 15% SDS-PAGE, transferred to
polyvinylidene fluoride membranes (Sigma-Aldrich; Merck KGaA) and
blocked in fat-free milk overnight at 4°C. The membranes were
subsequently incubated with primary antibodies for 2 h at 25°C and
then with secondary antibodies for 1 h at 37°C, and were visualized
using an enhanced chemiluminescence kit (EMD Millipore, Billerica,
MA, USA) and signals were semi-quantified by densitometry (Quantity
One software, version 4.62; Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Statistical analysis
Data were presented as the mean ± standard deviation
with three repeats and were analyzed using GraphPad Prism 5
software (GraphPad Software, Inc., La Jolla, CA, USA). Differences
were assessed using a one-way analysis of variance, followed by
Tukey's post hoc test. Survival analysis was performed by the
Kaplan-Meier method, and subjected to the log rank test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Galectin-3 overexpression inhibits the
viability of cardiomyocytes
To investigate the role of galectin-3 in
cardiomyocytes, a galectin-3 overexpression vector was constructed;
the galectin-3 coding sequence was cloned into a pCDNA3.1(+)
vector, and the empty vector was used as NC. At 48 h after
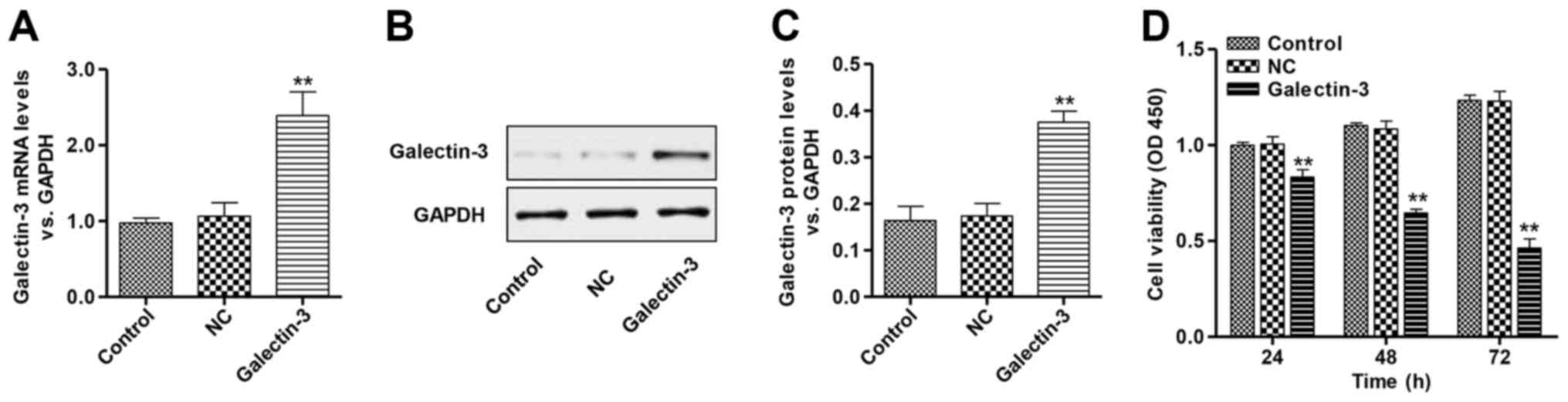
transfection, the expression of galectin-3 was determined using
RT-qPCR (Fig. 1A) and western
blotting (Fig. 1B and C). The
overexpression vector significantly increased the expression of
galectin-3 in cardiomyocytes at the mRNA and protein levels.
To determine the impact of galectin-3 overexpression
on the viability of cardiomyocytes, the cell viability of
galectin-3-overexpressing cardiomyocytes was analyzed by CCK-8
assay. Cell viability was significantly reduced in the
galectin-3-overexpressing cardiomyocytes compared with the control
and NC groups (Fig. 1D). There was
no significant difference between the control and NC groups. These
data suggest galectin-3 may have a role in the cell viability of
cardiomyocytes.
Galectin-3 overexpression induces cell
cycle arrest and apoptosis of cardiomyocytes
The potential inhibitory effect of galectin-3
overexpression on cell cycle progression was investigated. As
presented in Fig. 2A,
overexpression of galectin-3 resulted in a lower number of cells in
the G0-G1 phase (54.54±4.18%) compared with
the NC group (69.11±3.17%). Furthermore, there was a higher number
of cells in the S (26.94±1.39%) and G2-M phases
(16.64±1.52%), compared with the corresponding NC groups (S phase,
19.59±2.24%; G2-M phase, 9.86±1.46%). These data suggest
that galectin-3 overexpression may induce cell cycle arrest at S
and G2-M phases, which may be associated with reduced
cell viability in galectin-3-overexpressing cardiomyocytes.
To assess the effects of galectin-3 overexpression
on cell apoptosis, Annexin V/PI staining was performed (Fig. 2B). The ratio of cells undergoing
apoptosis was significantly increased to 27.17±2.32% in
galectin-3-overexpressing cardiomyocytes, compared with the NC
group (4.33±1.31%). These results indicate that galectin-3 may
promote apoptosis in cardiomyocytes.
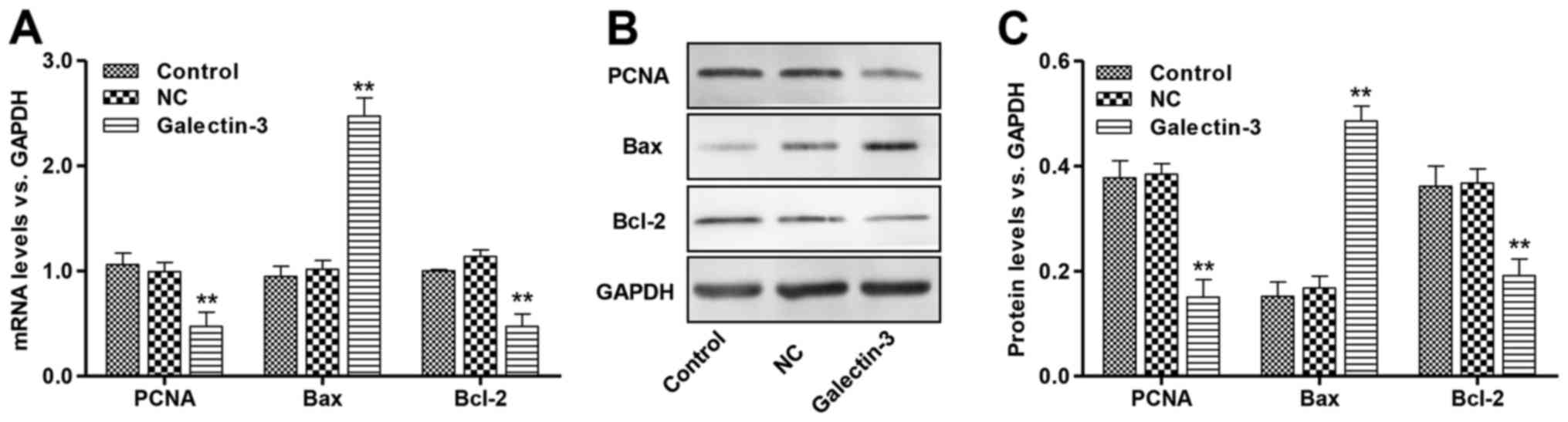
Effects of galectin-3 overexpression
on PCNA, Bcl-2 and Bax expression in cardiomyocytes
PCNA is a protein that is synthesized in early
G1 and S phases of the cell cycle and functions in cell
cycle progression, DNA replication and DNA repair (20). The proteins of the Bcl-2 family
perform critical roles in the regulation of apoptosis by
functioning as promoters (i.e., Bax) or as inhibitors (i.e., Bcl-2)
of cell death progression (21).
RT-qPCR and western blot analysis were performed to detect the mRNA
and protein expression levels of PCNA, Bcl-2 and Bax. Galectin-3
overexpression resulted in a marked reduction in the levels of PCNA
and Bcl-2, with a concomitant increase in the level of Bax compared
with the corresponding NC groups (Fig.
3). These results demonstrate that galectin-3 overexpression
may decrease PCNA expression and decrease the ratio of Bcl-2/Bax,
which may contribute to the observed increase in cell
apoptosis.
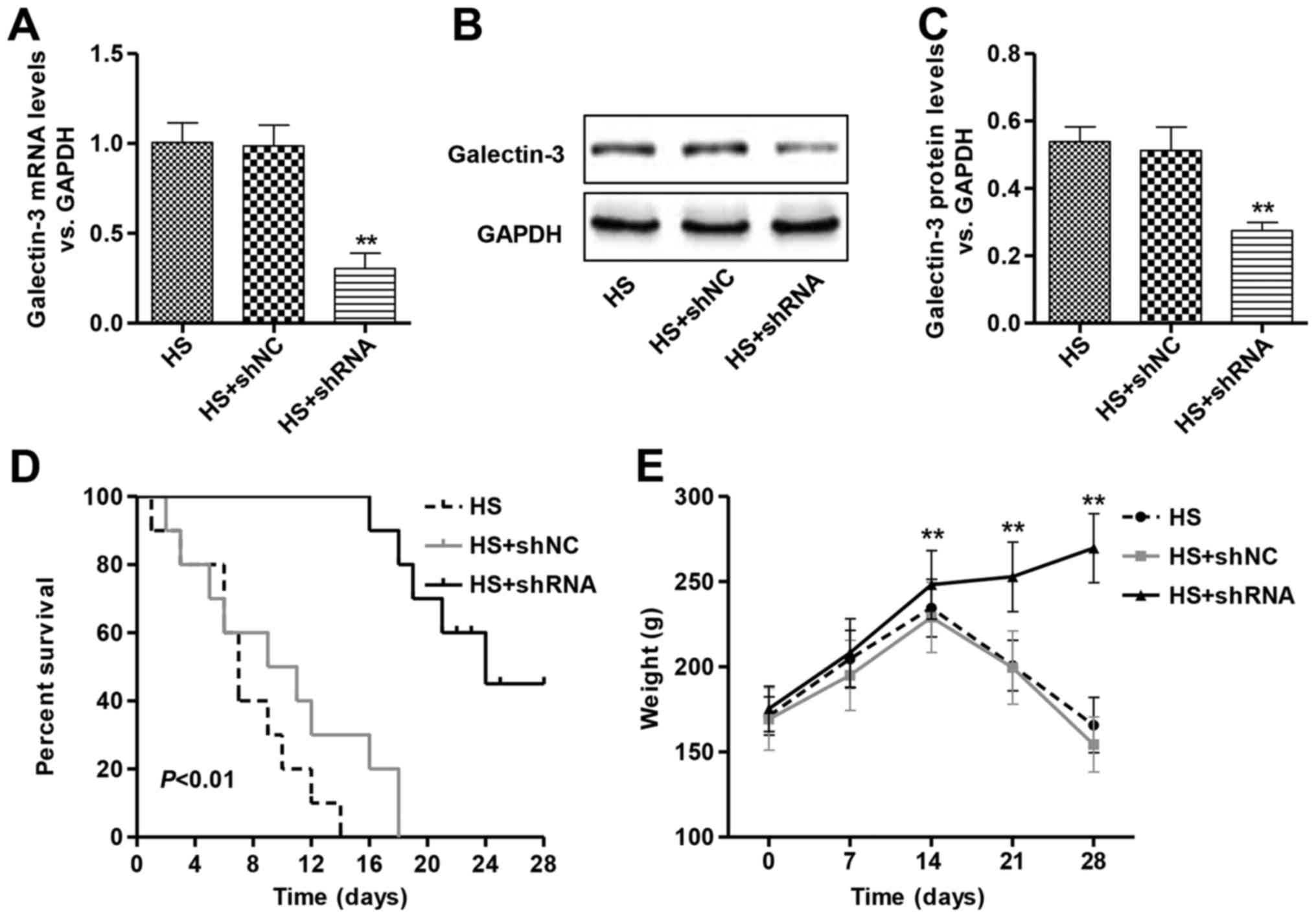
Therapeutic silencing of galectin-3
improves cardiomyocyte survival during heart failure
The potential therapeutic impact of galectin-3
inhibition was investigated in vivo. Dahl salt-sensitive
rats were fed a HS diet (8.0% NaCl), which causes the rats to
progress to a model of heart failure. After 1 week on the HS diet,
rats were intravenously injected with galectin-3 shRNA or shNC,
stably expressed in the pLKO.1 lentiviral vector. Knockdown of
galectin-3 significantly decreased the expression levels of
galectin-3 in the cardiac tissue of rats with heart failure
(Fig. 4A-C). After 4 weeks on the
HS diet, the control and shNC rats started to exhibit signs of
discomfort and were sacrificed; however, intravenous injection of
galectin-3 shRNA significantly increased the survival rate
(Fig. 4D). As a surrogate
indicator of overall health, body weight was regularly monitored
for the duration of the study (1).
All the rats gained weight together until day 14 of the HS diet,
whereas rats in the control and shNC groups after day 14 of HS diet
exhibited significantly reduced weight gain, compared with the rats
injected with galectin-3 shRNA (Fig.
4E).
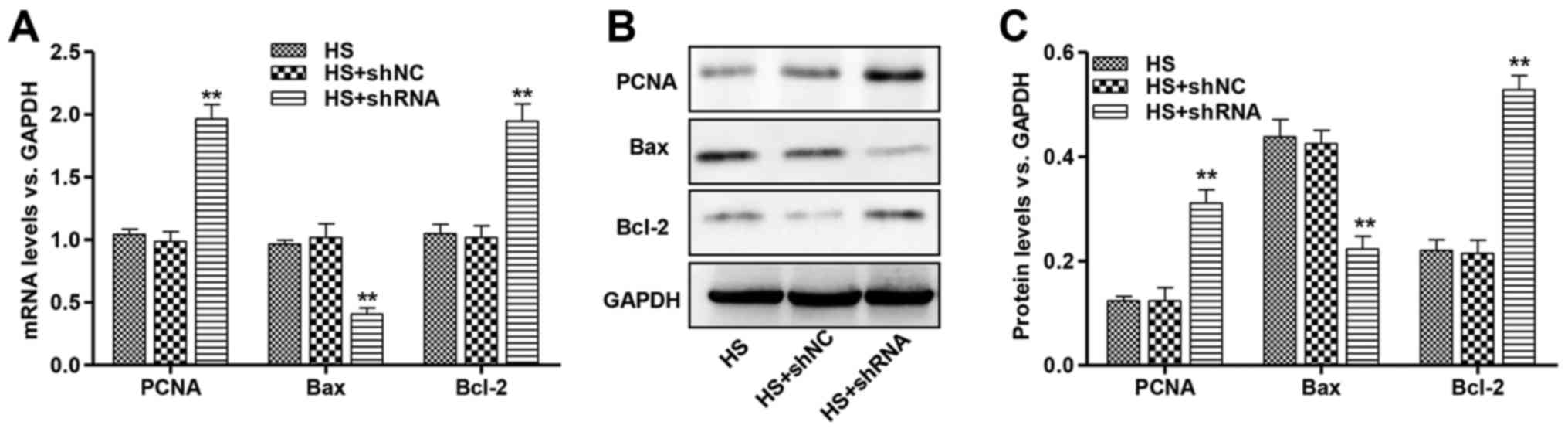
Effect of galectin-3 silencing on
PCNA, Bcl-2 and Bax expression in Dahl hypertensive rats
To investigate the regulation of proliferation- and
apoptosis-associated genes, PCNA, Bcl-2 and Bax mRNA and protein
levels were assessed in the cardiac tissue of Dahl hypertensive
rats. PCNA and Bcl-2 mRNA levels were significantly increased in
HS-induced rats injected with galectin-3 shRNA (Fig. 5A). Furthermore, treatment with
galectin-3 shRNA significantly decreased the Bax mRNA expression
level in HS-induced rats. The altered regulation of these proteins
was confirmed by western blot analysis (Fig. 5B and C). These results demonstrate
that galectin-3 knockdown may increase PCNA and the Bcl-2/Bax
ratio, which may lead to an increase in cardiomyocyte survival.
Discussion
Galectin-3 is expressed in various tissues and cell
types and is involved in several biological processes, including
cell proliferation, cell cycle, apoptosis and angiogenesis
(22). An increased concentration
of galectin-3 has been identified in patients with HF and this
protein may be promising for high-risk patient identification
(23). The present study
demonstrated that higher levels of galectin-3, a marker of cardiac
fibrosis (24), are associated
with the development and progression of HF. To the best of our
knowledge, this is the first study to report the association of
galectin-3 with cardiomyocyte proliferation and apoptosis in
vitro, and with survival in vivo. The present study has
also indicated that the HF-promoting galectin-3 mechanisms may be
associated with decreased expression of PCNA and Bcl-2, and
increased expression of Bax.
Experimental evidence has suggested that galectin-3
is the most predominantly overexpressed gene in transgenic Ren-2
rats that progress to HF (7). The
present study revealed that overexpression of galectin-3 in
cardiomyocytes resulted in suppression of cell proliferation in a
time-dependent manner, accompanied by apoptosis and cell cycle
arrest at S and G2-M phases. These data suggest that
galectin-3 overexpression may contribute to cardiac dysfunction by
inducing cardiomyocyte cell cycle arrest and apoptosis. In
agreement with these findings, infusion of galectin-3 into the
pericardial space leads to cardiac dysfunction in rats, a process
that appears to be mediated via the transforming growth
factor-β/SMAD family member 3 signaling pathway (8). However, several studies have
indicated that galectin-3 overexpression regulates the cell cycle
and apoptosis via alterations in the expression levels of cell
cycle regulators, including cyclin D1. Notably, the
growth-promoting activity of galectin-3 is predominantly dependent
on cyclin D1 promoter activity (25,26).
The expression of PCNA and Bcl-2 were significantly decreased in
galectin-3 overexpressing cardiomyocytes, whereas the Bax
expression was elevated, suggesting that galectin-3 may arrest the
cell cycle and induce apoptosis via regulation of the PCNA and
Bcl-2/Bax pathways.
Experimental evidence for the involvement of
galectin-3 in HF development came from studies that galectin-3
inhibition in mice resulted in a marked reduction of cardiac
fibrosis, and thus, may be beneficial in the prevention of HF
(8,27). Similarly, in the present study, the
therapeutic effects of galectin-3 inhibition in the Dahl
hypertensive rats provided evidence that intravenous tail vein
injections of galectin-3-shRNA is sufficient to deliver
galectin-3-shRNA effectively to the heart in vivo, and that
galectin-3 inhibition results in the increased survival and weight
gain of Dahl hypertensive rats, compared with rats without
galectin-3 inhibition. Cell cycle and apoptosis-associated
proteins, including PCNA, Bcl-2 and Bax, were also detected in Dahl
hypertensive rats. The results indicated that galectin-3 inhibition
significantly increased the expression of PCNA and Bcl-2, and
decreased the expression of Bax, compared with Dahl hypertensive
rats without galectin-3 inhibition. These results suggested that
PCNA and the Bcl-2/Bax pathway were also involved in the galectin-3
silencing-dependent promotion of the survival and weight gain of
Dahl hypertensive rats.
Subsequent pharmacokinetic and efficacy studies in
larger mammals are required to establish whether inhibition of
galectin-3 is able to establish a comparable therapeutic effect in
larger animals. In addition, therapeutic galectin-3 inhibition
would likely involve combination with the current standard
treatments in used for patients with HF; thus, it will be important
to assess whether galectin-3 inhibition, in conjunction with
current treatments, adds to the beneficial effects of these drugs.
Taken together, this study demonstrates that overexpression of
galectin-3 in cardiomyocytes inhibits cell proliferation, arrests
the cell cycle and induces apoptosis. Galectin-3 inhibition in Dahl
hypertensive rats increased survival and further validates
galectin-3 as a potential target for cardiac disease therapy.
References
|
1
|
Montgomery RL, Hullinger TG, Semus HM,
Dickinson BA, Seto AG, Lynch JM, Stack C, Latimer PA, Olson EN and
van Rooij E: Therapeutic inhibition of miR-208a improves cardiac
function and survival during heart failure. Circulation.
124:1537–1547. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mozaffarian D, Benjamin EJ, Go AS, Arnett
DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ,
Howard VJ, et al: Heart disease and stroke statistics-2015 update:
A report from the American heart association. Circulation.
131:e29–e322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guha K and McDonagh T: Heart failure
epidemiology: European perspective. Curr Cardiol Rev. 9:123–127.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu SS, Kong LZ, Gao RL, Zhu ML, Wang W,
Wang YJ, Wu ZS, Chen WW and Liu MB; Editorial Board, : Outline of
the report on cardiovascular disease in China, 2010. Biomed Environ
Sci. 25:251–256. 2012.PubMed/NCBI
|
|
5
|
Bui AL, Horwich TB and Fonarow GC:
Epidemiology and risk profile of heart failure. Nat Rev Cardiol.
8:30–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Spinelli L, Stabile E, Giugliano G,
Morisco C, Giudice CA, Imbriaco M, Santoro M, Esposito G and
Trimarco B: Intramyocardial dissecting hematoma in anterior wall ST
elevation myocardial infarction: Impact on left ventricular
remodeling and prognosis. Int J Cardiovasc Imaging. Aug
1–2017.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sharma UC, Pokharel S, van Brakel TJ, van
Berlo JH, Cleutjens JP, Schroen B, André S, Crijns HJ, Gabius HJ,
Maessen J and Pinto YM: Galectin-3 marks activated macrophages in
failure-prone hypertrophied hearts and contributes to cardiac
dysfunction. Circulation. 110:3121–3128. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu YH, D'Ambrosio M, Liao TD, Peng H,
Rhaleb NE, Sharma U, André S, Gabius HJ and Carretero OA:
N-acetyl-seryl-aspartyl-lysyl-proline prevents cardiac remodeling
and dysfunction induced by galectin-3, a mammalian
adhesion/growth-regulatory lectin. Am J Physiol Heart Circ Physiol.
296:H404–H412. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de Boer RA, Yu L and van Veldhuisen DJ:
Galectin-3 in cardiac remodeling and heart failure. Curr Heart Fail
Rep. 7:1–8. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Henderson NC, Mackinnon AC, Farnworth SL,
Poirier F, Russo FP, Iredale JP, Haslett C, Simpson KJ and Sethi T:
Galectin-3 regulates myofibroblast activation and hepatic fibrosis.
Proc Natl Acad Sci USA. 103:pp. 5060–5065. 2006; View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Henderson NC, Mackinnon AC, Farnworth SL,
Kipari T, Haslett C, Iredale JP, Liu FT, Hughes J and Sethi T:
Galectin-3 expression and secretion links macrophages to the
promotion of renal fibrosis. Am J Pathol. 172:288–298. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Van den Borne SW, Diez J, Blankesteijn WM,
Verjans J, Hofstra L and Narula J: Myocardial remodeling after
infarction: The role of myofibroblasts. Nat Rev Cardiol. 7:30–37.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
de Boer RA, Voors AA, Muntendam P, van
Gilst WH and van Veldhuisen DJ: Galectin-3: A novel mediator of
heart failure development and progression. Eur J Heart Fail.
11:811–817. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Anand IS, Rector TS, Kuskowski M, Adourian
A, Muntendam P and Cohn JN: Baseline and serial measurements of
galectin-3 in patients with heart failure: Relationship to
prognosis and effect of treatment with valsartan in the Val-HeFT.
Eur J Heart Fail. 15:511–518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wei F, Wang TZ, Zhang J, Yuan ZY, Tian HY,
Ni YJ, Zhuo XZ, Han K, Liu Y, Lu Q, et al: Mesenchymal stem cells
neither fully acquire the electrophysiological properties of mature
cardiomyocytes nor promote ventricular arrhythmias in infarcted
rats. Basic Res Cardiol. 107:2742012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rodenbaugh DW, Wang W, Davis J, Edwards T,
Potter JD and Metzger JM: Parvalbumin isoforms differentially
accelerate cardiac myocyte relaxation kinetics in an animal model
of diastolic dysfunction. Am J Physiol Heart Circ Physiol.
293:H1705–H1713. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
National Institutes of Health, . Guide for
the Care and Use of Laboratory AnimalsNIH publication no. 85_/23.
National Institutes of Health; Rockville, MD: 1996
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang XH, Zhuo XZ, Ni YJ, Gong M, Wang TZ,
Lu Q and Ma AQ: Improvement of cardiac function and reversal of gap
junction remodeling by neuregulin-1β in volume-overloaded rats with
heart failure. J Geriatr Cardiol. 9:172–179. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Niwa M, Miwa Y, Kuzuya T, Iwasaki K,
Haneda M, Ueki T, Katayama A, Hiramitsu T, Goto N, Nagasaka T, et
al: Stimulation index for PCNA mRNA in peripheral blood as immune
function monitoring after renal transplantation. Transplantation.
87:1411–1414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hetz C: BCL-2 protein family. Essential
regulators of cell death. Preface. Adv Exp Med Biol. 687:vii–viii.
2010.PubMed/NCBI
|
|
22
|
Hrynchyshyn N, Jourdain P, Desnos M,
Diebold B and Funck F: Galectin-3: A new biomarker for the
diagnosis, analysis and prognosis of acute and chronic heart
failure. Arch Cardiovasc Dis. 106:541–546. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gaggin HK and Januzzi JL Jr: Biomarkers
and diagnostics in heart failure. Biochim Biophys Acta.
1832:2442–2450. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ho JE, Liu C, Lyass A, Courchesne P,
Pencina MJ, Vasan RS, Larson MG and Levy D: Galectin-3, a marker of
cardiac fibrosis, predicts incident heart failure in the community.
J Am Coll Cardiol. 60:1249–1256. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yoshii T, Fukumori T, Honjo Y, Inohara H,
Kim HR and Raz A: Galectin-3 phosphorylation is required for its
anti-apoptotic function and cell cycle arrest. J Biol Chem.
277:6852–6857. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin HM, Pestell RG, Raz A and Kim HR:
Galectin-3 enhances cyclin D(1) promoter activity through SP1 and a
cAMP-responsive element in human breast epithelial cells. Oncogene.
21:8001–8010. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu L, Ruifrok WP, Meissner M, Bos EM, van
Goor H, Sanjabi B, van der Harst P, Pitt B, Goldstein IJ, Koerts
JA, et al: Genetic and pharmacological inhibition of galectin-3
prevents cardiac remodeling by interfering with myocardial
fibrogenesis. Circ Heart Fail. 6:107–117. 2013. View Article : Google Scholar : PubMed/NCBI
|