Introduction
Renal cell carcinoma (RCC) is the most frequent
neoplasm of the adult kidney and accounts for almost 2–3% of all
human malignancies (1). Among the
three major different pathological subtypes of RCC, clear cell RCC
is the predominant cancer type and accounts for almost 85% of all
RCC (2). Currently, surgical
resections are the only potentially curative treatments for
patients with early-stage RCC (3).
However, ~30% of RCC patients have developed distant metastases at
the time of diagnosis, which indicates a relatively poor long-term
prognosis (4). For RCC patients in
late stages, conventional treatments are limited and therapeutic
efficacy is also generally unsatisfactory (5). The tyrosine kinase inhibitors
including sorafenib and sunitinib, which are available for advanced
RCCs, also have limited effect in improving the 5-year survival
rate of patients with RCC (6).
Therefore, investigating the underlying molecular mechanisms
responsible for RCC progression and screening out novel biomarkers
for prognostic prediction of RCC patients are of great importance
for RCC management.
It has been demonstrated that RNA splicing serves a
critical role in eukaryotic gene expression (7). Increasing evidence has demonstrated
that alternations of activities or multiple mutations of
splicing-related elements could be involved in tumorigenesis and
malignant progression (8,9). Ubiquitin specific peptidase 39
(USP39), also known as Sad1p in yeast and a 65 kDa SR-related
protein in human, is implicated in the assembly of the mature
spliceosome complex (10,11). Although USP39 is a member of the
de-ubiquitylation family (12) and
contains a central zinc finger domain and two ubiquitin C-terminal
hydrolase domains, its de-ubiquitinating enzyme activity is
completely deficient (10,13). A previous study demonstrated that
USP39 could maintain the integrity of mitotic spindle checkpoint
and supported cellular cytokinesis through the splicing of Aurora B
and other mRNAs (13).
Additionally, a growing number of researchers have reported the
pivotal role of USP39 in cancer development and progression. Wang
et al (14) identified that
USP39 was significantly upregulated in breast cancer tissues when
compared with normal breast tissues, indicating that USP39 may
serve as a tumorigenic factor in this malignant tumor type.
Upregulation of USP39 has also been identified to be involved in
the tumorigenesis of human hepatocellular carcinoma (HCC) (15), medullary thyroid carcinoma (MTC)
(16) and oral squamous cell
carcinoma (17). A recent study
indicated that increased expression of USP39 promoted the
progression of prostate cancers by enhancing the transcriptional
elongation and maturation of epidermal growth factor receptor
(EGFR) mRNA, and predicted a poor outcome in patients with prostate
cancer (18). However, the
biological functions of USP39 in the development of human RCCs and
its underlying molecular mechanisms remain to be elucidated.
The current study inhibited the expression of USP39
in human RCC cell lines by RNA interference (RNAi) technology and
then assessed the cell growth, cell cycle, apoptosis, invasion and
metastasis capacity of human RCC cell lines. The findings revealed
that silencing of USP39 markedly suppressed RCC cell proliferation
and invasion, and induced cell cycle arrest and apoptosis. In
addition, depletion of USP39 suppressed the activation of Akt and
extracellular signal regulated kinase (ERK) signaling pathway.
Taken together, the data suggest that USP39 may be a promising
prognostic biomarker and potential therapeutic target for human
RCC.
Materials and methods
Cell lines and cell culture
The human RCC cell lines A498 and OSRC-2 were
obtained from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). The A498 cell line was maintained in MEM media
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and the
OSRC-2 cell line was cultured in RPMI-1640 media (Gibco; Thermo
Fisher Scientific, Inc.) at 37°C in 5% CO2. The media
were supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo
Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin.
Cell transfection and gene
silencing
Synthetic small interfering RNAs (siRNAs;
5′-AAGTTGCCTCCATATCTAATC-3′) targeting USP39 and negative control
were purchased from Shanghai Biotend Biotechnology Co., Ltd.
(Shanghai, China). The transfection of siRNAs was performed
according to the manufacturer's protocol. Briefly, RCC cell lines
were incubated in 12-well plate (4.5×105 cells/well) at
37°C for 12 h and then transfected with siRNA using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). At 12 h following transfection, fresh medium was
added to the plate wells and the RCC cells were maintained for
subsequent experiments.
Western blotting
The cultured cells were lysed in Triton lysis buffer
(Pierce; Thermo Fisher Scientific, Inc.) and centrifuged at 12,000
× g for 15 min at 4°C. Protein concentrations were detected by
using the BCA assay kit (Thermo Fisher Scientific, Inc.) according
to manufacturer's protocol. Total protein (40 µg) was separated
using 12% SDS-PAGE and then transferred to polyvinylidene fluoride
membranes. The protein on the membranes was then blocked using 5%
bovine serum albumin solution (cat. no. A7906; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) for 2 h at room temperature. Membranes
were then incubated for 12 h at 4°C with the following specific
primary antibodies: Anti-GAPDH (1:1,000; cat. no. 2118), Akt
(1:1,000; cat. no. 2920), phosphorylated (p)-Akt (1:1,000; cat. no.
4060), ERK1/2 (1:1,000; cat. no. 4695), p-ERK1/2 (1:1,000; cat. no.
4370) and poly-ADP ribose polymerase [PARP; full length (1:1,000;
cat. no. 9532) and cleaved forms (1:1,000; cat. no. 5625)] were
obtained from Cell Signaling Technology Inc. (Danvers, MA, USA).
Anti-cyclin D1 (1:1,000; cat. no. ab16663), cyclin E1 (1:1,000;
cat. no. ab3927), caspase-3 (1:1,000; cat. no. ab13585), caspase-8
(1:1,000; cat. no. ab25901) and caspase-9 (1:1,000; cat. no.
ab32539) were purchased from Abcam (Cambridge, MA, USA). The
following horseradish peroxidase-conjugated secondary antibodies
were purchased from Pierce (Thermo Fisher Scientific, Inc.) and
were then incubated with membranes for 1 h at room temperature:
Goat anti-mouse IgG (H+L; 1:2,000; cat. no. 32230) and goat
anti-rabbit IgG (H+L; 1:2,000; cat. no. 32260). The immunocomplexes
were visualized using a GeneGnome HR scanner (Synoptics Ltd.,
Cambridge, UK) at a wavelength of 420 nm.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using the TRIzol reagent
(Takara Bio, Inc., Otsu, Japan) according to the manufacturer's
instructions. The RT reaction was performed using 2 µg total RNA in
a reaction mixture containing 2 µl oligo dT primers (50 µM), 4 µl
of 5X Moloney-Murine Leukemia Virus buffer (M-MLV), 1 µl dNTPs (10
mM), 0.5 µl RNasin, 0.5 µl M-MLV RT (RNase H-) and nuclease-free
water in a total volume of 20 µl. The reaction mixture was
incubated at 42°C for 30 min, 75°C for 15 min, and then cooled on
ice in accordance with the M-MLV RT protocol. RT reagents were
obtained from Promega Corporation (Madison, WI, USA). mRNA levels
were determined by RT-qPCR using SYBR Premix Ex Taq (Takara Bio,
Inc.) according to the manufacturer's instructions. All experiments
were performed in triplicate, and the mRNA level of GAPDH was used
as an endogenous reference control. qPCR was performed at 94°C for
10 min, followed by 40 cycles of denaturation at 94°C for 15 sec,
annealing at 55°C for 30 sec, extension at 72°C for 30 sec and a
final extension at 72°C for 10 min. The following primers were
used: USP39 forward, 5′-GCCAGCAGAAGAAAAAGAGC-3′ and reverse,
5′-GCCATTGAACTTAGCCAGGA-3′; GAPDH forward,
5′-TGGGCTACACTGAGCACCAG-3′ and reverse, 5′-AAGTGGTCGTTGAGGGCAAT-3′.
Forward and reverse primers were mixed and diluted to 2.5 µM. The
PCR reaction mixture contained 0.8 µl primers, 5 µl cDNA (30
ng/µl), 10 µl 2X SYBR Premix Ex Taq (Takara Bio, Inc.) and 4.2 µl
RNA-free water, in a total volume of 20 µl. The data were analyzed
relative to controls. All assays were performed on an ABI 7300
system (Applied Biosystems; Thermo Fisher Scientific, Inc.). Each
experiment was performed in triplicate, and GAPDH expression was
used for normalization. Fold change relative to mean value was
determined using the 2−ΔΔCq method (19).
Cell proliferation assay
The cell proliferation assay was performed using
Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan) as previously described (20). Briefly, 5,000 cells per well were
seeded in triplicates into 96-well plates and were incubated at
37°C overnight. The cells were subsequently transfected with siRNAs
as aforementioned and maintained in 100 µl fresh cultural medium
following a 48 h incubation at 37°C. Subsequently, each well was
mixed with 10 µl CCK-8 and incubated at 37°C for an additional 1 h,
and the optical density values of each well were detected at an
absorbance of 450 nm using a microplate reader (Synergy HT; BioTek
Instruments, Inc., Winooski, VT, USA).
Cell invasion assay
The in vitro cell invasion assay was
performed using Transwell filter chambers (Costar; Corning
Incorporated, Corning, NY, USA) according to manufacturer's
protocols. Briefly, 1×105 siRNA-transfected cells were
suspended in DMEM containing 0.5% FBS and plated into the upper
Matrigel-coated invasion chambers (BD Biosciences, Franklin Lakes,
NJ, USA). Then, 500 µl culture medium supplemented with 10% FBS was
added to the lower chambers. The transmigrated cells were fixed and
stained with crystal violet at 37°C for 1 h after 24 h invasion.
The invaded cell number was counted and averaged in 6
randomly-selected fields using a Olympus IX73 inverted microscope
(Olympus Corporation, Tokyo, Japan; magnification ×200).
Wound-healing assay
The cells were incubated in 12-well plates to reach
confluence at 37°C. Then, the cells were maintained in culture
medium containing 0.1% FBS for another 24 h and the wound was
scratched with a sterile plastic pipette tip in the center of the
cell monolayer. Following three washes with phosphate buffer saline
(PBS) to remove any floating cells, the cells were maintained in
culture medium supplemented with 0.5% FBS. At the indicated time
points, images of cells were captured using an inverted light
microscope (Olympus Corporation, Tokyo, Japan) and the migration
rates were determined by calculating the distance of wound via
ImageJ software (version 1.49; National Institutes of Health,
Bethesda, MD, USA).
Cell cycle assay
The cells were maintained in culture medium until
the cell confluence reached ~80%. Then the cells were washed with
PBS and fixed in ice-cold 75% ethanol at −20°C for 12 h. The fixed
cells were collected and washed with PBS twice, then stained with
binding solution containing 50 mg/ml propidium iodide (PI; cat. no.
P1304MP; Invitrogen; Thermo Fisher Scientific, Inc.) and 0.5 mg/ml
RNase A for 30 min at room temperature in the dark. Following
incubation, the cells were re-suspended in PBS and the cell cycle
distribution was analyzed by using a FACScan flow cytometer (BD
Biosciences, San Jose, CA, USA) and BD CellQuest Pro Software
(version 5.1; BD Biosciences).
Cell apoptosis assay
Cell apoptosis were determined by using the Annexin
V-FITC Apoptosis Detection kit (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. Annexin
V/PI double staining was analyzed by flow cytometry within 1 h.
Briefly, ~2×105 cells were collected and re-suspended in
300 µl 1X binding buffer containing 5 µl Annexin V and 5 µl PI for
30 min in the dark. The apoptotic rate was quantified and results
were presented as the percentage of apoptotic cells at early stage
(Annexin V-positive and PI-negative) and late stage (Annexin
V/PI-double positive) using BD CellQuest Pro software (version 5.1;
BD Biosciences).
Statistical analysis
All data presented are expressed as mean ± standard
deviation. The Student's t-test was used to determine statistical
significance. Data analysis was performed using SPSS version 16
(SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Silencing of USP39 inhibits cell
proliferation of RCC cells
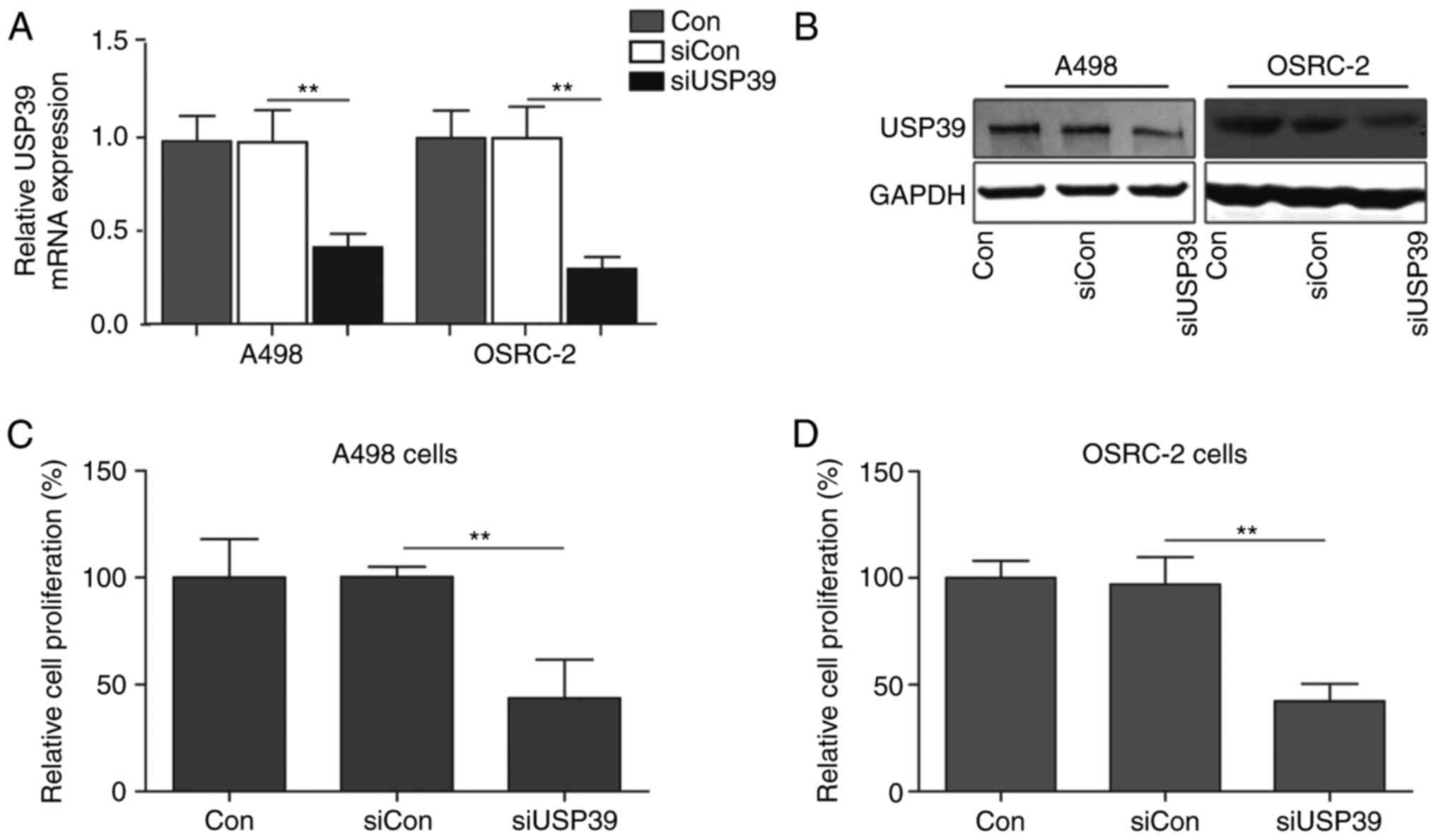
To determine the functional roles of USP39 in RCC,
the expression of USP39 in two RCC cell lines (A498 and OSRC-2) was
inhibited using a siRNA-based knockdown approach. The knockdown
efficiency of USP39 was verified by western blotting and RT-qPCR
(Fig. 1A and B). To determine
whether USP39 siRNA-mediated gene silencing may influence RCC cell
proliferation and CCK-8 assays were performed. A498 cells treated
with siRNA targeting USP39 (siUSP39) exhibited a significantly
reduced cell proliferation when compared with control or cells
treated with scrambled siRNA (siCon) (P<0.01; Fig. 1C). Following incubation for 48 h,
the relative proliferation rate of USP39-silenced A498 cells was
reduced when compared with control and siCon-transfected cells.
Similarly, silencing of USP39 also suppressed cell proliferation
rates of OSRC-2 cells under normal culture conditions (Fig. 1D), confirming that silencing of
USP39 impaired cell proliferative capacity in RCC cells.
Depletion of USP39 suppresses the
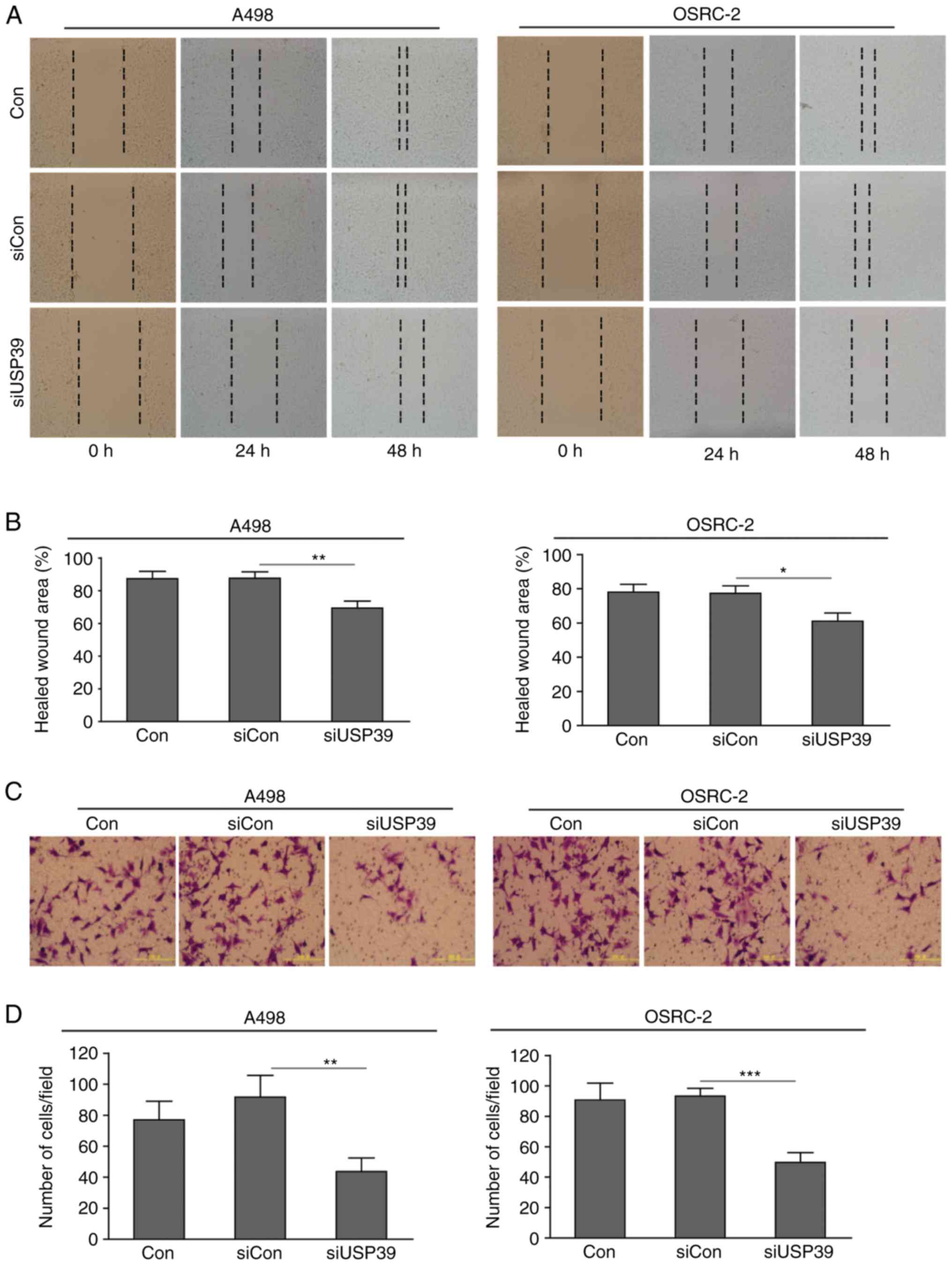
migratory and invasive capacity of RCC cells
It has been demonstrated that RCC cells are
characterized by a marked distant metastatic potential, which
primarily relies on cancer cell invasion and migration (4). Due to the oncogenic role of USP39 in
tumor progression, the present study investigated whether USP39 is
required for RCC cell invasion and migration to occur, using
wound-healing assays to identify cell migration. Depletion of USP39
contributed to markedly reduced cell migration, as revealed by the
smaller healed wound area in A498 and OSRC-2 cells transfected with
siUSP39 at indicated time points (Fig.
2A and B). Matrigel invasion assays were also used to determine
the effect of USP39 on cell invasive ability. As expected,
downregulation of USP39 markedly attenuated cell invasive capacity
when compared with control or siCon-transfected cells (Fig. 2C and D). These data suggested that
knockdown of USP39 inhibited cell migration and invasion in RCC
cells.
Downregulation of USP39 impairs cell
cycle progression at the G2/M phase
In order to identify the underlying mechanism of
USP39-mediated cell proliferation, the cell cycle phases of RCC
cells following USP39 depletion were assessed using flow cytometry.
As presented in Fig. 3A, the cell
population in the G0/G1 phase was markedly reduced from 56.0±2.1%
in the control or 53.7±2.4% in siCon-treated cells to 42.0±1.7% in
siUSP39-transfected cells. By contrast, the cell percentage of
cells in the G2/M phase was increased from 12.3±1.2% in the control
cells to 24.3±2.6% in siUSP39-treated cells. Similarly, it was also
determined that ~66.7±1.5% of cells were at the G0/G1 phase in
siUSP39-transfected OSRC-2 cells, which was markedly lower compared
with control cells (74.2±2.5%) and siCon-treated cells (74.5±2.8%;
Fig. 3B). As aforementioned, an
increase in cell percentage of G2/M-phase was observed in
siUSP39-transfected cells when compared with control cells. In
mammalian cells, the G2/M transition primarily depends on the
activity of the cyclin B1/cyclin-dependent kinase (CDK) 1
complexes. Therefore, the expression levels of the G2/M
phase-related cyclin proteins following USP39 depletion were
determined. Western blotting revealed that silencing of USP39
reduced the expression levels of cyclin B1, D1 and E1 in RCC cells
(Fig. 3C). The findings suggested
that knockdown of USP39 induced G2/M phase arrest in RCC cells,
implying that the proliferation suppression may be associated with
impaired cell cycle progression.
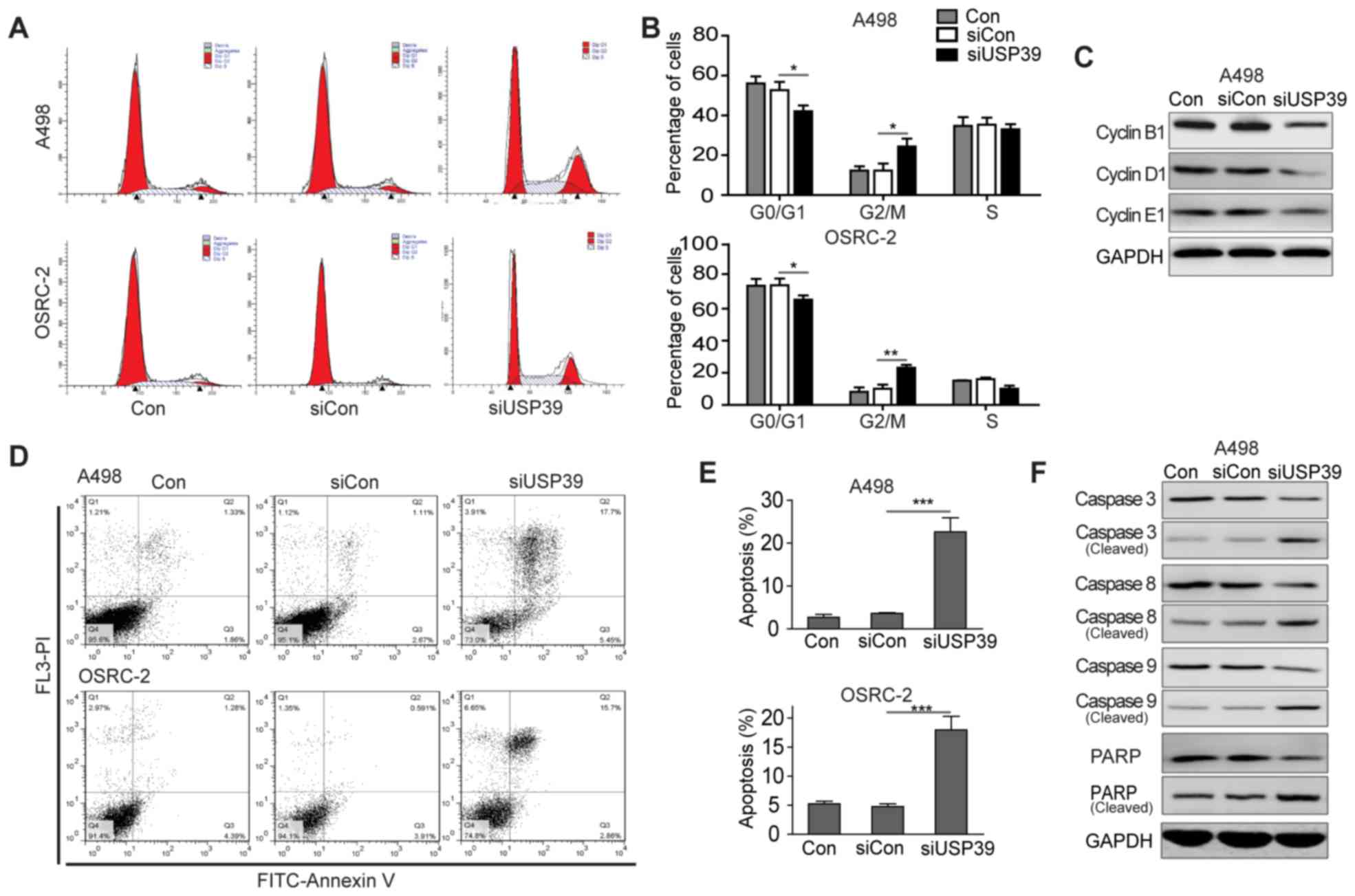 | Figure 3.Downregulation of USP39 impaired cell
cycle progression at G2/M phase and induced apoptosis in RCC cells.
(A) Cell cycle distribution of RCC cells transfected with siCon or
siUSP39 was analyzed by flow cytometric assay with PI staining.
Representative fluorescence-activated cell sorting histograms of
RCC cell cycle are presented. (B) Quantification of the percentage
of RCC cells at different cell cycle phases (G0/G1, S and G2/M;
*P<0.05, **P<0.01). Experiments were performed in triplicate
and data are presented as the mean ± standard deviation. (C) The
expression levels of cyclin B1, cyclin D1 and cyclin E1 proteins in
RCC cells following transfection with siCon or siUSP39 was
determined by western blot analysis. GAPDH protein was used as an
internal control. (D) Cell apoptosis assay of RCC cells transfected
with siCon or siUSP39 was analyzed by flow cytometric assay with
Annexin V/PI staining. Representative images of RCC cells are
shown. (E) The percentage of apoptotic cells in RCC cells following
transfection with siCon or siUSP39 was assessed (**P<0.01,
***P<0.001). Experiments were performed in triplicate and data
are presented as the mean ± standard deviation. (F) The expression
levels of common apoptosis-related proteins (caspase-3, 8, 9, PARP
and their cleaved forms) in RCC cells following transfection with
siCon or siUSP39 was detected by western blot analysis. GAPDH
protein was used as an internal control. USP39, ubiquitin specific
peptidase 39; si, small interfering; RCC, renal cell carcinomas;
Con, control; PI, propidium iodide; FASC, PARP, poly ADP ribose
polymerase. |
Knockdown of USP39 induces apoptosis
in RCC cells
To further investigate the effect of USP39 on cell
apoptosis, Annexin V/PI double staining was performed on RCC cells
transfected with siCon or siUSP39. According to the Annexin V/PI
plots from gated cells, the percentages of early apoptotic cells
(Annexin V+/PI−) and late apoptotic cells
(Annexin V+/PI+) were determined. Silencing
of USP39 markedly increased the populations of apoptotic cells
(early and late apoptosis), when compared with the control group
(Fig. 3D and E). To support these
findings, the expression levels of several apoptosis-associated
proteins, such as caspase-3, −8, −9, and PARP, were subsequently
detected by western blotting in the RCC cells following USP39
silencing. The cells transfected with siUSP39 exhibited increased
expression levels of cleaved caspase-3, −8, and −9, and PARP, which
represent the active forms (Fig.
3F). Together, the data indicated that depletion of USP39 could
induce cell apoptosis and alter the expression profiles of
pro-apoptotic proteins in RCCs.
Knockdown of USP39 blocked the Akt and
ERK signaling pathways in RCC cells
The underlying mechanism by which USP39 promotes
cell growth and cancer progression was investigated. Due to the
essential role of extracellular ERK and Akt signaling pathways in
the maintenance of malignant cell survival and proliferation
(21), the present study
investigated whether USP39 silencing may inhibit the activation of
these two signaling pathways. Therefore, the phosphorylated forms
of Akt and ERK in RCC cells transfected with siCon or siUSP39 were
analyzed using western blotting. Knockdown of USP39 contributed to
marked inhibition of Akt phosphorylation at the Ser473 site and ERK
phosphorylation at the Thr202/Tyr204 when compared with the siCon
transfection groups (Fig. 4).
These findings suggested that expression of USP39 may promote
cancer progression by activating the Akt and ERK signaling
axis.
Discussion
At present, molecular-targeted therapies are a
promising option for patients with unresectable RCC (22). Previous studies in molecular
biology have contributed to an increased understanding of the
underlying molecular mechanisms of RCC tumorigenesis (1,23).
Novel therapeutic approaches against specific targets in RCC have
demonstrated promising clinical activity in RCC patients (24). To the best of our knowledge, the
present study provides novel evidence that USP39, a spliceosome
factor, may have a critical role in RCC progression and
metastasis.
It has been previously established that USP39 is an
essential component of the spliceosome, which directly participates
in the pre-mRNA splicing of several oncogenes including Aurora B
and RB1 (13). This indicates that
USP39 may have a growth-promoting function by controlling the
process of mRNA splicing. In line with previous observations, the
expression level of USP39 has been associated with cell
proliferation in multiple malignancies (14–17).
The present study demonstrated for the first time, to the best of
the authors' knowledge, that silencing of USP39 via siRNA
significantly suppressed RCC cell proliferation in
vitro.
Previous studies have demonstrated that inhibition
of USP39 via siRNA may block the cell cycle distribution of human
MTC and HCC cell lines (15,16)
and this conclusion was further supported by the findings of the
present study. The whole cell cycle is divided into three periods:
Interphase (including G1, S and G2 phases), the mitotic (M) phase
and cytokinesis. As USP39 has been identified to be involved in the
maintenance of spindle checkpoint and cytokinesis, the present
study investigated the functional role of USP39 in the mitotic (M)
phase during cell division. Flow cytometric analysis revealed that
the cell cycle of RCC cells was impeded at the G2/M phase following
silencing of USP39, which was in accord with a previous study on
USP39 in prostate cancer cells (18). In cells with a nucleus, the cell
cycle process is regulated by cyclin proteins that may directly
activate CDKs (25). Among these
family members, cyclin B1 has been identified to partner with CDK1
and form the CDK1/cyclin B kinase complex, which facilitates the
entrance into mitosis, thus promoting the G2/M phase transition
(26). Therefore, a USP39-mediated
G2/M phase arrest of RCC was also identified to be accompanied with
the downregulation of cyclin B1 expression in the present study. It
was also observed that depletion of USP39 decreased the cell
proportions of RCC cells in the G1 and S phases. Unlike cyclin B1,
cyclin D1 and cyclin E1 are pivotal regulatory subunits of CDK2/4/6
and are able to interact with these kinases to promote the M/G1 and
G1/S phase transitions (27,28).
The expression levels of cyclin D1 and cyclin E1 were reduced in
siUSP39-transfected RCC cells, confirming the reduced cell
percentages of USP39-silenced RCC cells observed in the G1 and S
phases. Therefore, it is possible to that depletion of USP39 may
suppress RCC cell proliferation by inducing cell cycle arrest at
G2/M phase. The present study determined that knockdown of USP39
had a significant pro-apoptotic effect in human RCC, which may
partly account for the inhibition of RCC cell proliferation. A
previous study demonstrated that cell apoptosis is a highly
regulated and controlled biological process (29). Intrinsic and extrinsic pathways may
initiate cell apoptosis by activating multiple apoptosis-associated
proteins, including caspase families. Once these proteases or
enzymes are activated, the intracellular components are degraded
and cells were programmed to die in a controlled manner (30). PARP is another important protein
that has the ability to induce programmed cell death. As PARP may
be inactivated by caspase cleavage, the cleaved form of PARP is
also considered to be the biomarker of apoptosis (31). In the present study, the expression
levels of cleaved-caspase-3 −8 and −9, and PARP were elevated in
RCC cells following depletion of USP39, confirming the increased
cell apoptotic rates in USP39-silencing RCC cells.
In conclusion, the possible mechanisms by which
USP39 regulates a series of biological processes was investigated.
Huang et al (18)
identified EGFR to be a downstream target of USP39, whereas
knockdown of USP39 suppressed the transcriptional elongation and
maturation of EGFR mRNA. In addition, highly conserved signaling
pathways including mitogen-activated protein kinase/ERK and
PI3K/Akt axes are of great importance in internalizing the effects
of external growth factors and of membrane tyrosine kinases, and
have a key role in multiple cellular processes including cell
division, apoptosis and mRNA transcription (32,33).
The present study determined that silencing of USP39 impaired the
activation of the ERK and Akt pathways, indicating the relevance
between USP39 expression and the activities of the ERK and Akt
pathways. Therefore, specific molecular inhibitors targeting these
two pathways may be effective therapeutic treatments for RCC
patients with high expression levels of USP39.
Glossary
Abbreviations
Abbreviations:
|
USP39
|
ubiquitin specific peptidase 39
|
|
RCC
|
renal cell carcinomas
|
|
MTC
|
medullary thyroid carcinoma
|
|
HCC
|
human hepatocellular carcinoma
|
|
RNAi
|
RNA interference
|
|
FBS
|
fetal bovine serum
|
|
CDK
|
cyclin-dependent kinase
|
References
|
1
|
Rini BI, Campbell SC and Escudier B: Renal
cell carcinoma. Lancet. 373:1119–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Leibovich BC, Lohse CM, Crispen PL,
Boorjian SA, Thompson RH, Blute ML and Cheville JC: Histological
subtype is an independent predictor of outcome for patients with
renal cell carcinoma. J Urol. 183:1309–1315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tyritzis SI, Papadoukakis S, Katafigiotis
I, Adamakis I, Anastasiou I, Stravodimos KG, Alamanis C,
Mitropoulos D and Constantinides CA: Implementation and external
validation of Preoperative Aspects and Dimensions Used for an
Anatomical (PADUA) score for predicting complications in 74
consecutive partial nephrectomies. BJU Int. 109:1813–1818. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ljungberg B, Campbell SC, Choi HY, Jacqmin
D, Lee JE, Weikert S and Kiemeney LA: The epidemiology of renal
cell carcinoma. Eur Urol. 60:615–621. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cohen DD, Matin SF, Steinberg JR, Zagone R
and Wood CG: Evaluation of the intact specimen after laparoscopic
radical nephrectomy for clinically localized renal cell carcinoma
identifies a subset of patients at increased risk for recurrence. J
Urol. 173:1487–1491. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Escudier B, Albiges L and Sonpavde G:
Optimal management of metastatic renal cell carcinoma: Current
status. Drugs. 73:427–438. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Valadkhan S: The spliceosome: Caught in a
web of shifting interactions. Curr Opin Struct Biol. 17:310–315.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu S and Cheng C: Alternative RNA
splicing and cancer. Wiley interdisciplinary reviews RNA.
4:547–566. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Srebrow A and Kornblihtt AR: The
connection between splicing and cancer. J Cell Sci. 119:2635–2641.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lygerou Z, Christophides G and Séraphin B:
A novel genetic screen for snRNP assembly factors in yeast
identifies a conserved protein, Sad1p, also required for pre-mRNA
splicing. Mol Cell Biol. 19:2008–2020. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Makarova OV, Makarov EM and Lührmann R:
The 65 and 110 kDa SR-related proteins of the U4/U6.U5 tri-snRNP
are essential for the assembly of mature spliceosomes. EMBO J.
20:2553–2563. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Clague MJ, Barsukov I, Coulson JM, Liu H,
Rigden DJ and Urbé S: Deubiquitylases from genes to organism.
Physiol Rev. 93:1289–1315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
van Leuken RJ, Luna-Vargas MP, Sixma TK,
Wolthuis RM and Medema RH: Usp39 is essential for mitotic spindle
checkpoint integrity and controls mRNA-levels of aurora B. Cell
Cycle. 7:2710–2719. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang H, Ji X, Liu X, Yao R, Chi J, Liu S,
Wang Y, Cao W and Zhou Q: Lentivirus-mediated inhibition of USP39
suppresses the growth of breast cancer cells in vitro. Oncol Rep.
30:2871–2877. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yuan X, Sun X, Shi X, Jiang C, Yu D, Zhang
W, Guan W, Zhou J, Wu Y, Qiu Y and Ding Y: USP39 promotes the
growth of human hepatocellular carcinoma in vitro and in vivo.
Oncol Rep. 34:823–832. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
An Y, Yang S, Guo K, Ma B and Wang Y:
Reduced USP39 expression inhibits malignant proliferation of
medullary thyroid carcinoma in vitro. World J Surg Oncol.
13:2552015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li KY, Zhang J, Jiang LC, Zhang B, Xia CP,
Xu K, Chen HY, Yang QZ, Liu SW and Zhu H: Knockdown of USP39 by
lentivirus-mediated RNA interference suppresses the growth of oral
squamous cell carcinoma. Cancer Biomark. 16:137–144. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang Y, Pan XW, Li L, Chen L, Liu X, Lu
JL, Zhu XM, Huang H, Yang QW, Ye JQ, et al: Overexpression of USP39
predicts poor prognosis and promotes tumorigenesis of prostate
cancer via promoting EGFR mRNA maturation and transcription
elongation. Oncotarget. 7:22016–22030. 2016.PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Method. 25:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Zhang JW, Zhang SS, Song JR, Sun K, Zong
C, Zhao QD, Liu WT, Li R, Wu MC and Wei LX: Autophagy inhibition
switches low-dose camptothecin-induced premature senescence to
apoptosis in human colorectal cancer cells. Biochem Pharmacol.
90:265–275. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yajima I, Kumasaka MY, Thang ND, Goto Y,
Takeda K, Yamanoshita O, Iida M, Ohgami N, Tamura H, Kawamoto Y and
Kato M: RAS/RAF/MEK/ERK and PI3K/PTEN/Akt signaling in malignant
melanoma progression and therapy. Dermatol Res Pract.
2012:3541912012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vakkalanka BK and Rini BI: Targeted
therapy in renal cell carcinoma. Curr Opin Urol. 18:481–487. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Keefe SM, Nathanson KL and Rathmell WK:
The molecular biology of renal cell carcinoma. Semin Oncol.
40:421–428. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mellado B and Gascón P: Molecular biology
of renal cell carcinoma. Clin Transl Oncol. 8:706–710. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nigg EA: Cyclin-dependent protein kinases:
Key regulators of the eukaryotic cell cycle. BioEssays. 17:471–480.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sartor H, Ehlert F, Grzeschik KH, Müller R
and Adolph S: Assignment of two human cell cycle genes, CDC25C and
CCNB1, to 5q31 and 5q12, respectively. Genomics. 13:911–912. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baldin V, Lukas J, Marcote MJ, Pagano M
and Draetta G: Cyclin D1 is a nuclear protein required for cell
cycle progression in G1. Genes Dev. 7:812–821. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hwang HC and Clurman BE: Cyclin E in
normal and neoplastic cell cycles. Oncogene. 24:2776–2786. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
O'Rourke MG and Ellem KA: John Kerr and
apoptosis. Med J Aust. 173:616–617. 2000.PubMed/NCBI
|
|
30
|
Rathore S, Datta G, Kaur I, Malhotra P and
Mohmmed A: Disruption of cellular homeostasis induces organelle
stress and triggers apoptosis like cell-death pathways in malaria
parasite. Cell Death Dis. 6:e18032015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Agarwal A, Mahfouz RZ, Sharma RK, Sarkar
O, Mangrola D and Mathur PP: Potential biological role of poly
(ADP-ribose) polymerase (PARP) in male gametes. Reprod Biol
Endocrinol. 7:1432009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rao VN and Reddy ES: elk-1 proteins
interact with MAP kinases. Oncogene. 9:1855–1860. 1994.PubMed/NCBI
|
|
33
|
Freeman-Cook KD, Autry C, Borzillo G,
Gordon D, Barbacci-Tobin E, Bernardo V, Briere D, Clark T, Corbett
M, Jakubczak J, et al: Design of selective, ATP-competitive
inhibitors of Akt. J Med Chem. 53:4615–4622. 2010. View Article : Google Scholar : PubMed/NCBI
|