Introduction
The cardiovascular system provides blood flow to
tissues, which delivers oxygen and nutrients to maintain normal
metabolism for cells (1).
Restriction of cardiac blood flow results in myocardial ischemia,
which can trigger ischemia/reperfusion (I/R) injury, such as
myocardial infarction and hemorrhage shock (2). The decrease in myocardial oxygen
tension and rise in carbon dioxide tension contribute to the
initiation of cell death by increasing generation of reactive
oxygen species (ROS), disruption of calcium homeostasis, depletion
of glycogen stores, mitochondrial injury and oxidative damage to
DNA (3–5). The apoptosis of cardiomyocytes serves
a vital role in the development of various biological and
pathological processes, including ischemic injuries (6). Therefore, maintenance of normal
myocardial cell function is of great significance for the treatment
of myocardial ischemic injuries.
MicroRNAs (miRs) are a class of small non-coding RNA
(18–25 nucleotides) molecules that regulate the translation of
messenger RNAs (mRNAs) by binding to the 3′-untranslated regions
(3′-UTR) of target mRNAs (7).
Multiple studies have demonstrated that the pathogenic change in
various tissues of humans has been associated with abnormal miR
expression (8,9). Notably, miRs have provided novel
insights into hypoxia-induced myocardial cell dysfunction (10). It has been reported that miR-7a/b
attenuates post-myocardial infarction remodeling and protects H9c2
cardiomyoblast against hypoxia-induced apoptosis, involving
specificity protein 1 and poly (ADP-ribose) polymerase-1 (11). miR-221 is involved in survival of
hypoxia preconditioned ventricular myocytes through the DNA
damage-inducible transcript 4 protein/mechanistic target of
rapamycin complex 1 and tumor protein p53-inducible nuclear protein
1/p62 signaling pathways (12).
miR-21 and miR-146a synergistically decrease apoptosis under
ischemia/hypoxic conditions in cardiomyocytes (13). However, the role of miRNAs in
regulating hypoxia-induced myocardial cell injury has not been
completely clarified and requires further investigation. Previous
studies indicated that miR-150 protects the heart from injury by
inhibiting monocyte accumulation and cell death in a mouse model of
acute myocardial infarction (14,15).
However, the specific role of miR-150 in hypoxia-induced myocardial
cell dysfunction remains unknown.
Glucose-regulated protein-94 (GRP94) is a most
abundant glycoprotein in the endoplasmic reticulum (ER), and is
involved in the maintenance of cell survival by protecting against
stresses due to Ca2+ depletion from the ER (16,17).
A previous study demonstrated that GRP94 is expressed in the
sarcoplasmic reticulum (SR) of adult rabbit cardiomyocytes
(18). GRP94 is reported to
protect cardiomyocytes from both ischemia and calcium overload
(19). However, the underlying
molecular mechanisms of GRP94 in hypoxia-induced myocardial cell
dysfunction have not been previously addressed. The present study
demonstrated that miR-150 could regulate GRP94 expression by
targeting to the 3′-UTR of GRP94, suggesting that a
post-translational mechanism may exist for regulating GRP94
expression by miR-150 in hypoxia-treated cardiomyocytes, which
might provide a potential therapeutic treatment for I/R injury.
Materials and methods
Cell culture treatments
Human cardiomyocytes (HCMs) were commercially
obtained from ScienCell Research Laboratories (Carlsbad, CA, USA)
and were maintained in Dulbecco's modified Eagle's medium (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (cat. no. C0225; Beyotime Institute of
Biotechnology, Haimen, China) at 37°C with 5% CO2, 94%
N2 and 1% O2 for 3, 6, 12, 24 and 48 h.
MTT assay
HCM viability was assessed using an MTT Cell
Proliferation/Viability Assay kit (R&D Systems, Inc.,
Minneapolis, MN, USA) according to the manufacturer's protocol. The
purple formazan was dissolved in dimethyl sulfoxide and the optical
density value was read at 490 nm on a microplate reader (MD
SpectraMax M5; Molecular Devices, LLC, Sunnyvale, CA, USA).
Apoptosis assay by Annexin V/propidium
iodide (PI) and terminal deoxynucleotidyl transferase-mediated dUTP
nick end labeling (TUNEL) staining
HCMs were harvested and washed with PBS. The
percentage of normal (non-apoptotic) and apoptotic cells was
measured by double supravital staining with Annexin-V and PI, using
an Annexin V-FITC Apoptosis Detection kit (Beyotime Institute of
Biotechnology, Haimen, China). Flow cytometric analysis used a
Cytomics FC500 flow cytometer with CXP software (version 5.0;
Beckman Coulter, Inc., Brea, CA, USA). Additionally,
hypoxia-induced apoptosis was assessed by TUNEL staining. The TUNEL
assay was performed using a TUNEL Apoptosis kit (Beyotime Institute
of Biotechnology) according to the manufacturer's protocol.
Measurement of lactate dehydrogenase
(LDH) activity
The LDH activity in the culture medium was detected
using an LDH assay kit (Nanjing Jiancheng Bioengineering Institute,
Nanjing China) for the measurement of cell injury, according to the
manufacturer's protocol.
Caspase-3 activity and cell apoptosis
assay
HCMs lysates were prepared and incubated with an
anti-caspase 3 antibody (cat. no. sc7272; 1:200; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) at 37°C for 1 h.
Immunocomplexes were incubated with peptide substrate in assay
buffer for 2 h at 37°C. Release of p-nitroaniline was measured at
405 nm using an ELISA reader (MD SpectraMax M5) according to the
manufacturer's protocol.
miR target prediction
The potential binding site between miR-124 and MCP-1
were obtained using online predict software (miRanda-mirSVR;
http://www.microrna.org), miRDB (http://www.mirdb.org/miRDB/) and TargetScan
(http://www.targetscan.org/).
Retrovirus package, transduction and
transfection
Control short hairpin (sh)RNA and specific sh-RNAs
targeting GRP94 (cat. no. sc-44304) were purchased from Santa Cruz
Biotechnoogy, Inc. (Dallas, TX, USA), and the corresponding
sequences were cloned into the pSIREN-RetroQ plasmid (Addgene,
Inc.) for retrovirus production with HCMs.
For transfection, the FAM modified
2′-OMe-oligonucleotides were chemically synthesized and purified by
high-performance liquid chromatography (Shanghai GenePharma, Co.,
Ltd., Shanghai, China). The miR-150 mimics were composed of RNA
duplexes with the following sequence: 5′-UCUCCCAACCCUUGUACCAGUG-3′.
The sequences of miR-150 inhibitor and scramble oligonucleotides
were as follows: 5′-CACUGGUACAAGGGUUGGGAGA-3′; and
5′-CCGAAACCUCGGUUGAUUGCGG-3′. Cells were transfected using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) at
a final concentration of 100 nM. At 24 h post-transfection, the
culture medium was changed. After 48 h, cells were harvested for
analysis.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA extraction was performed using TRIzol reagent,
according to the manufacturer's protocol (Invitrogen; Thermo Fisher
Scientific, Inc.). Synthesis of cDNA was performed by RT reactions
with 4 µg total RNA using Moloney murine leukemia virus reverse
transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.) with
oligo dT (15) primers (Fermentas;
Thermo Fisher Scientific, Inc.) as described by the manufacturer.
miR-150 level was quantified by the mirVana qRT-PCR miRNA detection
kit (Ambion; Thermo Fisher Scientific, Inc.) in conjunction with
qPCR with SYBR Green (Bio-Rad Laboratories, Inc., Hercules, CA,
USA; 95°C for 5 min, followed by 35 cycles of 95°C for 15 sec, 60°C
for 30 sec, and 72°C for 30 sec). After circle reaction, the
quantitation cycle (Cq) was determined and relative miR-150 level
was calculated based on the Cq values and normalized to U6 level in
each sample using the 2−∆∆Cq method (20). PCR was performed with the following
primers: GRP94, forward 5′-AGCAAGACGTGTTCGATTC-3′ and reverse
5′-CCTCAATTTTGTCAAGGGTG-3′; GAPDH, forward
5′-GCACCGTCAAGCTGAGAAC-3′ and reverse
5′-TGGTGAAGACGCCAGTGGA-3′.
Western blotting
HCMs were extracted in NP-40 buffer, followed by
5–10 min boiling and centrifugation (12,000 × g for 15 min at 4°C)
to obtain the supernatant. Samples containing 50 µg protein were
separated by 10% SDS-PAGE and transferred to nitrocellulose
membranes (Bio-Rad Laboratories, Inc.). After blocking with 5%
(w/v) non-fat dry milk in TBS and 0.1% (w/v) Tween 20 (TBST), the
membranes were incubated with an anti-GRP94 primary antibody (cat.
no. sc-32249; 1:500; Santa Cruz Biotechnology, Inc.) at room
temperature for 2 h and normalized to β-actin (cat. no. sc-130065;
1:2,000; Santa Cruz Biotechnology, Inc.) at room temperature for 2
h. After three washes with TBST, the membranes were next incubated
with a horseradish peroxidase-conjugated secondary antibody (cat.
no. sc516102; 1:10,000; Santa Cruz Biotechnology, Inc.) at room
temperature for 2 h and subsequently visualized with
chemiluminescence (Thermo Fisher Scientific, Inc.). Signals were
assessed using Quantity One® software (version 4.5; Bio
Rad Laboratories, Inc.).
Statistical analysis
Data are presented mean ± standard error for each
group. All statistical analyses were performed by using PRISM
version 6.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Inter-group differences were analyzed by one-way analysis of
variance followed by Tukey's post hoc test to compare the group
means. P<0.05 was considered to indicate a statistically
significant difference.
Results
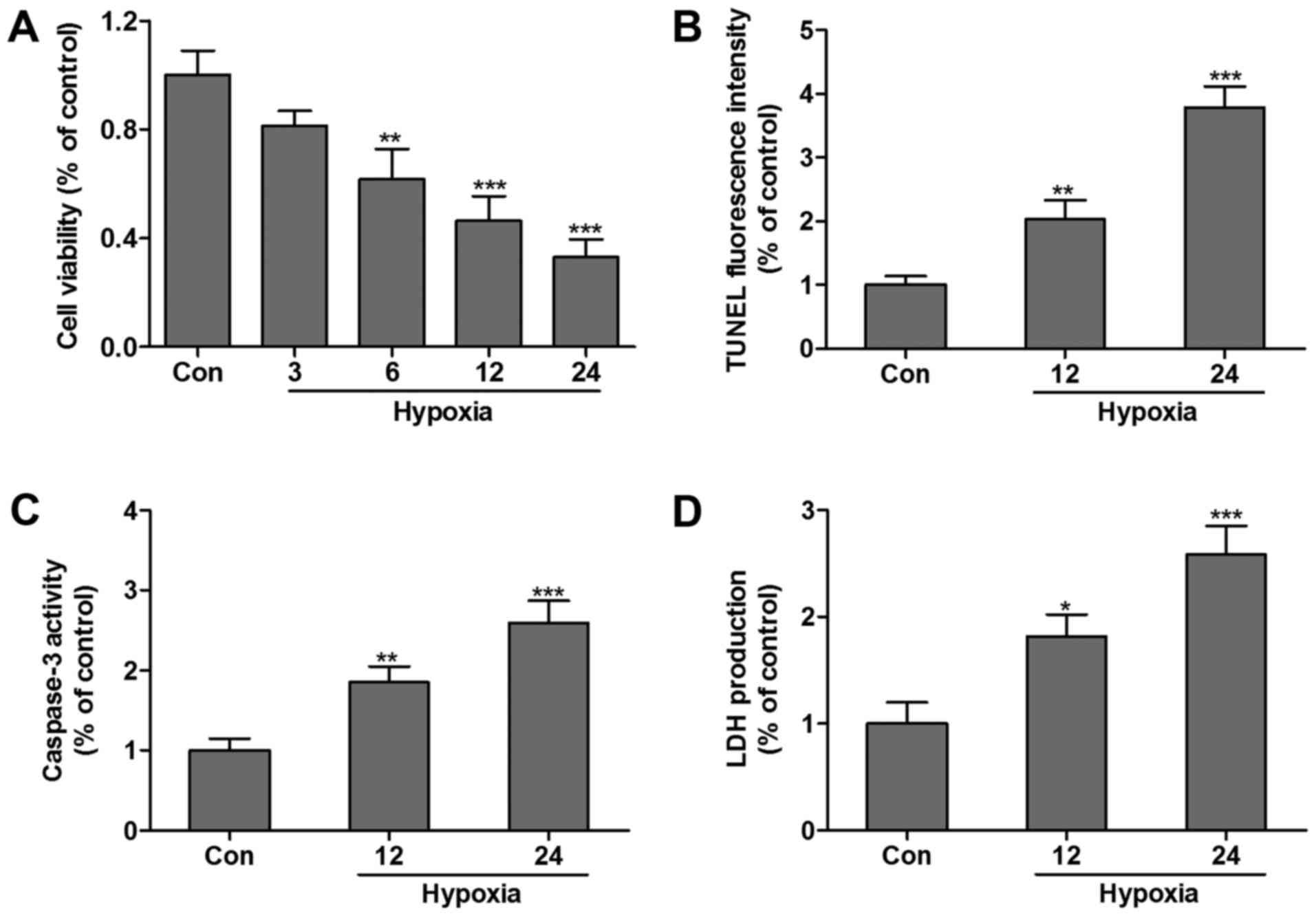
Hypoxia induces HCM death
The effect of hypoxia-induced HCMs apoptosis was
examined by the MTT assay and TUNEL assay. Compared with the
control group, the cell viability was significantly decreased
following exposure to hypoxia in a time-dependent manner (Fig. 1A). TUNEL analysis was performed,
and the results demonstrated that the relative percentage of living
cells after hypoxia treatment for 12 or 24 h markedly decreased
compared with the control group (Fig.
1B). Furthermore, caspase-3 (Fig.
1C) and LDH (Fig. 1D) levels
were increased in HCMs subjected to hypoxia for 12 or 24 h, which
indicated that HCMs suffered hypoxia-associated injuries.
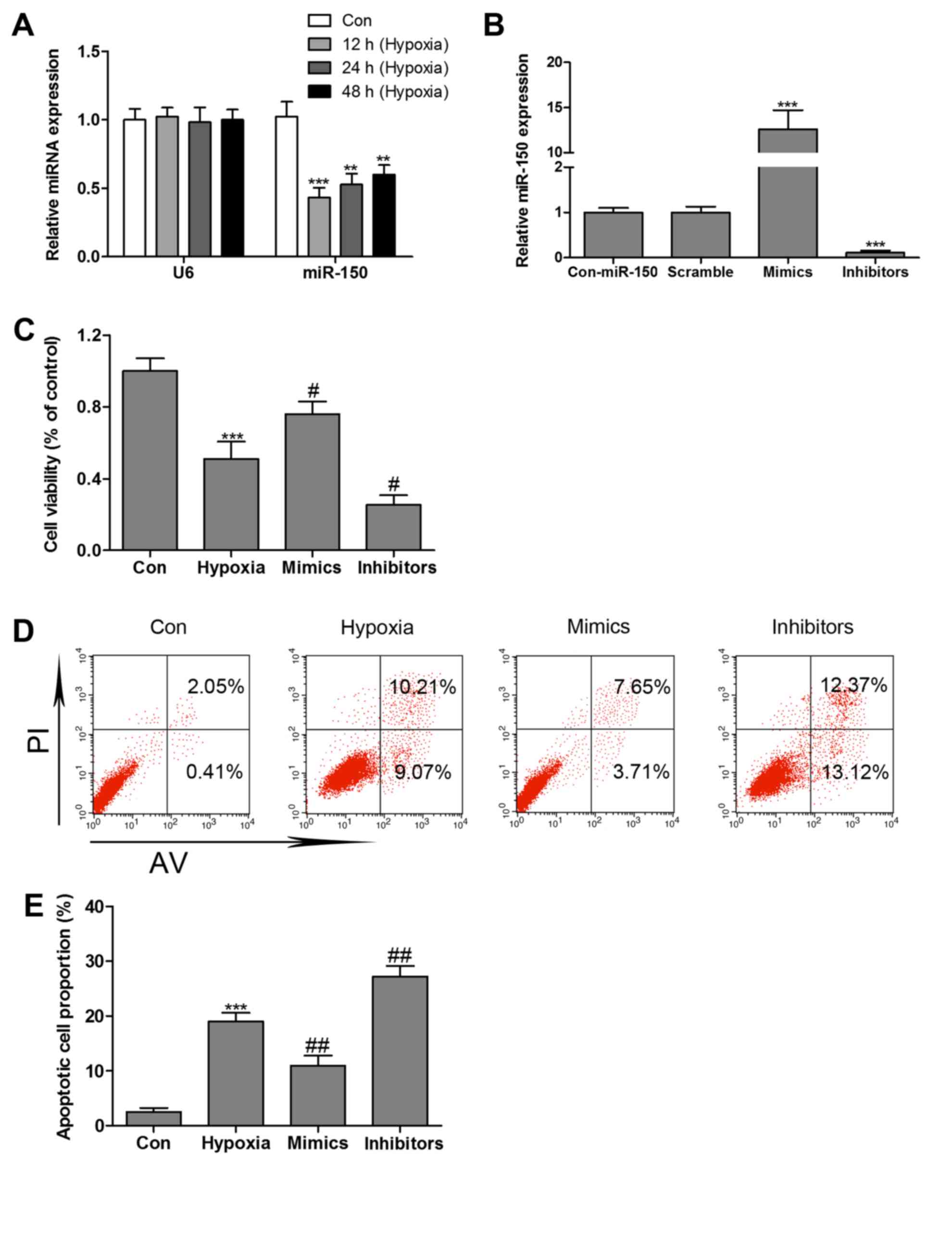
Hypoxia inhibits miR-150 expression in
HCMs
The levels of miR-150 was detected by RT-qPCR assay,
and the results demonstrated that miR-150 expression levels in the
hypoxia-treated group decreased by nearly 56, 48 and 40% at 12, 24
and 48 h, respectively, vs. the control group (Fig. 2A). To validate whether miR-150
mimics or inhibitors stable expression in HCMs, miR-150 mimics or
inhibitors was transfected into HCMs by adenovirus-mediated gene
transfer. RT-qPCR confirmed the elevated level of miR-150 in HCMs
transfected with miR-150 mimics and the reduced level of miR-150 in
HCMs transfected with miR-150 inhibitors (Fig. 2B). As hypoxia treatment was already
observed to markedly inhibit miR-150 expression in HCMs, to
validate whether miR-150 regulates hypoxia-induced HCMs apoptosis,
MTT assay and Annexin V/PI double-staining followed by flow
cytometry analysis were performed. As presented in Fig. 2C, HCMs transfected with miR-150
mimics exhibited a significant increase in cell viability in the
presence of hypoxia. However, miR-150 inhibitors markedly
reinforced hypoxia-induced decrease in cell viability.
Overexpression of miR-150 markedly reversed hypoxia-induced HCMs
apoptosis, whereas miR-150 inhibitors significantly increased cell
apoptosis in the presence of hypoxia (Fig. 2D and E).
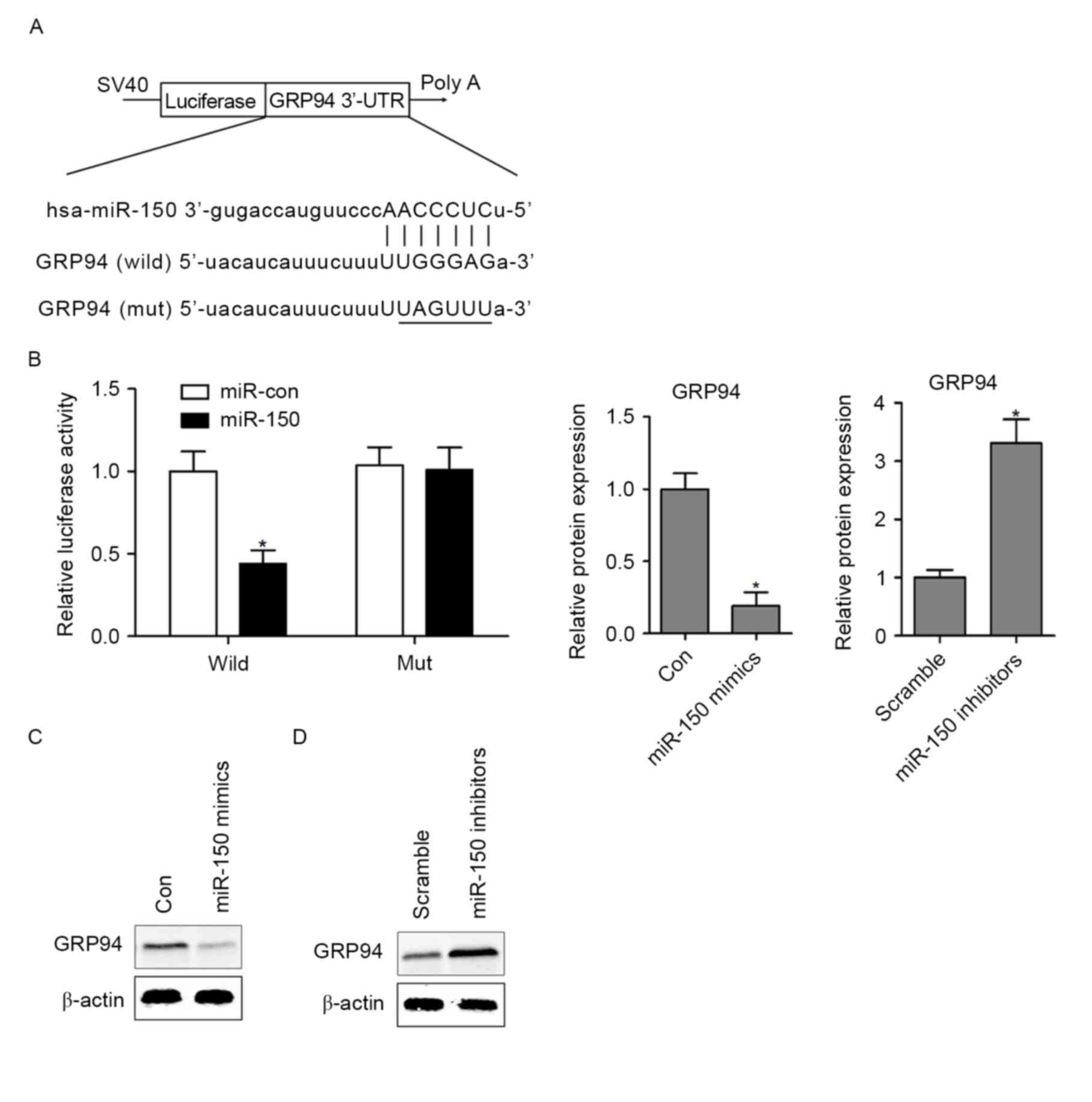
GRP94 is a direct target of
miR-150
To explore the molecular mechanism of miR-150 in
hypoxia-induced HCMs apoptosis, bioinformatics analysis indicated
that GRP94 is a potential target of miR-150 in mammals (Fig. 3A), which serves a critical role in
ER stress-induced cardiomyocyte apoptosis and heart failure
(21). To validate whether miR-150
regulates GRP94 directly through a putative binding site in HCMs,
the GRP94 3′-UTR (wild-type or mutant) in the predicted miRNA
binding site was cloned into the luciferase gene (pSiCHECK2).
Following cotransfection with the pSiCHECK2 vectors and miR-150
mimics or the control miRNA (miR-Con), the findings demonstrated
that overexpressing miR-150 in HCMs significantly decreased
luciferase activity in the wild-type GRP94 3′-UTR. However,
following transfection of miR-150 mimics into GRP94 mutant HCMs,
the luciferase activity did not exhibit a significant difference
compared with the control group (Fig.
3B). To further assess whether miR-150 regulates GRP94
expression in HCMs, the expression of GRP94 was assessed by western
blotting analysis in miR-150 mimic- or miR-150
inhibitor-transfected HCMs. The results indicated that
overexpressing miR-150 markedly decreased the protein expression of
GRP94 in HCMs (Fig. 3C). In
contrast, inhibition of miR-150 in HCMs resulted in upregulation of
GRP94 protein expression (Fig.
3D).
GRP94 is involved in the effect of
miR-150 on hypoxia-induced HCMs apoptosis
The mRNA (Fig. 4A)
and protein (Fig. 4B) expression
levels of GRP94 were significantly increased in hypoxia-injured
HCMs. To further understand the apoptosis mechanisms, sh-GRP94,
miR-150 mimics or miR-150 inhibitors was co-transfected into
hypoxia-injured HCMs. As presented in Fig. 4C, GRP94 knockout increased the cell
viability of hypoxia-impaired HCMs with miR-150 mimics or miR-150
inhibitors transfection. Furthermore, GRP94 knockout could inhibit
apoptosis and caspase-3 levels in hypoxia-impaired HCMs transfected
with miR-150 mimics or miR-150 inhibitors (Fig. 4D and E).
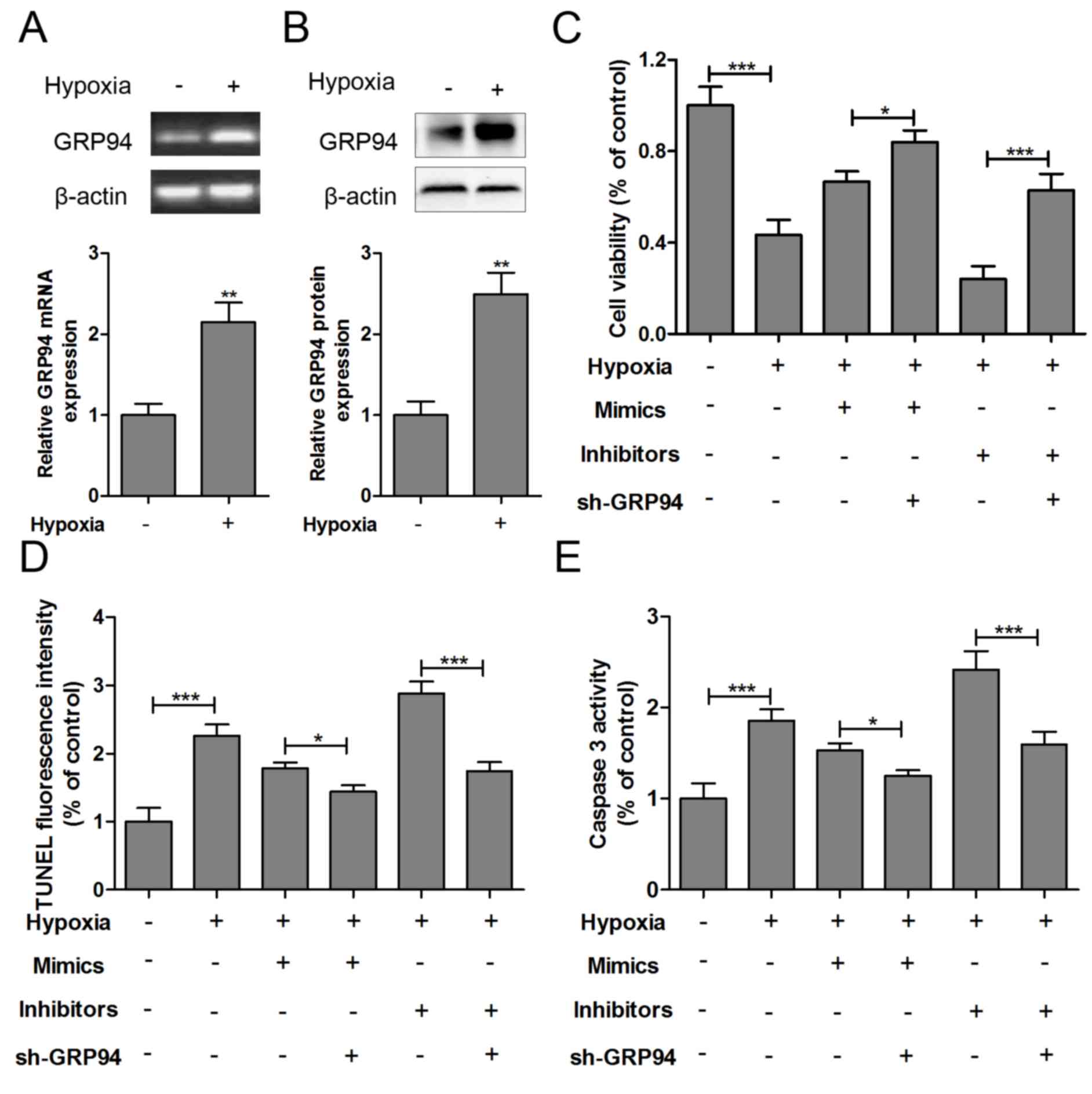 | Figure 4.GRP94 is involved in the effect of
miR-150 on hypoxia-induced HCM apoptosis. The (A) mRNA and (B)
protein expression levels of GRP94 were measured in HCMs exposed to
hypoxia for 24 h. sh-GRP94, miR-150 mimics or miR-150 inhibitors
was co-transfected into hypoxia-injured HCMs, and (C) cell
viability, (D) TUNEL analysis and (E) caspase-3 levels were
measured 12 h after treatment. Data are expressed as the mean ±
standard error (n=3/group). *P<0.05, **P<0.01, ***P<0.001
vs. control group. HCM, human cardiomyocyte; sh, short hairpin;
GRP94, glucose-regulated protein 94; miR, microRNA; TUNEL, terminal
deoxynucleotidyl transferase-mediated dUTP nick end labeling; Con,
control; wild, wild-type; mut, mutant. |
Discussion
The results of the present study demonstrated that
hypoxia directly induces HCM apoptosis, and the expression of
miR-150 was markedly decreased in hypoxia-injured HCMs. Therefore,
it was hypothesized that miR-150 may serve an important role in
hypoxia-induced HCM apoptosis. Notably, further study demonstrated
that overexpressing miR-150 significantly reversed hypoxia-induced
HCM death. In contrast, miR-150 inhibitors markedly reinforced a
hypoxia-induced decrease in cell apoptosis. The molecular mechanism
findings indicated that GRP94 as a direct target of miR-150 was
involved in hypoxia-induced HCM apoptosis. These findings suggested
that downregulation of GRP94 expression by miR-150 or GRP94
knockout protected against hypoxia-induced HCMs apoptosis.
miR-150 is strictly expressed in cardiomyocytes and
cardiac fibroblasts, and serves a pivotal role of cardiomyocyte
survival during cardiac injury (14,22).
Overexpressing miR-150 prevents high glucose-induced cardiomyocyte
hypertrophy by targeting the transcriptional co-activator p300
(23). In contrast, miR-150
aggravates H2O2-induced cardiac myocyte
injury by downregulating the c-myb gene (24). Furthermore, the expression of
miR-150 in cardiac tissues is decreased in myocardial infarction
and cardiac fibrosis induced by pressure overload (25). In addition, adenoviral
overexpression of miR-150 causes a reduction in cardiomyocyte size
(26). However, the potential
roles of miR-150 have not been evaluated in hypoxia-induced HCM
apoptosis. The present study further validated that miR-150
expression is decreased in hypoxia-injured HCMs. miR-150 deficiency
reinforced hypoxia-induced HCM apoptosis. These findings suggested
that downregulation of miR-150 at least partially promotes the
progression of cardiovascular diseases. A previous study verified
that miR-150 expression is inhibited in hypoxia-induced hepatocytes
(27). These research outputs
suggested that miR-150 may be a novel anti-hypoxic miRNA, and
reveal that miR-150 negatively regulates its target gene GRP94.
GRP94 as a molecular chaperone is involved in the
maintenance of cell survival and has been demonstrated to serve
important roles in ER stress-induced myocardial apoptosis (28). A previous study revealed GRP94 as
the only SR calcium-binding protein that is increased during
chronic atrial fibrillation (29).
However, the upstream signals that regulate GRP94 expression in
HCMs remain unclear. The present study demonstrated that miR-150 is
capable of targeting the 3′-UTR of GRP94 to regulate
hypoxia-induced HCMs apoptosis. The results also revealed that
GRP94 knockout suppressed cell apoptosis of hypoxia-impaired HCMs
transfected with miR-150 mimics or miR-150 inhibitors, suggesting
that a post-transcriptional regulatory mechanism exists between
GRP94 and miR-150 in the context of hypoxia-induced HCM
apoptosis.
In conclusion, these results suggested that the
expression of miR-150 in HCMs is downregulated following hypoxia
treatment. Overexpressing miR-150 inhibited hypoxia-induced HCM
apoptosis by downregulating GRP94 expression. These findings might
provide novel insight for the therapy of hypoxia-induced myocardial
I/R injury.
References
|
1
|
Loor G and Schumacker PT: Role of
hypoxia-inducible factor in cell survival during myocardial
ischemia-reperfusion. Cell Death Differ. 15:686–690. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin KH, Kuo WW, Jiang AZ, Pai P, Lin JY,
Chen WK, Day CH, Shen CY, Padma VV and Huang CY:
Tetramethylpyrazine ameliorated hypoxia-induced myocardial cell
apoptosis via HIF-1α/JNK/p38 and IGFBP3/BNIP3 inhibition to
upregulate PI3K/Akt survival signaling. Cell Physiol Biochem.
36:334–344. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang M, Nguyen P, Jia F, Hu S, Gong Y, de
Almeida PE, Wang L, Nag D, Kay MA, Giaccia AJ, et al: Double
knockdown of prolyl hydroxylase and factor-inhibiting
hypoxia-inducible factor with nonviral minicircle gene therapy
enhances stem cell mobilization and angiogenesis after myocardial
infarction. Circulation. 124 11 Suppl:S46–S54. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liang W, Guo J, Li J, Bai C and Dong Y:
Downregulation of miR-122 attenuates hypoxia/reoxygenation
(H/R)-induced myocardial cell apoptosis by upregulating GATA-4.
Biochem Biophys Res Commun. 478:1416–1422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chang W, Song BW, Lim S, Song H, Shim CY,
Cha MJ, Ahn DH, Jung YG, Lee DH, Chung JH, et al: Mesenchymal stem
cells pretreated with delivered Hph-1-Hsp70 protein are protected
from hypoxia-mediated cell death and rescue heart functions from
myocardial injury. Stem Cells. 27:2283–2292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao Y, Xu L, Qiao Z, Gao L, Ding S, Ying
X, Su Y, Lin N, He B and Pu J: YiXin-Shu, a ShengMai-San-based
traditional Chinese medicine formula, attenuates myocardial
ischemia/reperfusion injury by suppressing mitochondrial mediated
apoptosis and upregulating liver-X-receptor α. Sci Rep.
6:230252016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Welten SM, Goossens EA, Quax PH and
Nossent AY: The multifactorial nature of microRNAs in vascular
remodelling. Cardiovasc Res. 110:6–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Anadol E, Schierwagen R, Elfimova N, Tack
K, Schwarze-Zander C, Eischeid H, Noetel A, Boesecke C, Jansen C,
Dold L, et al: Circulating microRNAs as a marker for liver injury
in human immunodeficiency virus patients. Hepatology. 61:46–55.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang WC, Chin TM, Yang H, Nga ME, Lunny
DP, Lim EK, Sun LL, Pang YH, Leow YN, Malusay SR, et al:
Tumour-initiating cell-specific miR-1246 and miR-1290 expression
converge to promote non-small cell lung cancer progression. Nat
Commun. 7:117022016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu Y, Zhu W, Wang Z, Yuan W, Sun Y, Liu H
and Du Z: Combinatorial microRNAs suppress hypoxia-induced
cardiomyocytes apoptosis. Cell Physiol Biochem. 37:921–932. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li R, Geng HH, Xiao J, Qin XT, Wang F,
Xing JH, Xia YF, Mao Y, Liang JW and Ji XP: miR-7a/b attenuates
post-myocardial infarction remodeling and protects H9c2
cardiomyoblast against hypoxia-induced apoptosis involving Sp1 and
PARP-1. Sci Rep. 6:290822016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen Q, Zhou Y, Richards AM and Wang P:
Up-regulation of miRNA-221 inhibits hypoxia/reoxygenation-induced
autophagy through the DDIT4/mTORC1 and Tp53inp1/p62 pathways.
Biochem Biophys Res Commun. 474:168–174. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang W, Tian SS, Hang PZ, Sun C, Guo J
and Du ZM: Combination of microRNA-21 and microRNA-146a attenuates
cardiac dysfunction and apoptosis during acute myocardial
infarction in mice. Mol Ther Nucleic Acids. 5:e2962016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang Y, Wang Y, Park KM, Hu Q, Teoh JP,
Broskova Z, Ranganathan P, Jayakumar C, Li J, Su H, et al:
MicroRNA-150 protects the mouse heart from ischaemic injury by
regulating cell death. Cardiovasc Res. 106:387–397. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu Z, Ye P, Wang S, Wu J, Sun Y, Zhang A,
Ren L, Cheng C, Huang X and Wang K: MicroRNA-150 protects the heart
from injury by inhibiting monocyte accumulation in a mouse model of
acute myocardial infarction. Circ Cardiovasc Genet. 8:11–20. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vitadello M, Ausma J, Borgers M, Gambino
A, Casarotto DC and Gorza L: Increased myocardial GRP94 amounts
during sustained atrial fibrillation: A protective response?
Circulation. 103:2201–2206. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Little E and Lee AS: Generation of a
mammalian cell line deficient in glucose-regulated protein stress
induction through targeted ribozyme driven by a stress-inducible
promoter. J Biol Chem. 270:9526–9534. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vitadello M, Colpo P and Gorza L: Rabbit
cardiac and skeletal myocytes differ in constitutive and inducible
expression of the glucose-regulated protein GRP94. Biochem J.
332:351–359. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vitadello M, Penzo D, Petronilli V,
Michieli G, Gomirato S, Menabò R, Di Lisa F and Gorza L:
Overexpression of the stress protein Grp94 reduces cardiomyocyte
necrosis due to calcium overload and simulated ischemia. FASEB J.
17:923–925. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu X, Kwak D, Lu Z, Xu X, Fassett J, Wang
H, Wei Y, Cavener DR, Hu X, Hall J, et al: Endoplasmic reticulum
stress sensor protein kinase R-like endoplasmic reticulum kinase
(PERK) protects against pressure overload-induced heart failure and
lung remodeling. Hypertension. 64:738–744. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Deng P, Chen L, Liu Z, Ye P, Wang S, Wu J,
Yao Y, Sun Y, Huang X, Ren L, et al: MicroRNA-150 inhibits the
activation of cardiac fibroblasts by regulating c-Myb. Cell Physiol
Biochem. 38:2103–2122. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Duan Y, Zhou B, Su H, Liu Y and Du C:
miR-150 regulates high glucose-induced cardiomyocyte hypertrophy by
targeting the transcriptional co-activator p300. Exp Cell Res.
319:173–184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li X, Kong M, Jiang D, Qian J, Duan Q and
Dong A: MicroRNA-150 aggravates H2O2-induced cardiac myocyte injury
by down-regulating c-myb gene. Acta Biochim Biophys Sin (Shanghai).
45:734–741. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Devaux Y, Vausort M, McCann GP, Zangrando
J, Kelly D, Razvi N, Zhang L, Ng LL, Wagner DR and Squire IB:
MicroRNA-150: A novel marker of left ventricular remodeling after
acute myocardial infarction. Circ Cardiovasc Genet. 6:290–298.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
van Rooij E, Sutherland LB, Liu N,
Williams AH, McAnally J, Gerard RD, Richardson JA and Olson EN: A
signature pattern of stress-responsive microRNAs that can evoke
cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA.
103:pp. 18255–18260. 2006; View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu ZY, Bai YN, Luo LX, Wu H and Zeng Y:
Expression of microRNA-150 targeting vascular endothelial growth
factor-A is downregulated under hypoxia during liver regeneration.
Mol Med Rep. 8:287–293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shi ZY, Liu Y, Dong L, Zhang B, Zhao M,
Liu WX, Zhang X and Yin XH: Cortistatin improves cardiac function
after acute myocardial infarction in rats by suppressing myocardial
apoptosis and endoplasmic reticulum stress. J Cardiovasc Pharmacol
Ther. Apr 18–2016.(Epub ahead of print).
|
|
29
|
Lai LP, Su MJ, Lin JL, Lin FY, Tsai CH,
Chen YS, Huang SK, Tseng YZ and Lien WP: Down-regulation of L-type
calcium channel and sarcoplasmic reticular Ca(2+)-ATPase mRNA in
human atrial fibrillation without significant change in the mRNA of
ryanodine receptor, calsequestrin and phospholamban: An insight
into the mechanism of atrial electrical remodeling. J Am Coll
Cardiol. 33:1231–1237. 1999. View Article : Google Scholar : PubMed/NCBI
|