Introduction
As a barrier between vessel lumen and surrounding
tissues, vascular endothelial cells (VEC) are involved in many
aspects of vascular biology, including flow adjustment, substance
exchange, prevention of lipid leakage, and inhibition of platelet
aggregation. Highly developed endoplasmic reticulum (ER), which
realizes secretion function of cells, is observed under electron
microscopy in VECs, especially in regenerated vessel endothelium.
This presence of the ER results in high sensitivity of VEC to ER
induction factors. ER stress is a newly discovered pathway that can
initiate the apoptotic process. Many pathophysiological processes
and diseases, such as atherosclerosis, diabetes,
ischemia/reperfusion and injury, are implicated in ER
stress-induced VEC apoptosis (1–3).
The ER is an important organelle in eukaryotes that
provides an environment for proteins to synthesize, fold, and
assemble, and offers storage sites for Ca2+. The ER is
also involved in regulation of Ca2+ balance and
metabolism of cholesterol and phospholipids (4,5).
Under the presence of induction factors, e.g. oxidative stress,
hypoxia, calcium dyshomeostasis, and virus infection, ER stress
tends to be induced by the massive accumulation of unfolded and
misfolded proteins (6–9). Unfolded protein response (UPR),
endoplasmic reticulum-overload response (EOR), and sterol
regulatory cascade are three signalling pathways that are activated
to help maintain cellular homeostasis. When severe stress is
continuous, cell apoptosis processes are eventually induced
(10).
UPR is the most important and most clearly studied
pathway (11,12). In the early stage of ER stress,
through the UPR pathway, misfolded proteins are eliminated and
correct protein folding is promoted to recover cellular
physiological function and to re-establish ER homeostasis. UPR is
mediated by binding immunoglobulin heavy chain protein
(BiP)/glucose-regulated protein 78 (GRP78) along with three ER
stress sensor proteins: Inositol requiring kinase (IRE), PERK, and
activating transcription factor 6 (ATF6) (13). ATF6 is a type 2 transmembrane
protein kinase embedded in the ER. In response to ER stress, ATF6
is transported from the ER to the Golgi apparatus and cleaved into
a transcriptionally active p50ATF6. p50ATF6 then combines with the
promoter of ER stress response elements in the nucleus, and induces
expression of ER stress-related genes such as those encoding C/EBP
homologous protein (CHOP)/growth arrest, DNA damage inducible gene
153 (GADD153), BiP/GRP78, X-box-binding protein 1 (XBP1), and
calretinin, which regulate cell survival or apoptosis (14–16).
Nakanishi et al (17) documented that ATF6 regulates ER
stress-induced apoptosis of myogenous cells by activating
caspase-12. Morishima et al (18) found that ATF6 in rat myoblasts
regulate cell apoptosis by specifically suppressing Mcl-1 and
up-regulating WBP1. The regulatory pathways of activated ATF6 in
different cells are not the same, so the mechanism and pathway in
ER stress-induced VEC apoptosis is still unclear. Therefore, the
present study used thapsigargin (TG) as an ER stress inducer to
investigate the role of ATF6 in VEC apoptosis in response.
Materials and methods
Recombinant plasmids construction
Recombinant plasmids ATF6 (1-366aa) and ATF5
(151-366aa) were purchased from Shanghai Transheep Biotechnology
Co. Ltd., Shanghai, China). ATF6 (1-366aa) was ATF6 high-expressed
plasmid, the specific sequences is
5′-CCCAAGCTTATGGGGGGAGCCGGCTGGGGT-3′ for sense primer and
5′-ACGCGTCGACGTTCTCTGACACAACTTCAT-3 for reverse primer. ATF6
(151-366aa) was plasmid without transcriptional activity, the
specific sequences is 5′-CCCAAGCTTATGGATAAGCCTGTCACTGGTCC-3′ for
sense primer and 5′-ACGCGTCGACGTTCTCTGACACAACTTCAT-3′ for reverse
primer.
Cell infection and treatment
VECs (HUVEC-12 cell line) were purchased from Bogoo
Biotechnology Co. Ltd. (Shanghai, China). Cells in logarithmic
growth phase were seeded into a 6-well plate and cultured for 24 h.
Transfection of recombinant plasmids of ATF6 (1-366aa+) and ATF6
(151-366aa) was performed with Invitrogen Lipofectamine™ LTX
according to the manufacturer's instructions (Thermo Fisher
Scientific Inc., New York, NY, USA). Two microgram of
Pires2-ZsGreen1-vector or pIRES2-ZsGreen1-ARHGAP18 (Sangon Biotech
Inc., Shanghai, China), 5 µl of Lipofectamine™ LTX (Thermo Fisher
Scientific Inc.) and 250 µl Opti-MEM (Shanghai Haoran Biological
Technology Co. Ltd., Shanghai, China) were mixed and incubated at
room temperature for 25 min. Five hundred microlitre of the mixture
was added to a 6-well plate with RPMI 1640 medium (Thermo Fisher
Scientific Inc.). Then, after 48 h, the transfected cells were
harvested for subsequent experiments. Western blotting was
performed to detect the expression of ATF6 to test transfection
efficiency.
CCK-8 assay
Cells in TG, ATF6 (151-366aa) + TG and ATF6
(1-366aa) + TG groups were treated with 1 µM TG for respectively
12, 24 and 48 h. Cell viability in each group was detected by using
CCK8 kit (Shanghai Genomeditech Co., Ltd., Shanghai, China). Cells
were seeded into 96-well plats at amount of 100 µl per well, then
were incubated at 37°C in 5% CO2 incubator for 4 h.
Cells were added by 10 µl each well CCK reagent, then incubated at
37°C in 5% CO2 incubator for 1–4 h. The optical density
(OD) was observed at 450 nm by a microplate reader (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Flow cytometry (FCM)
Cells in TG only, ATF6 (151-366aa) + TG, and ATF6
(1-366aa) + TG groups were treated with 1 µM TG for 48 h to induce
ER stress. Cells in these three experimental groups plus the normal
control group were seeded into 6-well plates at a density of
2×104 cells/well, and digested and collected with EDTA
free trypsin (Beijing Solarbio Technology Co., Ltd., Beijing,
China). The cells were then stained with Annexin V-FITC and
propidium iodide (Qcbio Science and Technologies Co., Ltd.,
Shanghai, China), and incubated at room temperature for 15 min in a
dark place. The cultures were then analysed by EPICS XL-MCL flow
cytometry (Beckman Coulter, Fullerton, CA, USA) at an excitation
wave length of 488 nm and an emission wavelength of 530 nm. The
experiment was run three times, and the apoptosis rate for every
group was calculated.
RT-PCR
RT-PCR and SYBR Green I chemistry (Beijing Solarbio
Technology Co., Ltd.) were applied to investigate the expression of
genes in the study. Cells in each group were seeded into 6-well
plates at a density of 2×104 cells/well, the total RNA
of cells were extracted with Trizol (Thermo Fisher Scientific
Inc.), purity and concentration of the extracted RNA were measured
on a UV spectrophotometer (Thermo Fisher Scientific Inc.). cDNA was
synthesized by reverse transcription, and fluorescence quantitative
detection of the target gene was performed afterwards. β-actin was
applied as the internal control to monitor the RT-PCR efficiency.
All RT reactions were performed in triplicate. The primers were
designed by Sangon Biotech Co., Ltd. The specific primer sequences
for each gene were listed as the follows: 5′GCTGGAAAGCAGCGCATGAA3′
and 5′GCGAGTCGCCTCTACTTCCC3′ for CHOP (product: 126 bp);
5′TGTCACTGCGGGAAGGTCTC3′ and 5′AGGTGCACACATCCTTGGCT3′ for Cyt C
(product: 144 bp); 5′GCCAGCAAACTGGTGCTCAA3′ and
5′CCAACCACCCTGGTCTTGGA3′ for Bax (product: 126 bp);
5′AGTGGGATGCGGGAGATGTG3′ and 5′GGTGGACCACAGGTGGCA3′ for Bcl-2
(product: 198 bp); 5′GCCCTGTCCTCCAGGTGAAA3′ and
5′CTGGGTCCGGGTGCAGTTTA3′ for Fas (product: 187 bp) and
5′GCCGGGACCTGACTGACTAC3′ and 5′GTCAGGCAGCTCGTAGCTCT3′ for β-actin
(product: 188 bp).
Western blotting
Cells in control, non-transfected ATF6 (151-366aa)
and ATF6 (1-366aa) groups were harvested and washed twice with PBS,
and protein lysed in ice-cold radioimmunoprecipitation assay buffer
(Whiga Technology Co., Ltd., Guangdong, China) with freshly added
0.01% protease inhibitor phenylmethanesulfonyl fluoride (PMSF)
(Sigma-Aldrich Co., LLC, Darmstadt, Germany). The cells were
incubated on ice for 30 min and then centrifuged at 10,000 × g for
5 min at 4°C. The supernatant (20-30 µg of protein) was collected,
resolved by 10% sodium dodecyl sulphate polyacrylamide gel
electrophoresis (Bio-Rad Laboratories, Inc.), and subsequently
transferred to a nitrocellulose membrane via western blotting
(Millipore, Shanghai, China). Then the expression of protein
cleaved-caspase-3, cleaved-caspase-9, cleaved-PARP,
cleaved-caspase-4, cleaved-caspase-12, CHOP, cyt c, Bax, Bcl-2,
pro-caspase-8, cleaved-caspase-8, Fas, phosphorylated-JNK, JNK and
NF-κB were detected. Protein loading was estimated using mouse
anti-β-actin monoclonal antibody (Beijing Solarbio Technology Co.,
Ltd.). Blottings were visualized using enhanced chemiluminescence
(Thermo Fisher Scientific Inc.).
Statistical analysis
All values were expressed as mean ± S.D. Differences
between groups were assessed by means of variance analysis and
student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
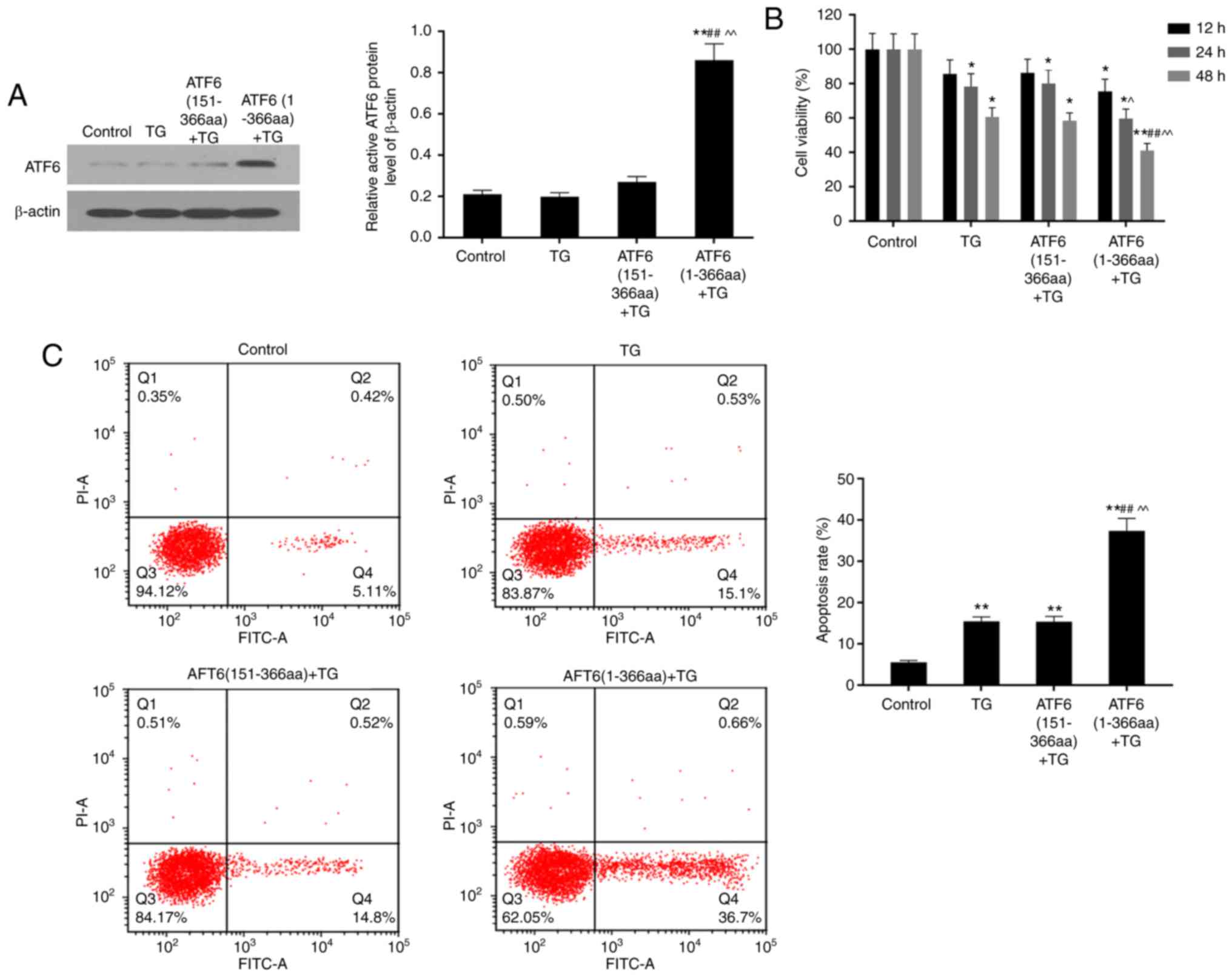
High expression of ATF6 detected in
ATF6(1-366aa)-transfected cells
In testing the transfection efficiency, expression
of ATF6 protein was detected through western blotting of ATF6
(151-3aa) and ATF6 (1-366aa) groups. The ATF6 protein was five
times more highly expressed in cells that were transfected with the
ATF6 (1-366aa) plasmid than in other groups (P<0.01) (Fig. 1A).
TG treatment for 48 h decreased
viability of VEC
Cell viabilities in TG, ATF6 (151-366aa) + TG and
ATF6 (1-366aa) + TG groups were inhibited under TG-induced ER
stress compared to that inthe control group. ATF6 (1-366aa) + TG
cells were the most sensitive to ER stress, showing the lowest
viability. Cell viability after TG treatment decreased in a
time-dependent manner. Furthermore, there was a significant
difference among groups when the cells were treated with TG for 48
h (P<0.01 or P<0.05) (Fig.
1B).
TG treatment significantly increased
cell apoptosis rate
Compared to that in the control group, cell
apoptosis rates in three TG-treated groups increased, particularly
in ATF6 (1-366aa)-transfected cells, with the highest significant
increase from 5.50±0.43 to 37.41±2.99 5% (P<0.01). Moreover, the
apoptosis rate in ATF6 (1-366aa) + TG group was 2.5 times higher
than that in the TG only and ATF6 (151-366aa) + TG groups. This
reflects that high expression of active ATF6 induces cell apoptosis
under ER stress (P<0.01) (Fig.
1C).
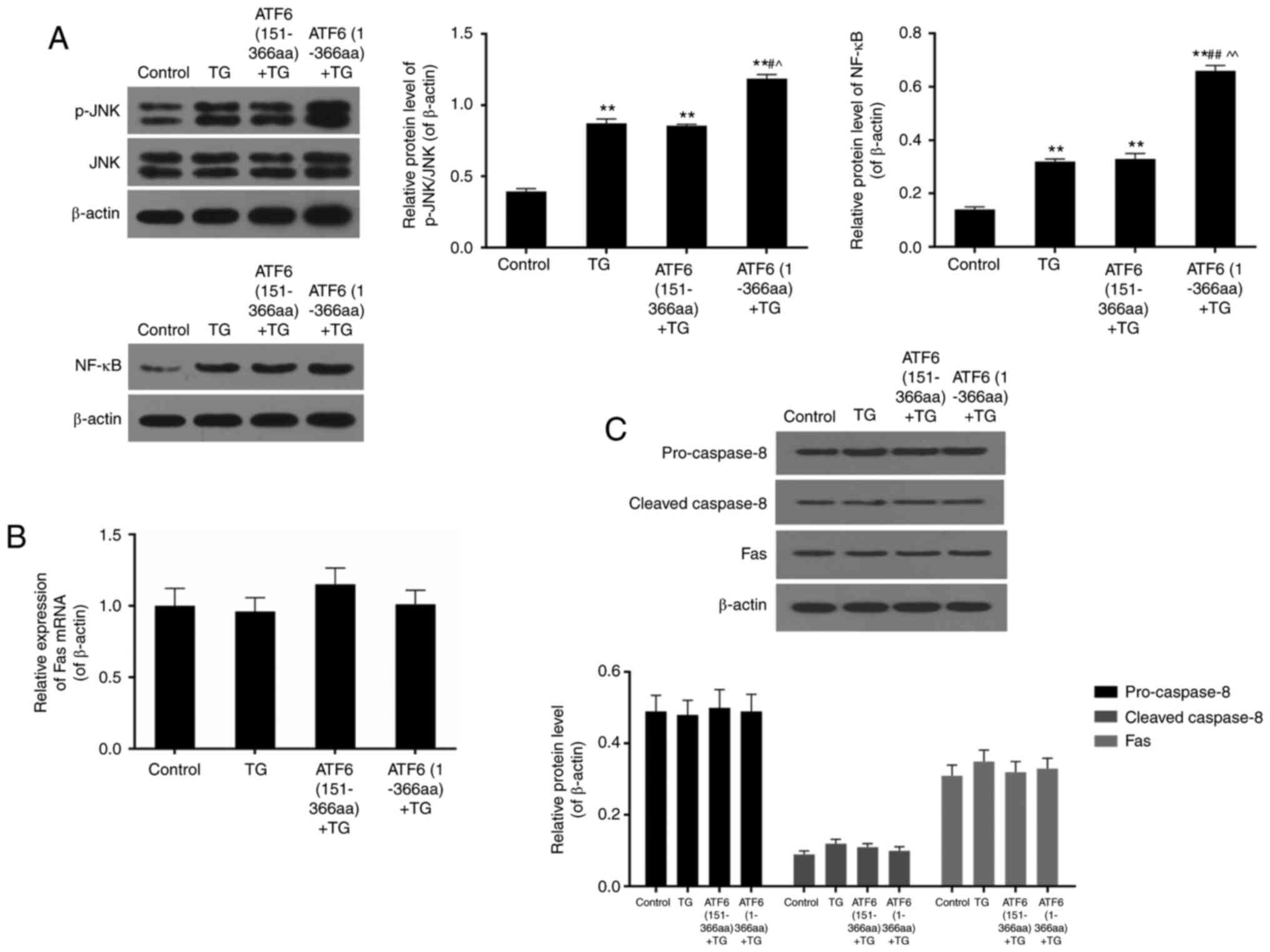
High expression of active ATF6
increased activation of caspase-3, caspase-4, caspase-9 and
caspase-12 and cleavage of PARP
TG treatment increased cleavage of caspase-3,
caspase-4, caspase-9, caspase-12, and PARP in VEC, when compared to
that in the control group (P<0.01). In ATF6 (1-366aa) + TG group
where active ATF6 was over-expressed, the protein levels of cleaved
caspase-3, caspase-4, caspase-9, caspase-12, and PARP were
significantly higher than those in TG only and ATF6 (151-366aa) +
TG groups (P<0.01) (Fig. 2A and
B).
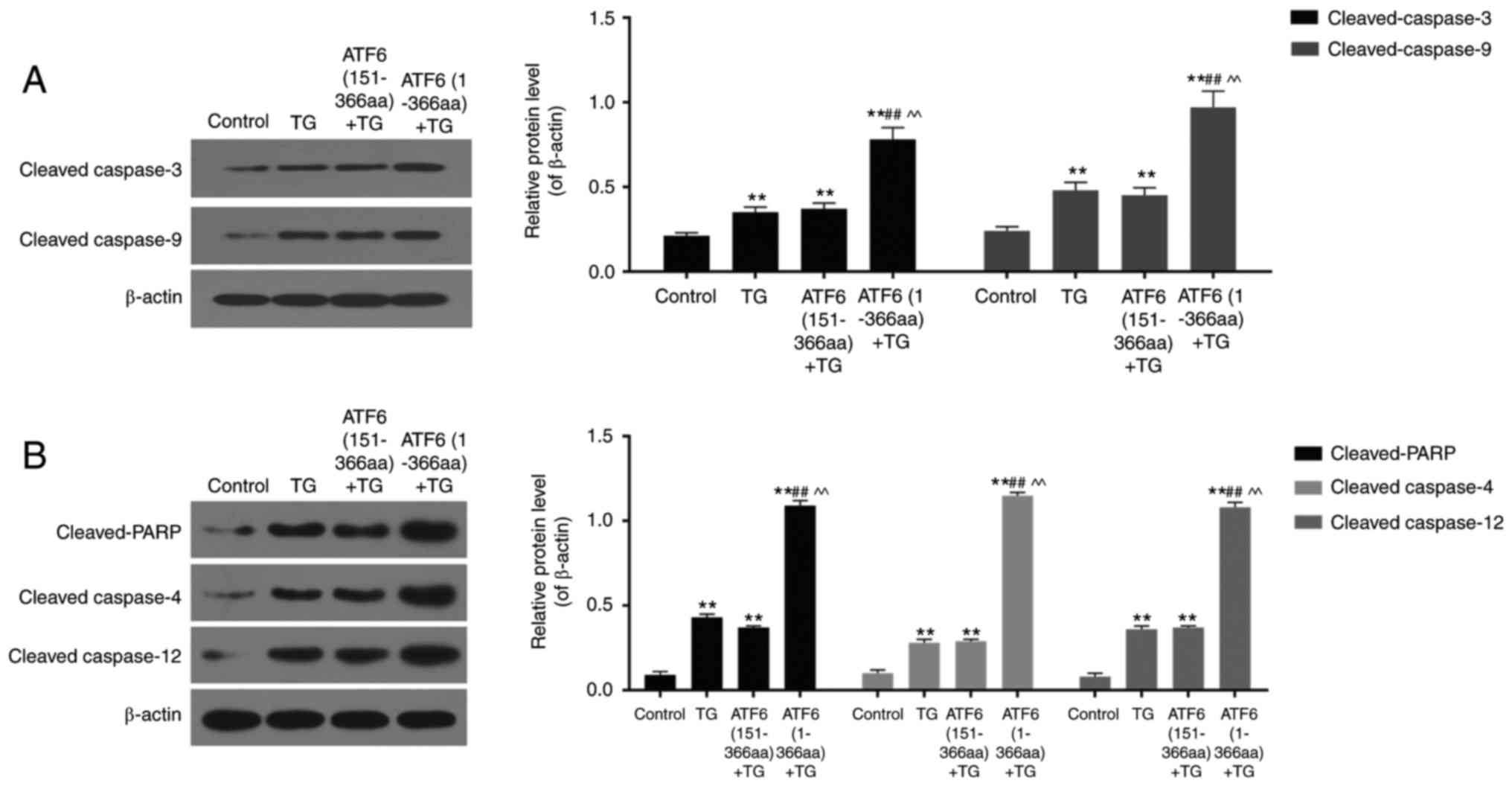 | Figure 2.Protein levels of cleaved caspase-3,
caspase-9, PARP, caspase-4 and caspase-12 in control, TG, ATF6
(151-366aa) + TG and ATF6 (1-366aa) + TG groups detected using
western blotting. (A and B) High expression of active ATF6
up-regulated protein levels of cleaved caspase-3, caspase-9, PARP,
caspase-4 and caspase-12 in ER stress. Data were presented as mean
± SD, n=3, **P<0.01 vs. control, ##P<0.01 vs. TG
treatment (1 µM for 48 h), ^^P<0.01 vs. ATF6
(151-366aa). |
High expression of active ATF6
increased expression of CHOP, cyt c, and Bax but led to decreased
Bcl-2 protein levels
RT-PCR and western blotting were conducted to
analyse the expression levels of apoptosis-related proteins,
including CHOP, cyt c, Bax, and Bcl-2. TG treatment of VECs for 48
h in TG, ATF6 (151-366aa) + TG, and ATF6 (1-366aa) + TG groups
increased mRNA and protein levels of CHOP and cyt c in comparison
with that in the control group (P<0.05). Expression of these
mRNA and proteins in ATF6 (1-366aa) + TG group was significantly
different from that in the TG and ATF6 (151-366aa) + TG groups
(P<0.01). Expression levels of Bax mRNA and protein were
up-regulated, while those of Bcl-2 were down-regulated by TG
treatment (P<0.01). mRNA expression and protein levels of CHOP
and Bax were nearly double in ATF6 over-expressing cells, while cyt
c levels were approximately three times higher. Up-regulation of
ATF6 led to a decreased expression of Bcl-2 (Fig. 3 A-E).
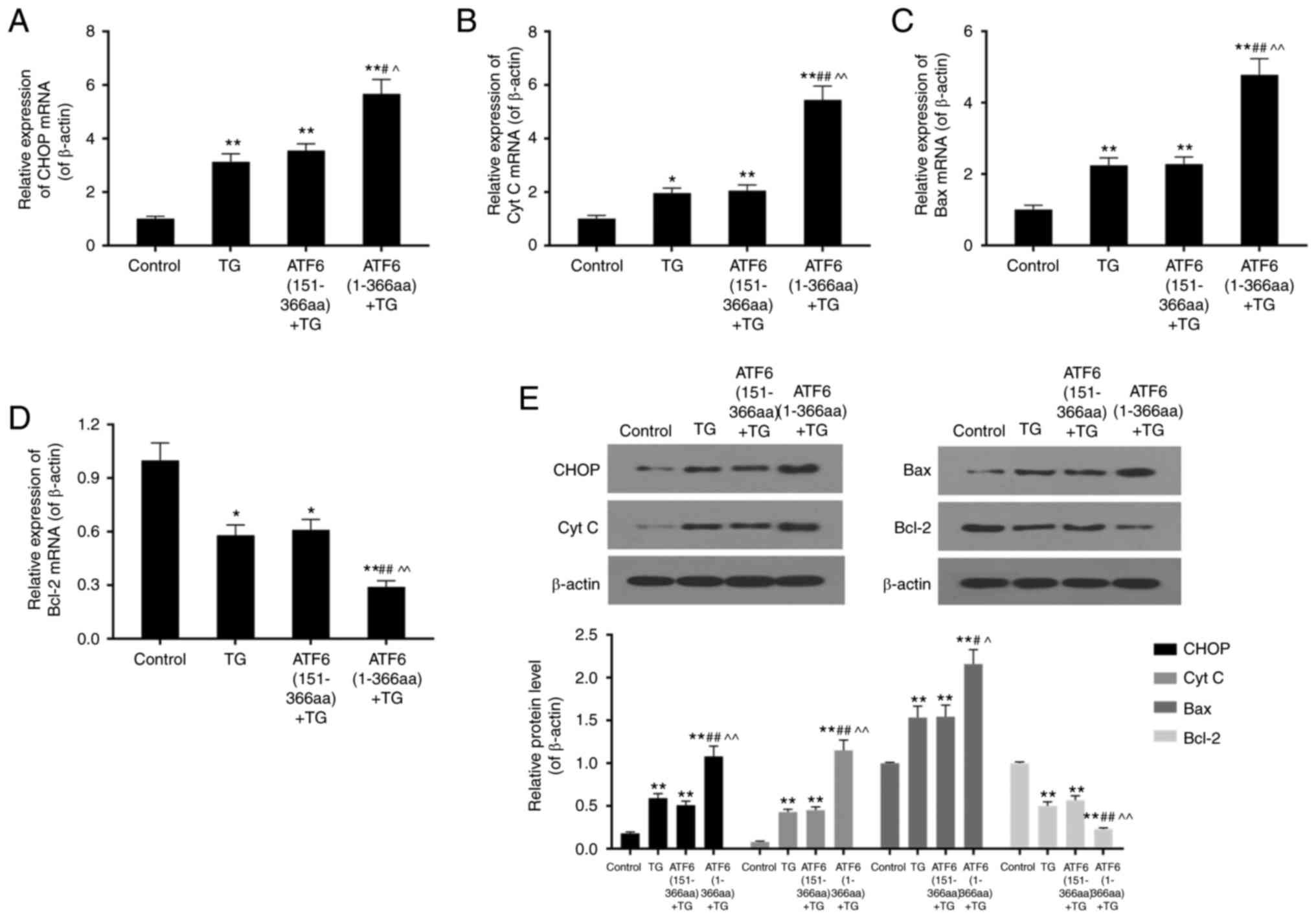 | Figure 3.mRNA expression and protein level of
apoptosis related genes CHOP, CytC, Bax, and Bcl-2 in control, TG,
ATF6 (151-366aa) + TG and ATF6 (1-366aa) + TG groups by means of
RT-PCR and western blotting. (A-C) High expression of active ATF6
up-regulated mRNA expression of CHOP, CytC, and Bax in ER stress.
(D) High expression of active ATF6 down-regulated mRNA expression
of Bcl-2 in ER stress. (E) High expression of active ATF6
up-regulated protein levels of CHOP, Cyt C and Bax, and down
regulated that of Bcl-2 in ER stress. Data were presented as mean ±
SD, n=3, *P<0.05 and **P<0.01 vs. control,
#P<0.05 and ##P<0.01 vs. TG treatment
(1 µM for 48 h), ^P<0.05 and ^^P<0.01
vs. ATF6 (151-366aa). |
High expression of active ATF6
activated JNK/NF-Κb pathway
The protein levels of total and phosphorylated JNK
as well as total NF-κB were detected by western blotting. The ratio
of p-JNK/JNK dramatically increased upon TG treatment (P<0.01).
Phosphorylation levels of JNK was further elevated when active ATF6
was up-regulated in ATF6 (1-366aa) + TG (P<0.01). High
expression of NF-κB was also observed in TG-treated groups,
especially in active ATF6-up-regulated cells where the protein
levels of NF-κB nearly doubled in comparison to that in the TG and
ATF6 (151-366aa) + TG groups (P<0.01) (Fig. 4A).
No significant effect of TG treatment
and up-regulation of ATF6 was found on expression of death
receptor-related genes
In the present study, Fas mRNA and protein were
determined using PCR and western blotting. The results showed no
significant difference in its expression among control, TG, ATF6
(151-366aa) + TG and ATF6 (1-366aa) + TG groups (P>0.05)
(Fig. 4B). Moreover, the protein
levels of pro-caspase-8 and cleaved-caspase-8 were also found to be
not affected by TG treatment and ATF6 expression (P>0.05)
(Fig. 4C).
Discussion
As VECs are involved in numerous aspects of vascular
biology, ER stress-induced VEC apoptosis may be related to numerous
pathophysiological processes and diseases (19). As a cell protection mechanism, ER
stress reactions reduce protein translation or increase expression
of ER BiP to protect cells from ER stress. However, recent research
found that if the stress levels are elevated, three pathways
including PERK, IRE-1 and ATF6 are activated to induce cell
apoptosis (20). ATF6 is one of
three sensor proteins in ER stress that induce expression of ER
BiP. It was shown that activated ATF6 can lead to natural apoptosis
of rat myocytes during cell differentiation, which means that
activated ATF6 is able to regulate cell apoptosis through
activating expression of caspase-12 (17). In rat myoblasts, it is theorised
that high expression of activated ATF6 may induce cell apoptosis
through down-regulating Mcl-1 and up-regulating WBP1. The pathways
of activated ATF6 in regulating cell apoptosis are probably
different in different cells (18). Therefore, ATF6 (1-366aa; ATF6
high-expressed plasmid) and ATF6 (151-366aa; plasmid without
transcriptional activity) were transfected into VEC to construct
ATF6 high-expression model, and TG was applied as an ER stress
inducer to explore the role of activated ATF6 in VEC under ER
stress.
Apoptosis is a self-destruction process controlled
by gene expression, which plays a crucial part in maintenance of
homeostasis (21). The
mitochondrial pathway and the death receptor-mediated pathway are
two classic cell apoptotic pathways that are regulated by caspase-9
and caspase-8, respectively. These pathways finally activate
caspase-3 to induce cell apoptosis (22). For example, when cells are
stimulated by drugs, nutrient deficiency, and oxidative stress,
caspase-9 and caspase-8 are cleaved and activated through diverse
pathways, which leads to caspase-3-induced cell apoptosis (23). Recent research reported that ER
stress could also initiate apoptotic pathways, although the
specific mechanism remained unclear (24).
It is believed that TG, a specific inducer of ER
stress, can stimulate ER stress and eventually induce apoptosis in
numerous types of cells (25). In
the present study, a reduction of VEC viability upon TG treatment
was observed, along with a significant increase in the rate of cell
apoptosis. In TG-treated VEC, cleaved-caspase-3 and
cleaved-caspase-12 levels were both elevated, which implies that TG
can induce apoptosis of VEC through ER stress.
Ito et al documented that in TG-treated mouse
embryonic fibroblasts, there is an increase in Ca2+
concentration, reduction of mitochondrial membrane potential,
release of cyt c from the mitochondria, cleavage of caspase-9 and
caspase-12, and activation of caspase-3, along with cell apoptosis.
This indicates that the mitochondrial pathway is possibly
implicated in ER stress-induced cell apoptosis (26). Mitochondria plays a crucial role in
the signal transduction process of cell apoptosis, and altering the
permeability of the mitochondrial inner membrane is a necessary and
sufficient requirement for cell apoptosis (27). In response to external stimuli,
permeability of the mitochondrial inner membrane is changed, and
pro-caspase-9 is activated by released cyt c to activate other
caspases such as caspase-3, which results in cell apoptosis
(28). Our study found that upon
TG treatment, cyt c expression was up-regulated with activation of
caspase-3, caspase-4, caspase-9, caspase-12, and PARP.
Over-expression of ATF6 further increased their protein levels as
well as that of CHOP. Moreover, activation of JNK/NF-κB was also
detected in ER stress-induced cell apoptosis, especially during
ATF6 over-expression, which further supports the role of the
mitochondrial pathway.
On the other hand, we found that in VEC with highly
expressed and activated ATF6, the expression of mRNA and protein
levels of death receptor-related proteins caspase-8 and Fas was not
significantly increased in comparison with that in the control
groups. This indicates that the death receptor-mediated pathway may
not take part in the ATF6-induced apoptotic process. After the cell
was stimulated for apoptosis, cyt c was rapidly released from the
mitochondria into the cytoplasm, which is a key event in cell
apoptosis (29). In the present
study, expression of cyt c and caspase-9 was significantly
increased in active ATF6 highly expressing cells, which supports
the relationship between ATF6 up-regulation and cyt c release and
subsequently, caspase-9 activation.
In mammalian cells, the Bcl-2 family plays an
important role in the mitochondrial pathway-mediated cell
apoptosis. Different family members play different roles in the
apoptotic process. The interaction of anti-apoptotic genes and
pro-apoptotic genes is influenced by different levels of reactive
oxygen species (ROS), so the relative ratio of pro-apoptotic genes
and anti-apoptotic genes of a cell is the key factor in deciding
cellular survival or death. At the centre of apoptosis regulation
by the Bcl-2 family, lie Bcl-2 and Bax. The expression of Bcl-2 and
Bax proteins was used asa marker for apoptosis in the present study
(30,31). In tumour cells, many
apoptosis-stimulating factors induce the mitochondrial
pathway-mediated cell apoptosis through down-regulating the
expression of apoptosis inhibitor proteins such as Bcl-2 and
Bcl-xL, or up-regulating expression of pro-apoptotic
proteins like Bax, Bad, and Bid (32). In the present study, we found that
the expression of Bax was significantly up-regulated when levels of
Bcl-2 were down-regulated in active ATF6 high expressing cells,
which indicated that active ATF6-mediated mitochondrial pathway
regulates cell apoptosis through down-regulation of the Bcl-2/Bax
ratio.
Fas and caspase-8 are two of main members in death
receptor signalling pathway. Transmembrane receptor Fas belongs to
tumor necrosis factor super family, and induces apoptosis of
numerous types of cells. The death domain in the intracellular
region of Fas plays a crucial part in transduction of apoptotic
signal. Research has reported that Fas is extensively expressed in
immune cells such as activated T cells, B cells, natural killer
cell and monocyte, and is also found in histocytes, endothelial
cells and epithelial cells (33).
Fas combines with Fas ligand (FasL), and links with the death
domain of Fas-associated death domain protein (FADD) (34). The death effector domain in the
n-terminal of FADD peptide chain can gather caspase-8 zymogen to
inform death inducing signalling complex (DISC) with Fas receptor,
leading to activation of caspase-8 and eventually resulting in
activation of caspase-3. Activated caspase-3, as the key effector
enzyme in apoptosis signal transduction, induces cell apoptosis at
the end (35,36). The result of the present study
manifested no significant difference in the levels of Fas as well
as pro- and cleaved-caspase-8 among experimental groups. It was
implied that ER stress-induced VEC apoptosis is likely not through
the death receptor signalling pathway, and ATF6 does not affect the
activation of the pathway.
The present study probed into the role of active
ATF6 in response to ER stress-induced VEC apoptosis, and
demonstrated that high expression of active ATF6 could aggravate
VEC apoptosis through mitochondrial apoptotic pathway. The main
limitation, however, is no comparison of the influences of high
expression of active ATF6 between cells with the coding plasmid but
no TG and those which are submitted to stressful condition. Whether
ATF6 alone is implicated in the cause of apoptosis in VEC or any
other cell type is required to be further studied.
References
|
1
|
Mannarino E and Pirro M: Endothelial
injury and repair: A novel theory for atherosclerosis. Angiology.
59 2 Suppl:69S–72S. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Luchetti F, Crinelli R, Cesarini E,
Canonico B, Guid L, Zerbinati C, Di Sario G, Zamai L, Magnani M,
Papa S and Iuliano L: Endothelial cells, endoplasmic reticulum
stress and oxysterols. Redox Biol. 13:581–587. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Battson ML, Lee DM and Gentile CL:
Endoplasmic reticulum stress and the development of endothelial
dysfunction. Am J Physiol Heart Circ Physiol. 312:H355–H367. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Verkhratsky A and Toescu EC: Endoplasmic
reticulum Ca(2+) homeostasis and neuronal death. J Cell Mol Med.
7:351–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marciniak SJ and Ron D: Endoplasmic
reticulum stress signaling in disease. Physiol Rev. 86:1133–1149.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Verras M, Papandreou I, Lim AL and Denko
NC: Tumor hypoxia blocks Wnt processing and secretion through the
induction of endoplasmic reticulum stress. Mol Cell Biol.
28:7212–7224. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kammoun HL, Hainault I, Ferré P and
Foufelle F: Nutritional related liver disease: Targeting the
endoplasmic reticulum stress. Curr Opin Clin Nutr Metab Care.
12:575–582. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fu S, Yang L, Li P, Hofmann O, Dicker L,
Hide W, Lin X, Watkins SM, Ivanov AR and Hotamisligil GS: Aberrant
lipid metabolism disrupts calcium homeostasis causing liver
endoplasmic reticulum stress in obesity. Nature. 473:528–531. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He S, Yaung J, Kim YH, Barron E, Ryan SJ
and Hinton DR: Endoplasmic reticulum stress induced by oxidative
stress in retinal pigment epithelial cells. Graefes Arch Clin Exp
Ophthalmol. 246:677–683. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu J and Kaufman RJ: From acute ER stress
to physiological roles of the unfolded protein response. Cell Death
Differ. 13:374–384. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Su HL, Liao CL and Lin YL: Japanese
encephalitis virus infection initiates endoplasmic reticulum stress
and an unfolded protein response. J Virol. 76:4162–4171. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pahl HL: Signal transduction from the
endoplasmic reticulum to the cell nucleus. Physiol Rev. 79:683–701.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Patil C and Walter P: Intracellular
signaling from the endoplasmic reticulum to the nucleus: The
unfolded protein response in yeast and mammals. Curr Opin Cell
Biol. 13:349–355. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bertolotti A, Zhang Y, Hendershot LM,
Harding HP and Ron D: Dynamic interaction of BiP and ER stress
transducers in the unfolded-protein response. Nat Cell Biol.
2:326–332. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schroder M and Kaufman RJ: The mammalian
unfolded protein response. Annu Rev Biochem. 74:739–789. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakanishi K, Sudo T and Morishima N:
Endoplasmic reticulum stress signaling transmitted by ATF6 mediates
apoptosis during muscle development. J Cell Biol. 169:555–560.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Morishima N, Nakanishi K and Nakano A:
Activating transcription factor-6 (ATF6) mediates apoptosis with
reduction of myeloid cell leukemia sequence 1 (Mcl-1) protein via
induction of WW domain binding protein 1. J Biol Chem.
286:35227–35235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Toya SP and Malik AB: Role of endothelial
injury in disease mechanisms and contribution of progenitor cells
in mediating endothelial repair. Immunobiology. 217:569–580. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thompson CB: Apoptosis in the pathogenesis
and treatment of disease. Science. 267:1456–1462. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Colell A, Ricci JE, Tait S, Milasta S,
Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A,
Waterhouse NJ, Li CW, et al: GAPDH and autophagy preserve survival
after apoptotic cytochrome c release in the absence of caspase
activation. Cell. 129:983–997. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rathmell JC and Kornbluth S: Filling a
GAP(DH) in caspase-independent cell death. Cell. 129:861–863. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Momoi T: Conformational diseases and ER
stress-mediated cell death: Apoptotic cell death and autophagic
cell death. Curr Mol Med. 6:111–118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feng XQ, You Y, Xiao J and Zou P:
Thapsigargin-induced apoptosis of K562 cells and its mechanism.
Zhongguo Shi Yan Xue Ye Xue Za Zhi. 14:25–30. 2006.(In Chinese).
PubMed/NCBI
|
|
26
|
Ito Y, Pandey P, Mishra N, Kumar S, Narula
N, Kharbanda S, Saxena S and Kufe D: Targeting of the c-Abl
tyrosine kinase to mitochondria in endoplasmic reticulum
stress-induced apoptosis. Mol Cell Biol. 21:6233–6242. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rizzuto R, Pinton P, Carrington W, Fay FS,
Fogarty KE, Lifshitz LM, Tuft RA and Pozzan T: Close contacts with
the endoplasmic reticulum as determinants of mitochondrial Ca2+
responses. Science. 280:1763–1766. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Adams JM and Cory S: Apoptosomes: Engines
for caspase activation. Curr Opin Cell Biol. 14:715–720. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hengartner MO: Apoptosis. Death cycle and
Swiss army knives. Nature. 391:441–442. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Al-Fatlawi AA, Al-Fatlawi AA, Irshad M,
Zafaryab M, Rizvi MM and Ahmad A: Rice bran phytic acid induced
apoptosis through regulation of Bcl-2/Bax and p53 genes in HepG2
human hepatocellular carcinoma cells. Asian Pac J Cancer Prev.
15:3731–3736. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Płonka J, Latocha M, Kuśmierz D and
Zielińska A: Expression of proapoptotic BAX and TP53 genes and
antiapoptotic BCL-2 gene in MCF-7 and T-47D tumour cell cultures of
the mammary gland after a photodynamic therapy with photolon. Adv
Clin Exp Med. 24:37–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Korsmeyer SJ, Shutter JR, Veis DJ, Merry
DE and Oltvai ZN: Bcl-2/Bax: A rheostat that regulates an
anti-oxidant pathway and cell death. Semin Cancer Biol. 4:327–332.
1993.PubMed/NCBI
|
|
33
|
Yamana K, Bilim V, Hara N, Kasahara T,
Itoi T, Maruyama R, Nishiyama T, Takahashi K and Tomita Y:
Prognostic impact of FAS/CD95/APO-1 in urothelial cancers:
Decreased expression of Fas is associated with disease progression.
Br J Cancer. 93:544–551. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ashkenazi A and Dixit VM: Death receptors:
Signaling and modulation. Science. 281:1305–1308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Scatena R, Bottoni P, Botta G, Martorana
GE and Giardina B: The role of mitochondria in pharmacotoxicology:
A reevaluation of an old, newly emerging topic. Am J Physiol Cell
Physiol. 293:C12–C21. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gehring S, Rottmann S, Menkel AR,
Mertsching J, Krippner-Heidenreich A and Lüscher B: Inhibition of
proliferation and apoptosis by the transcriptional repressor Mad1.
Repression of Fas-induced caspase-8 activation. J Biol Chem.
275:10413–10420. 2000. View Article : Google Scholar : PubMed/NCBI
|