Introduction
Laryngeal squamous cell carcinoma (LSCC), which
accounts for ~14% of head and neck squamous cell carcinomas (SCCs)
(1), is one of the most common
carcinomas of the head and neck (1). Key features of LSCC include rapid
progression, aggressive behavior, resistance to chemotherapy and
poor prognosis (2,3). Patients with advanced LSCC are
typically treated with a combination of surgery, chemotherapy and
radiation therapy (4). Despite
advances in treatment, the survival of patients with LSCC remains
low, primarily due to its resistance to anticancer drugs (2,3).
Therefore, it is imperative to identify novel methods to improve
therapeutic efficacy for this disease.
Overexpression of epidermal growth factor receptor
(EGFR) has been associated with tumor progression and resistance to
chemotherapy and radiotherapy (5).
EGFR and its ligands are overexpressed in head and neck SCCs
including LSCC (5). Cetuximab, an
anti-EGFR monoclonal humanized antibody, has been proved clinically
effective in patients with recurrent or metastatic head and neck
SCCs in combination with cisplatin-based chemotherapy (5,6). A
recent in vitro study has demonstrated that cetuximab
increases the therapeutic effect of cisplatin in LSCC cells
(5).
As the most commonly used chemotherapeutic agent for
the treatment of solid tumors, cisplatin has been proven effective
for treatment of LSCC (7).
However, the efficacy of cisplatin diminishes when LSCC develops
resistance to it during the treatment process (7). It has been demonstrated that
cisplatin has effects on multiple cellular targets in tumor cells
in addition to nuclear DNA (8–10).
Previous studies have revealed that cisplatin induces endoplasmic
reticulum (ER) stress (11–13).
ER stress-associated apoptosis is hypothesized to be a major
cisplatin-induced pathway, which contributes to cisplatin
cytotoxicity and is also involved in cisplatin resistance (14). Recent studies have suggested that
EGFR signaling is also involved in ER stress-associated apoptosis
(15,16). Miao et al (15) demonstrated that EGFR signaling
protected cardiomyocytes from ER stress-associated apoptosis.
However, Hong et al (16)
demonstrated that inhibiting EGFR signaling triggered ER stress,
which resulted in ER-mediated cell death.
ER is an essential subcellular compartment
responsible for the synthesis and folding of proteins (17). Various physiological and
pathological conditions may lead to ER stress, which results in an
accumulation of unfolded or misfolded proteins in the ER lumen
(18,19). This cellular stress subsequently
causes an activation of the unfolded protein response, which
induces the expression of chaperones and proteins involved in the
recovery process (20). Moderate
ER stress can be resolved and ER homeostasis restored to maintain
cell survival, whereas severe and prolonged ER stress may induce
cell apoptosis by activating downstream apoptotic signaling
pathways (20). A decisive factor
in this process is CCAAT/enhancer-binding protein homologous
protein (CHOP), also known as growth arrest and DNA damage
inducible gene (GADD153) (21).
CHOP exhibits pro-apoptotic activity and is critical for triggering
apoptosis in response to ER stress (22). Increased expression of CHOP
activates caspases, integrates mitochondrial events and amplifies
the death signal (21).
Thioredoxin domain-containing protein 5 (TXNDC5), a
member of the disulfide isomerase family, is primarily expressed in
the ER (23). It has been
demonstrated that TXNDC5 protects cells from ER stress-induced
apoptosis (24,25) by facilitating proteins to fold
correctly via formation of disulfide bonds through its thioredoxin
domains (26).
The present study for the first time, to the best of
the authors' knowledge, investigated the interaction among
cisplatin, cetuximab and TXNDC5 in ER stress-associated apoptosis
in LSCC cells.
Materials and methods
Cell culture and treatments
The AMC-HN-8 human LSCC cell line was purchased from
the Shanghai Institute of the Chinese Academy of Science (Shanghai,
China), and cultured in RPMI 1640 media supplemented with 10% fetal
bovine serum and 100 µM penicillin and streptomycin (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) in a humidified atmosphere
containing 5% CO2 at 37°C. The cells were treated with
cisplatin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at 5, 10,
20 and 40 µM and/or cetuximab (Merck KGaA) at 10, 50, 100 and 150
µg/ml for 12, 24, 36 and 48 h, in the presence or absence of 10 mM
reactive oxygen species (ROS) scavenger/antagonist N-acetylcysteine
(NAC; Sigma-Aldrich; Merck KGaA). For TXNDC5 knockdown, AMC-HN-8
cells were transduced with human ERp46/TXNDC5 lentiviral particles
(sc-60601-V; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), with
cells transduced with control short hairpin RNA (shRNA) lentiviral
particles (sc-108080; Santa Cruz Biotechnology, Inc.) as a control.
For TXNDC5 overexpression, human full length TXNDC5 cDNA clone
(SC109657) was purchased from Origene Technologies, Inc. (Beijing,
China) and subcloned into pcDNA 3.1 expression vector (V79020;
Thermo Fisher Scientific, Inc.); then AMC-HN-8 cells were
transfected with the TXNDC5-expressing vector using Lipofectamine
3000 transfection reagent (L3000008; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. Empty expression
vector was used as the control. The cells were subject to
subsequent experiments 24 h following
transduction/transfection.
Real-time reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA was prepared from AMC-HN-8 cells using TRIzol
reagent (Thermo Fisher Scientific, Inc.) followed by purification
with TURBO DNA-free System (Ambion, Austin, TX, USA). cDNA was
synthesized using SuperScript II reverse transcriptase (Thermo
Fisher Scientific, Inc.) and random hexamer primers (Thermo Fisher
Scientific, Inc.). RT-qPCR was performed using an ABI-PRISM 7700
Sequence Detection System (Applied Biosystems; Thermo Fisher
Scientific, Inc.) and the fluorescent dye SYBR Green Master Mix
(Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. The primers used were as follows: Forward,
5′-GGGTCAAGATCGCCGAAGTA-3′ and reverse, 5′GCCTCCACTGTGCTCACTGA3′
for TXNDC5; and forward, 5′CCCTGTAATTGGAATGAGTCCAC3′ and reverse,
5′GCTGGAATTACCGCGGCT3′ for 18S rRNA. The PCR amplification
condition was: Initial denaturation for 20 sec at 95°C, 40 cycles
of denaturation for 3 sec at 95°C, annealing for 30 sec at 60°C.
Relative quantification of the TXNDC5 expression level was
determined using the 2−ΔΔCq method (27) and normalized against that of 18S
rRNA in the same sample. Each experiment was repeated three
independent times in duplicate.
Western blot analysis
Whole cell lysates were extracted by incubating
AMC-HN-8 cells with lysis buffer [50 mM Tris-HCl (pH 7.2), 150 mM
NaCl, l% (v/v) Triton X-100, 1 mM sodium orthovanadate, 50 mM
sodium pyrophosphate, 100 mM sodium fluoride, 0.01% (v/v)
aprotinin, 4 µg/ml pepstatin A, 10 µg/ml leupeptin and 1 mM
phenylmethanesulfonyl fluoride; all Sigma-Aldrich; Merck KGaA] on
ice for 30 min and removing cell debris by centrifugation at 2,000
× g for 15 min at 4°C. For the detection of nuclear receptor
subfamily 4 group A member 1 (NR4A1), nuclear extracts were
prepared as previously described (28). Equal amount of proteins (7 µg) for
each sample were separated by 10% SDS-PAGE and blotted onto a
polyvinylidene difluoride microporous membrane (EMD Millipore,
Billerica, MA, USA). The membranes were blocked with 5% skimmed
milk powder in TBS containing 0.1% Tween-20 (SRE0031;
Sigma-Aldrich; Merck KGaA) for 2 h at room temperature, and
incubated for 1 h at room temperature with a 1:1,000 dilution of
goat anti-human ERp46/TXNDC5 polyclonal antibody (sc-49660), mouse
anti-mouse GADD153/CHOP monoclonal antibody (sc-7351), rabbit
anti-human cleaved caspase-3 p11 polyclonal antibody (sc-22171-R),
mouse anti-human β-actin monoclonal antibody (sc-130301), mouse
anti-human NR4A1/Nur77 monoclonal antibody (sc-365113), or goat
anti-human histone H3 polyclonal antibody (sc-8654; all Santa Cruz
Biotechnology, Inc.). Membranes were subsequently washed in TBST
(Sigma-Aldrich; Merck KGaA) for 5 min three times, and probed using
bovine anti-goat (sc-2378), bovine anti-mouse (sc-2371) or bovine
anti-rabbit (sc-2370; all Santa Cruz Biotechnology, Inc.)
horseradish peroxidase-conjugated secondary antibodies at a 1:5,000
dilution for 1 h at room temperature. Protein bands were visualized
by enhanced chemiluminescence (GE Healthcare Life Sciences, Little
Chalfont, UK) using the ChemiDoc Touch Imaging system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and Image Lab Touch software
version 4.1 (Bio-Rad Laboratories, Inc.) for image acquisition and
densitometric analysis. Three independent experiments were
performed.
Intracellular ROS detection
ROS were measured using the
Dichlorodihydrofluorescein Diacetate (DCFDA) Cellular Reactive
Oxygen Species Detection Assay kit (ab113851; Abcam, Cambridge,
UK), according to the manufacturer's protocol. Cells were plated at
7×104 cells/well in black 96-well plates and incubated
with 25 µM DCFDA for 45 min at 37°C. Fluorescence was detected
using a Victor3 1420 Multilabel Counter (PerkinElmer, Inc.,
Shanghai, China).
Caspase-3 activity assay
The activity of caspase-3 was determined using a
colorimetric Caspase-3 Assay kit (ab39401; Abcam, Cambridge, MA,
USA). The assays were performed in 96-well plates by incubating 20
µl cell lysate protein/sample in 70 µl reaction buffer [1% NP-40,
20 mM Tris-HCl (pH 7.5), 137 mM Nad and 10% glycerol] containing 10
µl caspase-3 substrate (2 mM). The lysates were subsequently
incubated at 37°C for 6 h, following which the samples were assayed
using an iMark Microplate Absorbance Reader (1681130; Bio-Rad
Laboratories, Inc.) at 405 nm. Each experiment was repeated three
independent times in duplicate.
Cellular apoptosis assay
AMC-HN-8 cells with or without TXNDC5 knockdown or
overexpression were cultured at 7×104 cells/well in
96-well tissue culture plates in the presence of cisplatin (40 µM)
and/or cetuximab (150 µg/ml) for 48 h at 37°C. Cellular apoptosis
was measured with a microplate reader-based TiterTACS in
situ apoptosis detection kit (4822–96-K; R&D systems, Inc.,
Minneapolis, MN, USA), according to manufacturer's protocol. Each
experiment was repeated three independent times in duplicate.
Luciferase reporter assay
AMC-HN-8 cells were transfected with a commercially
available human TXNDC5 gene promoter/pLightSwitch_Prom luciferase
reporter (S709642; SwitchGear Genomics, Menlo Park, CA, USA) using
Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific,
Inc.) for 24 h at 37°C, and subsequently treated with cisplatin
(Sigma-Aldrich; Merck KGaA) at 5, 10, 20 and 40 µM and/or cetuximab
(Merck KGaA) at 10, 50, 100 and 150 µg/ml for 48 h 37°C. Luciferase
assays were performed with the LightSwitch Luciferase Assay kit
(LS010; SwitchGear Genomics) according to the manufacturer's
protocol. Plasmid PRL-CMV (Promega Corporation, Madison, WI, USA)
encoding Renilla luciferase (at 1/5 molar ratio to the
reporter plasmid) was co-transfected with the reporter plasmid in
each transfection as an internal control for data normalization.
Each experiment was repeated three independent times in
duplicate.
Statistical analysis
Statistical analyses were performed with SPSS
software version 10.0 (SPSS Inc., Chicago, IL, USA). All data
values were expressed as the mean ± standard deviation. Comparisons
of means among multiple groups were performed using one-way
analysis of variance followed by post hoc pairwise comparisons
using Tukey's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Cisplatin and cetuximab increase and
decrease the expression of TXNDC5 in human LSCC cells,
respectively
To examine the effects of cisplatin and cetuximab on
the expression of TXNDC5 in LSCC cells, AMC-HN-8 human LSCC cells
were treated with cisplatin (5, 10, 20 and 40 µM) or cetuximab (10,
50, 100 and 150 µg/ml) for 12, 24, 36 and 48 h. As demonstrated in
Table I, cisplatin concentration-
and time-dependently increased the mRNA level of TXNDC5 in AMC-HN-8
cells, until it reached a plateau at 20–40 µM following 36–48 h of
treatment. As demonstrated in Table
II, cetuximab concentration- and time-dependently decreased the
mRNA level of TXNDC5 in AMC-HN-8 cells, until it reached a plateau
at 100–150 µg/ml following 36–48 h of treatment. Western blot
analyses confirmed that in AMC-HN-8 cells under 48 h of treatment,
cisplatin and cetuximab concentration-dependently increased and
decreased the protein levels of TXNDC5 until reaching a plateau at
20–40 µM and 100–150 µg/ml, respectively (Fig. 1).
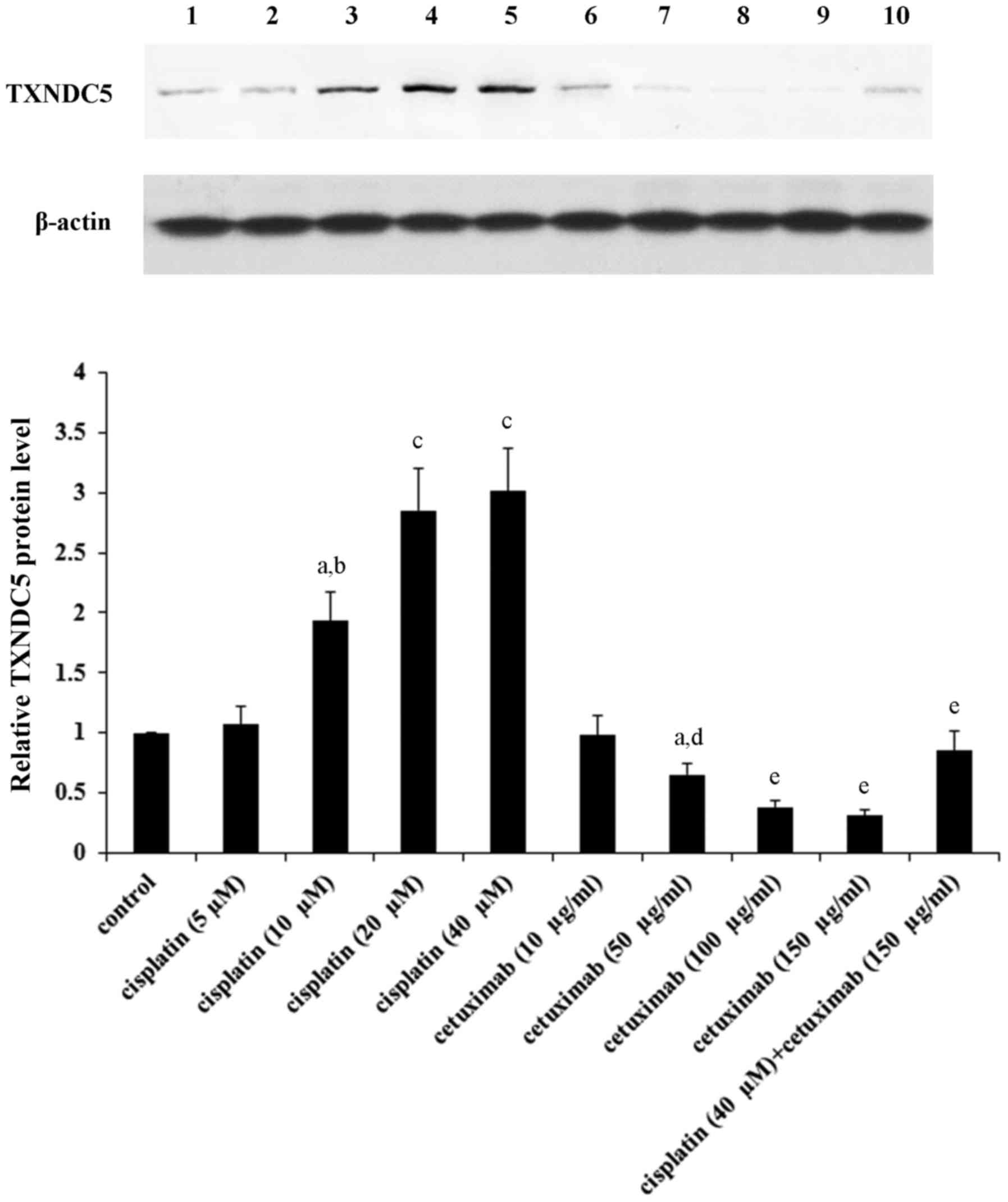 | Figure 1.TXNDC5 protein levels in laryngeal
squamous cell carcinoma cells in the presence of cisplatin and/or
cetuximab. AMC-HN-8 human LSCC cells were treated with cisplatin
(5, 10, 20 and 40 µM) and/or cetuximab (10, 50, 100 and 150 µg/ml)
for 48 h. Then cell lysates were subject to western blot analysis
for TXNDC5 expression. Lysates from untreated AMC-HN-8 cells were
used as a control. Lane 1, control; lane 2, cisplatin (5 µM); lane
3, cisplatin (10 µM); lane 4, cisplatin (20 µM); lane 5, cisplatin
(40 µM); lane 6, cetuximab (10 µg/ml); lane 7, cetuximab (50
µg/ml); lane 8, cetuximab (100 µg/ml); lane 9, cetuximab (150
µg/ml); lane 10, cisplatin (40 µM) + cetuximab (150 µg/ml). β-actin
was used as a loading control. Density of the TXNDC5 blot was
normalized against that of β-actin to obtain a relative density,
which was expressed as fold changes to that of control (designated
as 1). aP<0.05 vs. control; bP<0.05 vs.
cisplatin (5 µM); cP<0.05 vs. cisplatin (10 µM);
dP<0.05 vs. cetuximab (10 µg/ml);
eP<0.05 vs. cetuximab (50 µg/ml). TXNDC5, thioredoxin
domain-containing protein 5. |
 | Table I.TXNDC5 mRNA levels in LSCC cells
following treatment with cisplatin. |
Table I.
TXNDC5 mRNA levels in LSCC cells
following treatment with cisplatin.
|
| TXNDC5 mRNA levels
at time point, h |
|---|
|
|
|
|---|
| Cisplatin, µM | 12 | 24 | 36 | 48 |
|---|
| 5 | 1.01±0.03 | 1.03±0.03 | 1.05±0.04 | 1.02±0.02 |
| 10 |
1.19±0.06a |
1.55±0.08a,c |
1.97±0.11a,c,d |
2.07±0.12a,c,d |
| 20 |
1.68±0.08a,b |
2.39±0.13a–c |
2.94±0.15a–d |
3.06±0.15a–d |
| 40 |
1.76±0.09a,b |
2.50±0.14a–c |
3.04±0.15a–d |
3.19±0.16a–d |
| Table II.TXNDC5 mRNA levels in LSCC cells
following treatment with cetuximab. |
Table II.
TXNDC5 mRNA levels in LSCC cells
following treatment with cetuximab.
|
| TXNDC5 mRNA levels
at times point, h |
|---|
|
|
|
|---|
| Cetuximab,
µg/ml | 12 | 24 | 36 | 48 |
|---|
| 10 | 1.03±0.02 | 1.01±0.02 | 0.98±0.03 | 0.96±0.03 |
| 50 |
0.92±0.03a |
0.76±0.06a,c |
0.62±0.07a,c,d |
0.57±0.07a,c,d |
| 100 |
0.84±0.06a,b |
0.50±0.08a–c |
0.38±0.06a–d |
0.33±0.07a–d |
| 150 |
0.79±0.07a,b |
0.44±0.08a–c |
0.32±0.07a–d |
0.27±0.06a–d |
Cetuximab enhances cisplatin-induced
ER stress-associated apoptosis in LSCC cells by inhibiting
expression of TXNDC5
It has been reported that cisplatin induces ER
stress-associated apoptosis (20,21),
while TXNDC5 has been demonstrated to protect cells from ER
stress-induced apoptosis (24,25).
Recent studies have suggested that EGFR signaling is also involved
in ER stress-associated apoptosis (15,16).
To investigate the functional role of TXNDC5 in the effects of
cisplatin and cetuximab on ER stress in LSCC cells, TXNDC5 was
overexpressed and knocked down in AMC-HN-8 cells, respectively. As
demonstrated in Fig. 2, compared
with the controls, TXNDC5 was successfully overexpressed >3-fold
and knocked down by >80% in AMC-HN-8 cells. As demonstrated in
Fig. 3, compared with the control,
cisplatin (40 µM) increased the expression of TXNDC5 by ~3-fold,
which was completely eliminated by knocking down TXNDC5; however,
cetuximab (150 µg/ml) decreased the expression of TXNDC5 by ~70%,
which was completely reversed by overexpressing TXNDC5; combined
treatment with cisplatin (40 µM) and cetuximab (150 µg/ml) restored
the expression of TXNDC5 to the control level, compared with their
individual effect. The expression of CHOP, a decisive factor in
ER-stress-associated apoptosis (21,22),
was increased by ~2.5-fold by cisplatin; this effect of cisplatin
was more than doubled by TXNDC5 knockdown (Fig. 3). Cetuximab increased the
expression of CHOP by ~3-fold, which was completely eliminated by
TXNDC5 overexpression (Fig. 3).
Cisplatin and cetuximab demonstrated a combinatorial effect on
increasing the expression of CHOP, which was decreased by ~45% by
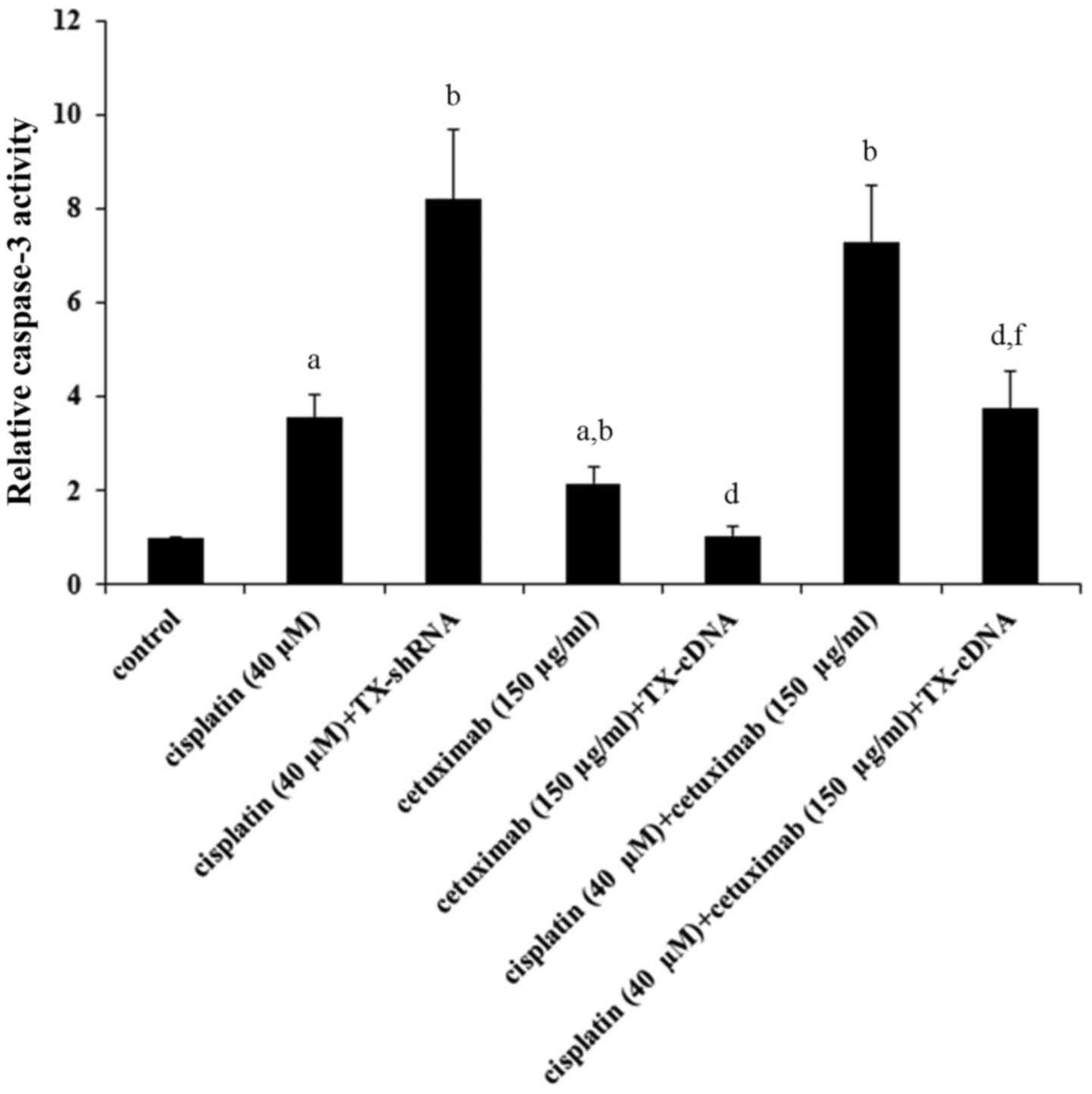
TXNDC5 overexpression. The level of cleaved caspase-3, a major
caspase activated in apoptotic cells (29), was increased by ~4-fold by
cisplatin; this effect of cisplatin was increased by >2-fold by
TXNDC5 knockdown (Fig. 3).
Cetuximab increased the level of cleaved caspase-3 by ~3-fold,
which was completely eliminated by TXNDC5 overexpression (Fig. 3). Cisplatin and cetuximab
demonstrated a combinatorial effect on increasing the level of
cleaved/activated caspase-3, which was decreased by ~50% by TXNDC5
overexpression. The findings were confirmed by directly measuring
the caspase-3 activity (Fig. 4),
which demonstrated a similar data trend.
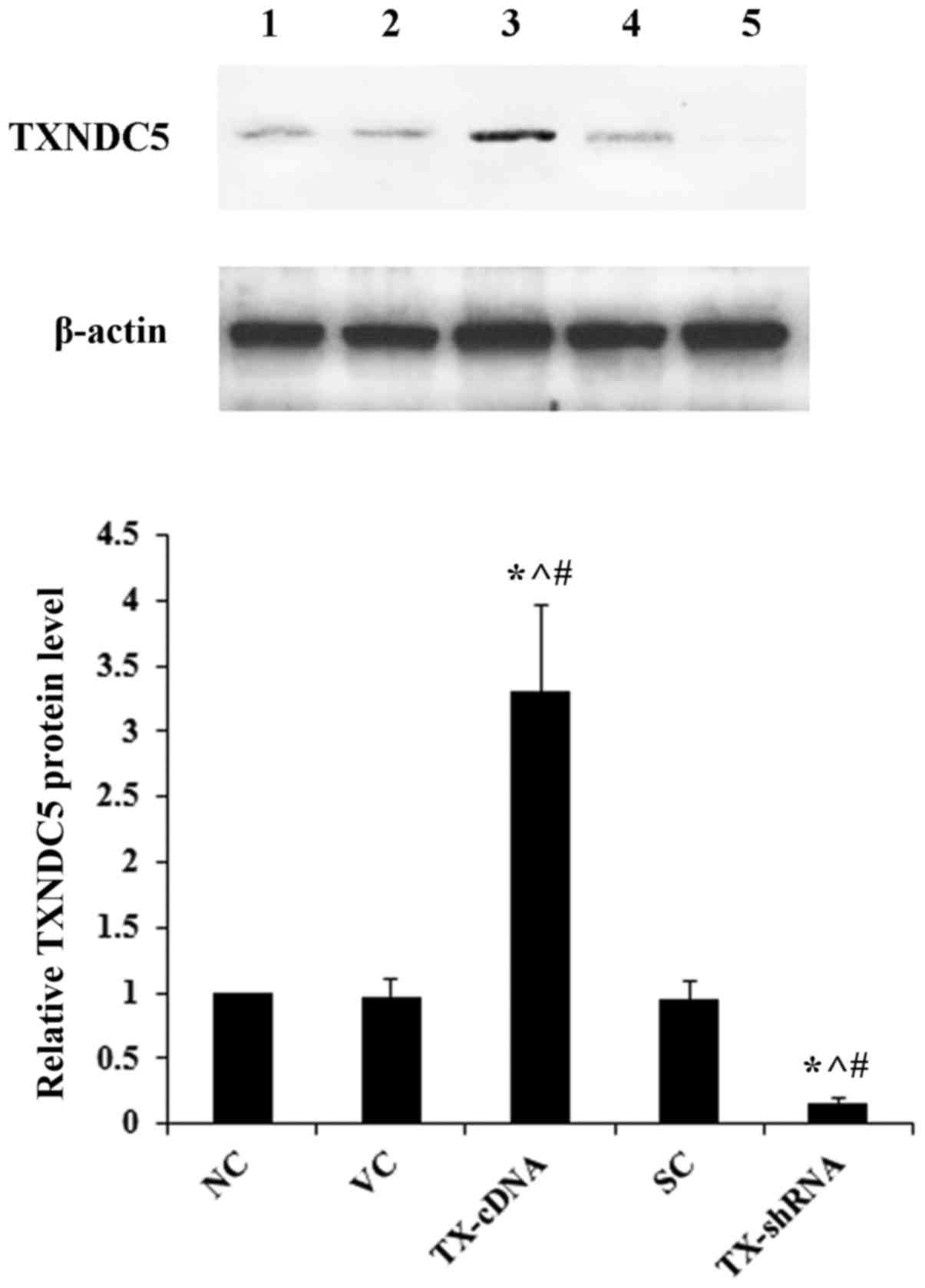 | Figure 2.Overexpression and knockdown of
TXNDC5 in laryngeal squamous cell carcinoma cells. AMC-HN-8 cells
were transfected with a human TXNDC5-cDNA expression vector or
transduced with human TXNDC5-shRNA lentiviral particles to
overexpress and knock down TXNDC5, respectively. The NC, cells
transfected with the VC, or cells transduced with SC were used as
controls. Lane 1, NC; lane 2, VC; lane 3, TX-cDNA; lane 4, SC; lane
5, TX-shRNA. β-actin was used as a loading control. Density of the
TXNDC5 blot was normalized against that of β-actin to obtain a
relative density, which was expressed as fold changes to that of NC
(designated as 1). *P<0.05 vs. NC; ^P<0.05 vs. VC;
#P<0.05 vs. SC. TXNDC5/TX, thioredoxin
domain-containing protein 5; shRNA, short hairpin RNA; NC,
untransduced/untransfected cells; VC, empty expression vector; SH,
control shRNA lentiviral particles. |
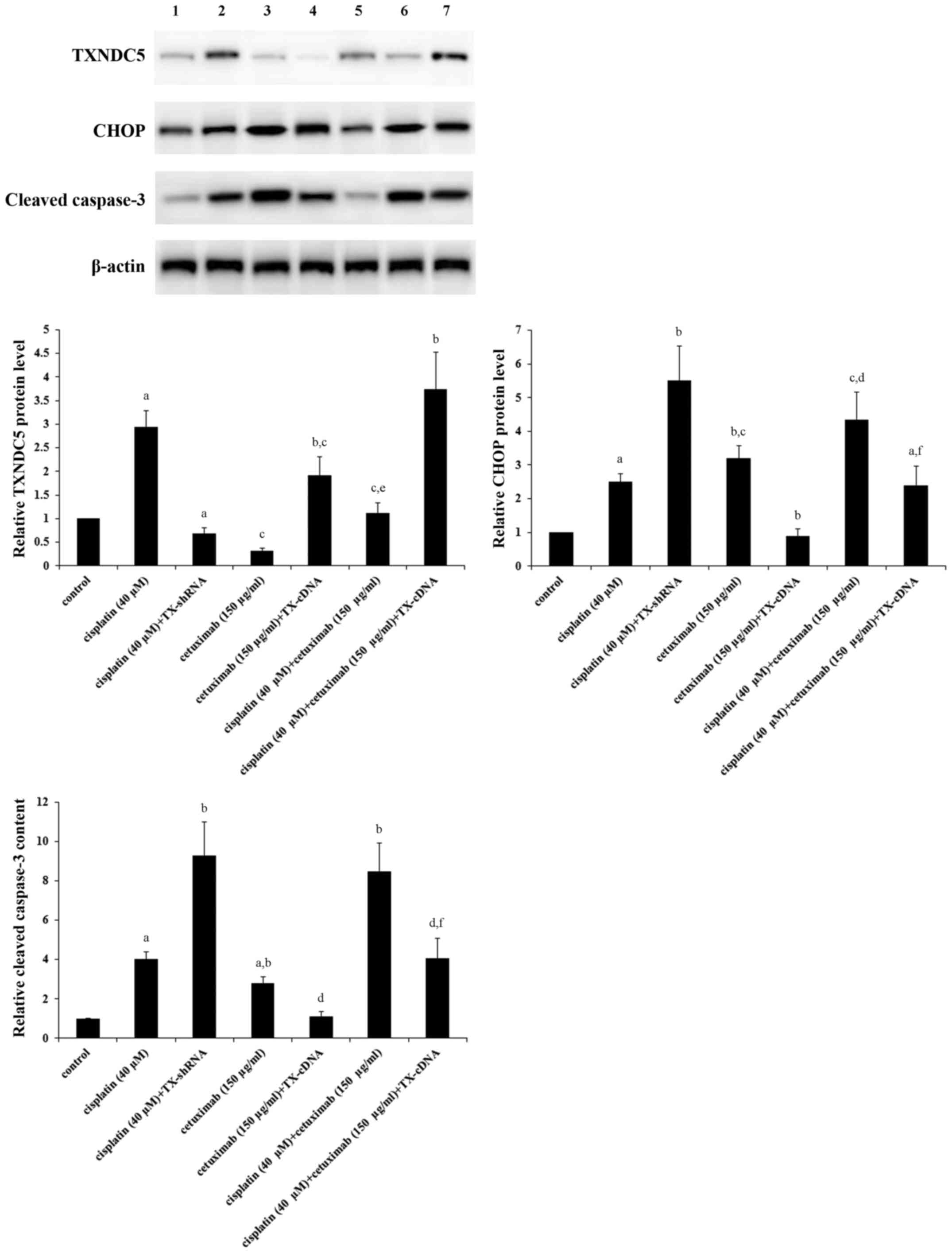 | Figure 3.Protein levels of TXNDC5, CHOP and
cleaved caspase-3 in laryngeal squamous cell carcinoma cells with
or without TXNDC5 overexpression or knockdown in the presence of
cisplatin and/or cetuximab. AMC-HN-8 cells with or without TXNDC5
overexpression (TX-cDNA) or knockdown (TX-shRNA) were treated with
cisplatin (40 µM) and/or cetuximab (150 µg/ml) for 48 h. The cell
lysates were subject to western blot analyses to determine the
protein levels of TXNDC5, CHOP and cleaved caspase-3. Lysates from
untreated AMC-HN-8 cells were used as a control. Lane 1, control;
lane 2, cisplatin (40 µM); lane 3, cisplatin (40 µM) + TX-shRNA;
lane 4, cetuximab (150 µg/ml); lane 5, cetuximab (150 µg/ml) +
TX-cDNA; lane 6, cisplatin (40 µM) + cetuximab (150 µg/ml); lane 7,
cisplatin (40 µM) + cetuximab (150 µg/ml) + TX-cDNA. β-actin
blotting was used as a loading control. Density of the TXNDC5, CHOP
or cleaved caspase-3 blot was respectively normalized against that
of β-actin to obtain a relative density, which was expressed as
fold changes to that of control (designated as 1).
aP<0.05 vs. control; bP<0.05 vs.
cisplatin (40 µM); cP<0.05 vs. cisplatin (40 µM) +
TX-shRNA; dP<0.05 vs. cetuximab (150 µg/ml);
eP<0.05 vs. cetuximab (150 µg/ml) + TX-cDNA;
fP<0.05 vs. cisplatin (40 µM) + cetuximab (150
µg/ml). TXNDC5/TX, thioredoxin domain-containing protein 5; CHOP,
CCAAT/enhancer-binding protein homologous protein; shRNA, short
hairpin RNA. |
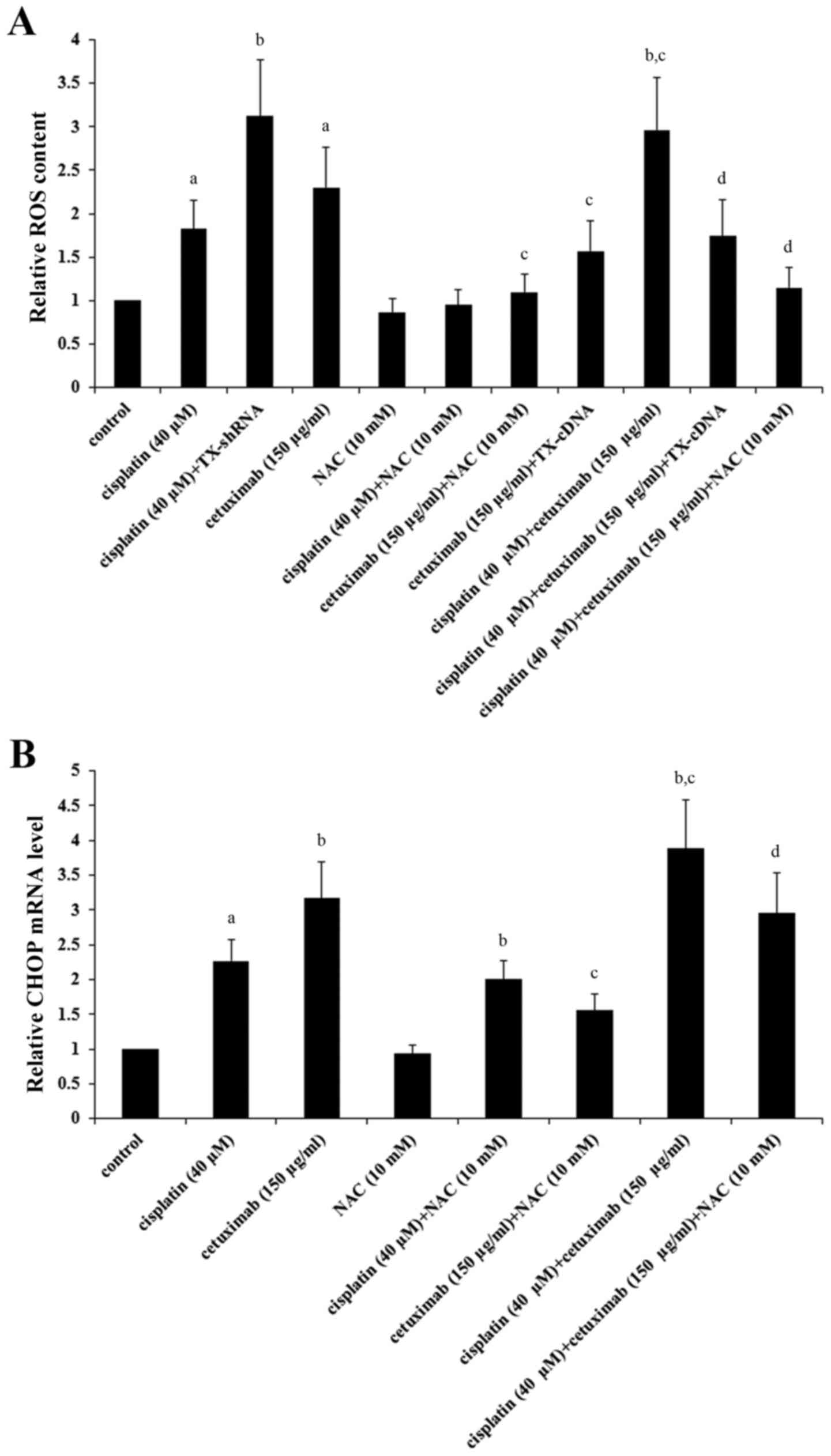
It has been reported that TXNCD5 maintains levels of
cellular reductants and that drug- or siRNA-induced downregulation
of TXNDC5 induces ROS, which in turn activates ER stress and CHOP
expression (30). Therefore, the
interactive effects of TXNDC5, cisplatin, cetuximab and ROS
scavenger/antagonist NAC on ROS production and CHOP expression in
LSCC cells were investigated. As demonstrated in Fig. 5A, cisplatin (40 µM) and cetuximab
(150 µg/ml) significantly induced ROS in AMC-HN-8 cells
(P<0.05), which was respectively enhanced by TNXDC5 knockdown
and inhibited by TXNDC5 overexpression; cisplatin and cetuximab
exhibited a combinatorial effect on inducing ROS, which was
significantly eliminated by TXNDC5 overexpression or NAC
(P<0.05). As demonstrated in Fig.
5B, NAC significantly decreased cisplatin- and
cetuximab-induced CHOP expression in addition to the combinatorial
promoting effect of cisplatin and cetuximab on CHOP expression in
AMC-HN-8 cells (P<0.05).
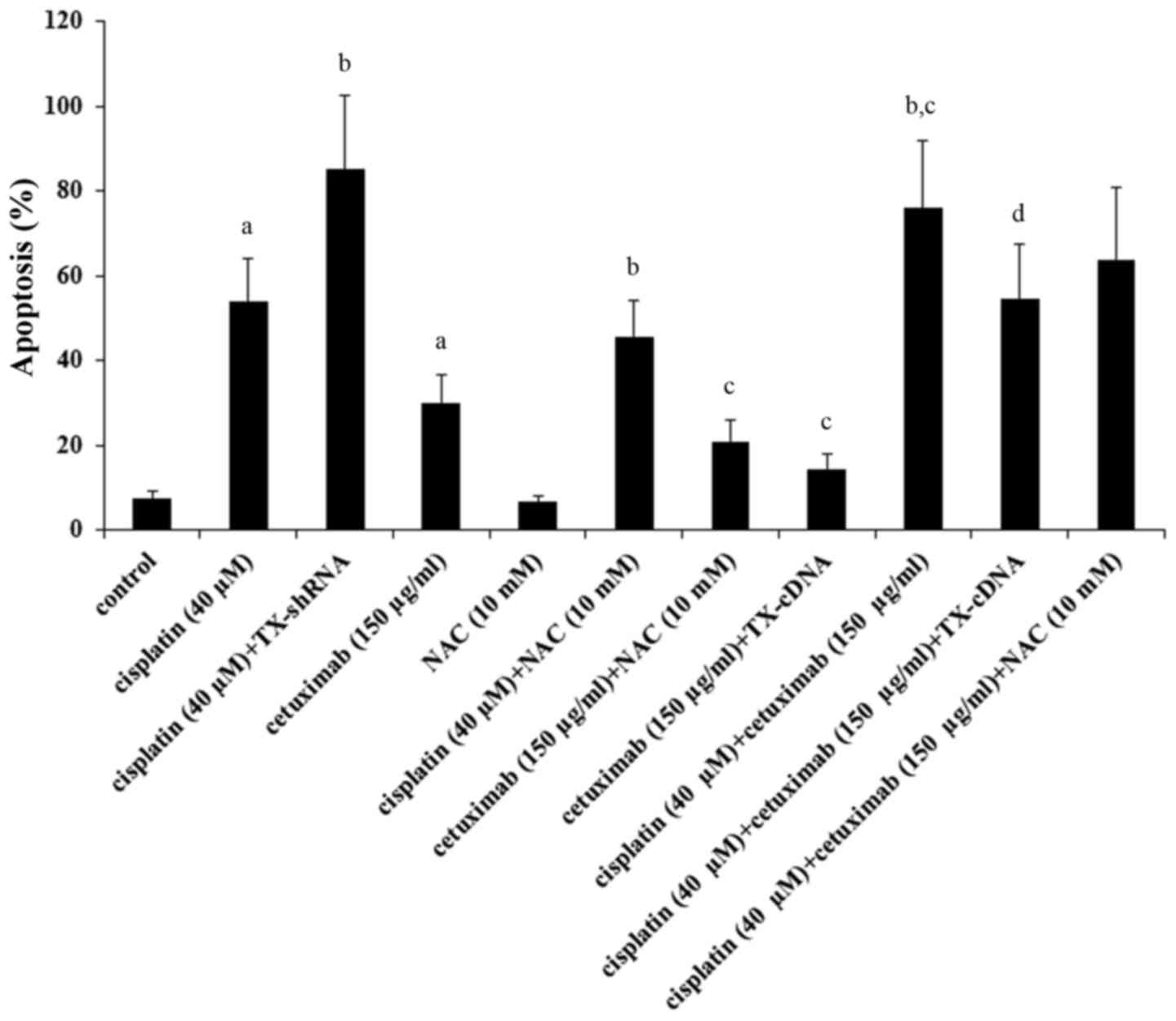
As demonstrated in Fig.
6, treatment with cisplatin (40 µM) for 48 h resulted in 56% of
apoptosis, which was increased to 82% by TXNDC5 knockdown and
decreased to 43% by NAC; treatment with cetuximab (150 µg/ml) for
48 h resulted in 30% apoptosis, which was brought down to 14.5% by
TXNDC5 and to 21% by NAC. Combined treatment with cisplatin (40 µM)
and cetuximab (150 µg/ml) resulted in 76% apoptosis, which was
brought down to 54% by TXNDC5 overexpression and to 63% by NAC.
Analyses of the data revealed that knockdown of TXNDC5 augmented
the apoptotic effect of cisplatin by ~54%
[(82–56%)/(56–7.7%)=53.8%; basal apoptosis level=7.7%];
overexpression of TXNDC5 reduced the apoptotic effect of cetuximab
by ~70% [(30–14.5%)/(30–7.7%)=69.5%]; the effect of TXNDC5 was
primarily mediated by inhibiting ROS production (~59%)
[(76–63%)/(76–54%)=59.1%].
The above findings suggested that cetuximab enhanced
the apoptotic effect of cisplatin on LSCC cells by promoting ER
stress-associated apoptosis primarily via inhibiting expression of
TXNDC5 and thereby increasing ROS production.
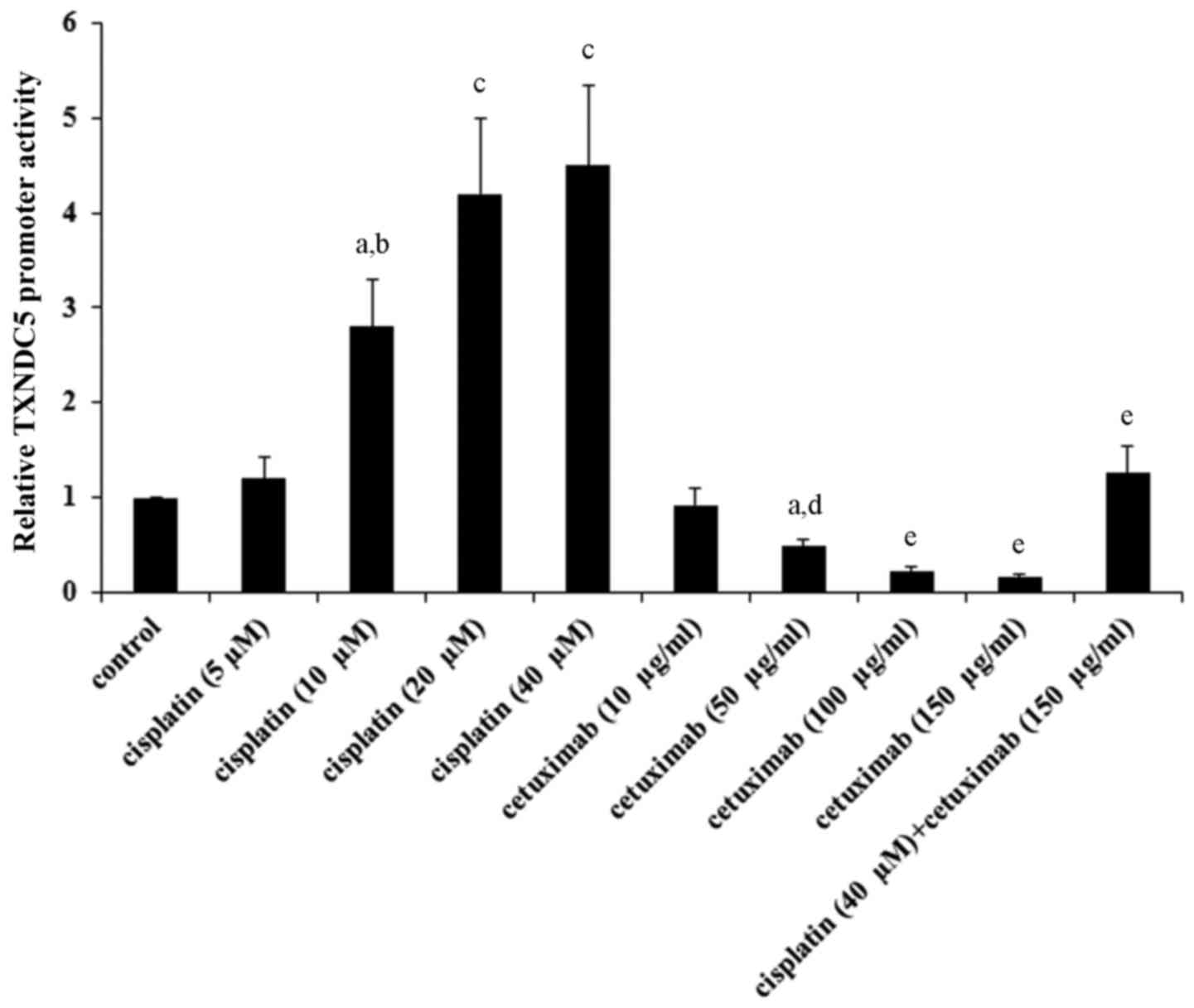
Cisplatin and cetuximab exhibit
opposing effects on TXNDC5 gene promoter
As indicated in Tables
I and II, cisplatin and
cetuximab exhibited opposing effects on the TXNDC5 mRNA level,
suggesting that the two drugs may affect the TXNDC5 gene promoter
activity. AMC-HN-8 cells were transfected with a human TXNDC5 gene
promoter/luciferase reporter, and the cells were treated with
cisplatin (5, 10, 20 and 40 µM) or cetuximab (10, 50, 100 and 150
µg/ml) for 48 h. As demonstrated in Fig. 7, cisplatin and cetuximab
concentration-dependently increased and decreased the TXNDC5
promoter activity, respectively. Combined treatment with cisplatin
(40 µM) and cetuximab (150 µg/ml) restored the TXNDC5 promoter
activity to the control level, compared with their individual
effect (Fig. 7). The findings
suggested that cisplatin and cetuximab exhibit opposing effects on
the TXNDC5 gene promoter.
As TXNDC5 expression is reportedly regulated by the
orphan nuclear receptor NR4A1 in cancer cell lines (30–32)
and NR4A1 also serves a role in cisplatin-induced responses
(33,34), it was subsequently examined whether
cisplatin and/or cetuximab exhibited an effect on NR4A1 levels in
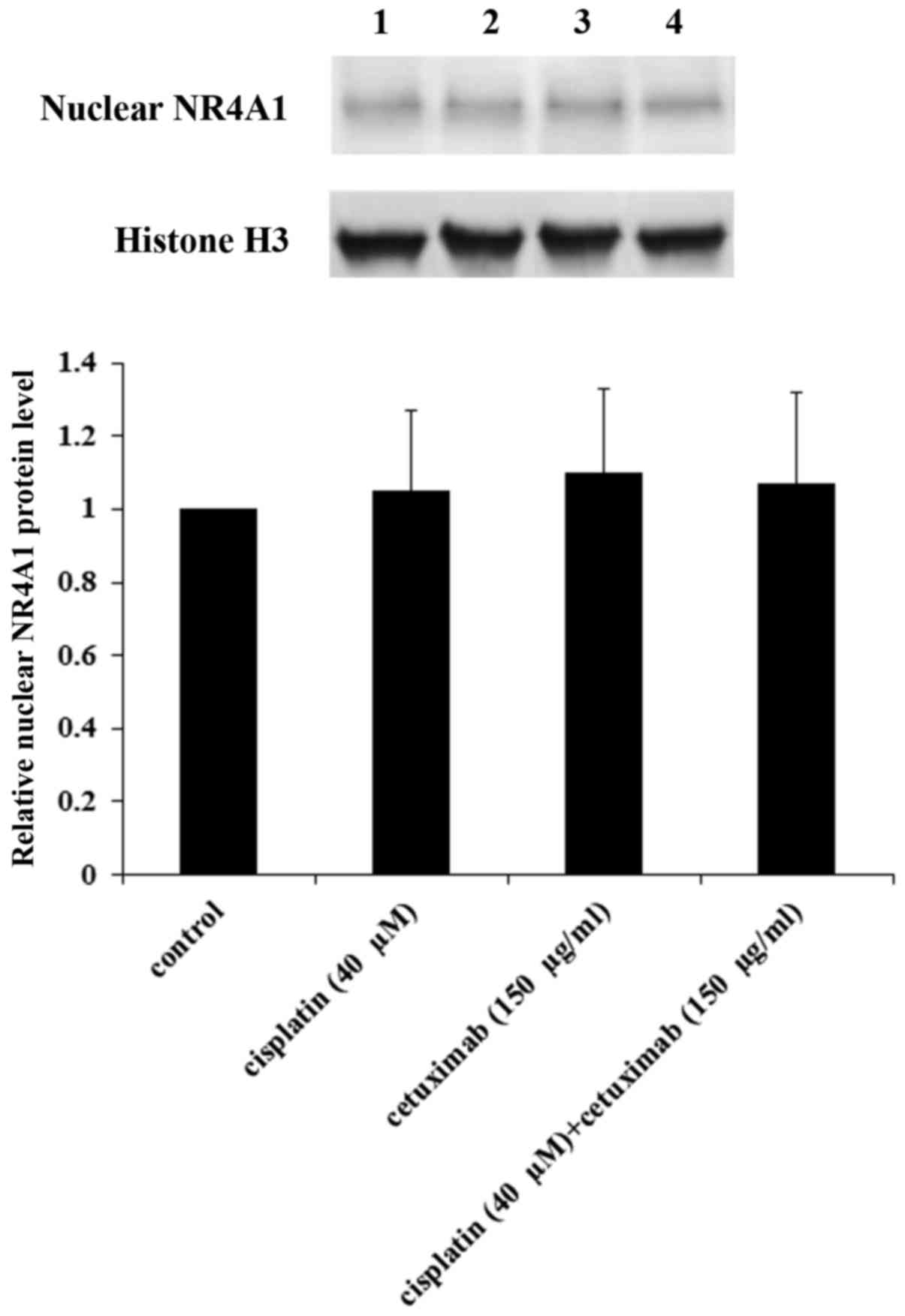
the nucleus of AMC-HN-8 cells. As demonstrated in Fig. 8, cisplatin and cetuximab
demonstrated no significant effects on the nuclear NR4A1 protein
level, suggesting that NR4A1 did not serve a role in the regulatory
effects of cisplatin and cetuximab on the TXNDC5 gene promoter.
Discussion
Cisplatin and cetuximab have been used for the
treatment of LSCC (5,6). It has been demonstrated that
cisplatin and inhibition of EGFR signaling may induce ER
stress-associated apoptosis (11–16).
However, ER protein TXNDC5 reportedly protects cells from ER
stress-induced apoptosis (24,25).
The present study provided the first evidence, to the best of the
authors' knowledge, that: i) While inducing ER stress-associated
apoptosis, cisplatin also induces expression of TXNDC5 in LSCC
cells; ii) cetuximab inhibits the expression of TXNDC5 in LSCC
cells; and iii) cetuximab enhances the apoptotic effect of
cisplatin on LSCC cells primarily via inhibiting expression of
TXNDC5.
As the previously widely used Hep-2 cell line has
been identified to be dominated by HeLa cell contamination rather
than LSCC cells (35), only the
AMC-HN-8 human LSCC cell line was used as a cell model in the study
(36). In agreement with previous
studies demonstrating that cisplatin induces ER stress-associated
apoptosis (14,21), cisplatin induced ER
stress-associated apoptosis in LSCC cells in the present study, as
evidenced by elevated levels of CHOP, caspase activity and
apoptosis. Cisplatin also induced TXNDC5, an ER protein protective
against ER stress-associated apoptosis (23–26);
this may be a protective response of LSCC cells to promote survival
under cisplatin-induced ER stress. The direct effect of this
response was to decrease cisplatin-induced apoptosis, which was
demonstrated by: i) The marked enhancement of the apoptotic effect
of cisplatin following TXNDC5 knockdown; and ii) cetuximab, which
inhibited the expression of TXNDC5, markedly enhanced the apoptotic
effect of cisplatin and this effect was eliminated by
overexpressing TXNDC5. Knockdown of TXNDC5 augmented the apoptotic
effect of cisplatin by ~54%, suggesting that ER stress-associated
apoptosis is a major mechanism underlying the apoptotic effect of
cisplatin on LSCC cells and that TXNDC5 is a critical factor in
this process.
It has been reported that cetuximab effectively
antagonizes EGFR signaling (5,6,37)
and that inhibition of EGFR signaling induces ER stress-associated
apoptosis (15,16). In agreement with these previous
reports, cetuximab induced ER stress-associated apoptosis in LSCC
cells in the present study, as evidenced by elevated levels of
CHOP, caspase activity and apoptosis. Overexpression of TXNDC5
reduced the apoptotic effect of cetuximab by ~70%, suggesting that
ER stress-associated apoptosis is a major mechanism underlying the
apoptotic effect of cetuximab on LSCC cells and that TXNDC5 is a
critical factor in this process.
In agreement with previous studies demonstrating
that TXNDC5 protects cells from ER stress-induced apoptosis
(24,25), the present study demonstrated by
overexpression and knockdown experiments that TXNDC5 is an
effective protective/survival factor against cisplatin- and
cetuximab-induced ER stress-associated apoptosis. Therefore, TXNDC5
may be a new potential therapeutic target for LSCC and other
cancers. In the present study, cetuximab enhanced the apoptotic
effect of cisplatin on LSCC cells primarily by inhibiting the
expression of TXNDC5 and the overexpression of TXNDC5 eliminated
the enhancing effect of cetuximab. It may be worthwhile to examine
whether cetuximab in combination with ER stress-inducing
chemotherapeutic agents other than cisplatin may benefit patients
with LSCC or other cancers. In addition, as radiotherapy is a major
treatment for a number of types of cancer, including advanced LSCC
(4), and induces ER stress in
tumor cells (38,39), cetuximab in combination with
radiotherapy may benefit patients with advanced LSCC or other
cancer types.
TXNDC5 reportedly facilitates the correct folding of
proteins via the formation of disulfide bonds through its
thioredoxin domains, thereby alleviating ER stress and protecting
cells from ER stress-associated apoptosis (24–26).
It has also been demonstrated that TXNCD5 maintains levels of
cellular reductants and that drug- or siRNA-induced downregulation
of TXNDC5 induces ROS, which in turn activates ER stress and CHOP
expression (30). Consistent with
previous reports, the present study identified that cisplatin and
cetuximab significantly induced ROS and CHOP expression in AMC-HN-8
cells, which were respectively enhanced by TNXDC5 knockdown and
inhibited by TXNDC5 overexpression; an ROS scavenger/antagonist
significantly decreased cisplatin- and cetuximab-induced ROS and
CHOP expression. Apoptosis analysis suggested that 59% of TXNDC5
overexpression-induced inhibition of the apoptotic effect of
cetuximab may be attributed to it inhibiting ROS production. The
present results suggest that cetuximab enhances cisplatin-induced
ER stress-associated apoptosis primarily by regulating
TXNDC5-mediated inhibition of ROS production. Nevertheless, the
other mechanisms involved in how TXNDC5 functions to lessen
cisplatin- and cetuximab-induced ER stress in LSCC cells remain to
be elucidated and require future studies. The present study
identified that cisplatin and cetuximab exhibited opposing effects
on the TXNDC5 gene promoter, suggesting that cisplatin and
cetuximab regulate the expression of TXNDC5 at the gene
transcription/promoter level. The orphan nuclear receptor NR4A1
reportedly regulates TNXDC5 expression (30–32)
and also serves a role in cisplatin-induced responses (33,34).
However, as cisplatin and cetuximab demonstrated no significant
effects on the nuclear NR4A1 protein level, it is unlikely that
NR4A1 mediates the regulatory effects of cisplatin and cetuximab on
the TXNDC5 gene promoter. It is hypothesized that the mechanisms
underlying transcriptional regulation of TXNDC5 expression may be
investigated in future studies.
In conclusion, the results of the present study
suggested that ER stress-associated apoptosis is a major mechanism
underlying the apoptotic effect of cisplatin and cetuximab on LSCC
cells. Cetuximab enhances cisplatin-induced ER stress-associated
apoptosis in LSCC cells primarily by inhibiting the expression of
TXNDC5 and thereby increasing ROS production. Cisplatin and
cetuximab exhibited stimulatory and inhibitory effects on the
TXNDC5 gene promoter, respectively. The present study presented a
novel understanding of the pharmacological effects of cisplatin and
cetuximab on LSCC. The present study also suggested that TXNDC5 may
be a potential novel therapeutic target for LSCC.
Acknowledgements
The present study was funded by the Science and
Technology Foundation of Hunan Province, China (grant no.
2015AK2056).
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mnejja M, Hammami B, Bougacha L, Chakroun
A, Charfeddine I, Khabir A, Boudaoura T and Ghorbel A: Occult lymph
node metastasis in laryngeal squamous cell carcinoma: Therapeutic
and prognostic impact. Eur Ann Otorhinolaryngol Head Neck Dis.
127:173–176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Amar A, Chedid HM, Franzi SA and Rapoport
A: Diagnostic and therapeutic delay in patients with larynx cancer
at a reference public hospital. Braz J Otorhinolaryngol.
76:700–703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hitt R, López-Pousa A, Martinez-Trufero J,
Escrig V, Carles J, Rizo A, Isla D, Vega ME, Marti JL, Lobo F, et
al: Phase III study comparing cisplatin plus fluorouracil to
paclitaxel, cisplatin, and fluorouracil induction chemotherapy
followed by chemoradiotherapy in locally advanced head and neck
cancer. J Clin Oncol. 23:8636–8645. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bussu F, Pozzoli G, Giglia V, Rizzo D,
Limongelli A, De Corso E, Graziani C, Paludetti G, Navarra P and
Almadori G: Effects of the administration of epidermal growth
factor receptor specific inhibitor cetuximab, alone and in
combination with cisplatin, on proliferation and apoptosis of Hep-2
laryngeal cancer cells. J Laryngol Otol. 128:902–908. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Burtness B, Goldwasser MA, Flood W, Mattar
B and Forastiere AA; Eastern Cooperative Oncology Group, : Phase
III randomized trial of cisplatin plus placebo compared with
cisplatin plus cetuximab in metastatic/recurrent head and neck
cancer: An Eastern Cooperative Oncology Group study. J Clin Oncol.
23:8646–8654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lv X, Song DM, Niu YH and Wang BS:
Inhibition of heme oxygenase-1 enhances the chemosensitivity of
laryngeal squamous cell cancer Hep-2 cells to cisplatin. Apoptosis.
21:489–501. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Macciò A and Madeddu C: Cisplatin: An old
drug with a newfound efficacy - from mechanisms of action to
cytotoxicity. Expert Opin Pharmacother. 14:1839–1857. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sancho-Martínez SM, Prieto-García L,
Prieto M, López-Novoa JM and López-Hernández FJ: Subcellular
targets of cisplatin cytotoxicity: An integrated view. Pharmacol
Ther. 136:35–55. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu F, Megyesi J and Price PM: Cytoplasmic
initiation of cisplatin cytotoxicity. Am J Physiol Renal Physiol.
295:F44–F52. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peyrou M, Hanna PE and Cribb AE:
Cisplatin, gentamicin, and p-aminophenol induce markers of
endoplasmic reticulum stress in the rat kidneys. Toxicol Sci.
99:346–353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mandic A, Hansson J, Linder S and Shoshan
MC: Cisplatin induces endoplasmic reticulum stress and
nucleus-independent apoptotic signaling. J Biol Chem.
278:9100–9106. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu H and Baliga R: Endoplasmic reticulum
stress-associated caspase 12 mediates cisplatin-induced LLC-PK1
cell apoptosis. J Am Soc Nephrol. 16:1985–1992. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu Y, Wang C and Li Z: A new strategy of
promoting cisplatin chemotherapeutic efficiency by targeting
endoplasmic reticulum stress. Mol Clin Oncol. 2:3–7. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miao Y, Bi XY, Zhao M, Jiang HK, Liu JJ,
Li DL, Yu XJ, Yang YH, Huang N and Zang WJ: Acetylcholine inhibits
tumor necrosis factor α activated endoplasmic reticulum apoptotic
pathway via EGFR-PI3K signaling in cardiomyocytes. J Cell Physiol.
230:767–774. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hong S, Gu Y, Gao Z, Guo L, Guo W, Wu X,
Shen Y, Sun Y, Wu X and Xu Q: EGFR inhibitor-driven endoplasmic
reticulum stress-mediated injury on intestinal epithelial cells.
Life Sci. 119:28–33. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Berridge MJ: The endoplasmic reticulum: A
multifunctional signaling organelle. Cell Calcium. 32:235–249.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jørgensen MM, Bross P and Gregersen N:
Protein quality control in the endoplasmic reticulum. APMIS Suppl.
1–91. 2003.
|
|
20
|
Xu Y, Li D, Zeng L, Wang C, Zhang L, Wang
Y, Yu Y, Liu S and Li Z: Proteasome inhibitor lactacystin enhances
cisplatin cytotoxicity by increasing endoplasmic reticulum
stress-associated apoptosis in HeLa cells. Mol Med Rep. 11:189–195.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang R, Wang R, Chen Q and Chang H:
Inhibition of autophagy using 3-methyladenine increases
cisplatin-induced apoptosis by increasing endoplasmic reticulum
stress in U251 human glioma cells. Mol Med Rep. 12:1727–1732. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nishitoh H: CHOP is a multifunctional
transcription factor in the ER stress response. J Biochem.
151:217–219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Horna-Terrón E, Pradilla-Dieste A,
Sánchez-de-Diego C and Osada J: TXNDC5, a newly discovered
disulfide isomerase with a key role in cell physiology and
pathology. Int J Mol Sci. 15:23501–23518. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sullivan DC, Huminiecki L, Moore JW, Boyle
JJ, Poulsom R, Creamer D, Barker J and Bicknell R: EndoPDI, a novel
protein-disulfide isomerase-like protein that is preferentially
expressed in endothelial cells acts as a stress survival factor. J
Biol Chem. 278:47079–47088. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Funkner A, Parthier C, Schutkowski M,
Zerweck J, Lilie H, Gyrych N, Fischer G, Stubbs MT and Ferrari DM:
Peptide binding by catalytic domains of the protein disulfide
isomerase-related protein ERp46. J Mol Biol. 425:1340–1362. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kojima R, Okumura M, Masui S, Kanemura S,
Inoue M, Saiki M, Yamaguchi H, Hikima T, Suzuki M, Akiyama S and
Inaba K: Radically different thioredoxin domain arrangement of
ERp46, an efficient disulfide bond introducer of the mammalian PDI
family. Structure. 22:431–443. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Johnson DR, Levanat S and Bale AE: Direct
molecular analysis of archival tumor tissue for loss of
heterozygosity. Biotechniques. 19:190–192. 1995.PubMed/NCBI
|
|
29
|
Woo M, Hakem R, Soengas MS, Duncan GS,
Shahinian A, Kägi D, Hakem A, McCurrach M, Khoo W, Kaufman SA, et
al: Essential contribution of caspase 3/CPP32 to apoptosis and its
associated nuclear changes. Genes Dev. 12:806–819. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee SO, Jin UH, Kang JH, Kim SB, Guthrie
AS, Sreevalsan S, Lee JS and Safe S: The orphan nuclear receptor
NR4A1 (Nur77) regulates oxidative and endoplasmic reticulum stress
in pancreatic cancer cells. Mol Cancer Res. 12:527–538. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hedrick E, Lee SO, Doddapaneni R, Singh M
and Safe S: Nuclear receptor 4A1 as a drug target for breast cancer
chemotherapy. Endocr Relat Cancer. 22:831–840. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hedrick E, Lee SO, Kim G, Abdelrahim M,
Jin UH, Safe S and Abudayyeh A: Nuclear receptor 4A1 (NR4A1) as a
drug target for renal cell adenocarcinoma. PLoS One.
10:e01283082015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yao LM, He JP, Chen HZ, Wang Y, Wang WJ,
Wu R, Yu CD and Wu Q: Orphan receptor TR3 participates in
cisplatin-induced apoptosis via Chk2 phosphorylation to repress
intestinal tumorigenesis. Carcinogenesis. 33:301–311. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin H, Lin Q, Liu M, Lin Y, Wang X, Chen
H, Xia Z, Lu B, Ding F, Wu Q and Wang HR: PKA/Smurf1
signaling-mediated stabilization of Nur77 is required for
anticancer drug cisplatin-induced apoptosis. Oncogene.
33:1629–1639. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
ATCC, . http://atcc.org/Products/All/CCL-23.aspx#characteristicsJuly
1–2016
|
|
36
|
Xu L, Chen Z, Xue F, Chen W, Ma R, Cheng S
and Cui P: MicroRNA-24 inhibits growth, induces apoptosis, and
reverses radioresistance in laryngeal squamous cell carcinoma by
targeting X-linked inhibitor of apoptosis protein. Cancer Cell Int.
15:612015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mendelsohn J and Baselga J: The EGF
receptor family as targets for cancer therapy. Oncogene.
19:6550–6565. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Saglar E, Unlu S, Babalioglu I, Gokce SC
and Mergen H: Assessment of ER Stress and autophagy induced by
ionizing radiation in both radiotherapy patients and ex vivo
irradiated samples. J Biochem Mol Toxicol. 28:413–417. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhu H, Abulimiti M, Liu H, Su XJ, Liu CH
and Pei HP: RITA enhances irradiation-induced apoptosis in
p53-defective cervical cancer cells via upregulation of IRE1α/XBP1
signaling. Oncol Rep. 34:1279–1288. 2015. View Article : Google Scholar : PubMed/NCBI
|