Introduction
Cerebral hypoxic ischemia is a leading cause of
numerous neurological diseases, including cerebral epilepsy, palsy
and cognitive disabilities associated with high mortality and
morbidity (1–3). Matsumoto et al (4) reported that ischemia may cause
neuronal cell apoptosis in the central nervous system due to an
excess of L-glutamate. However, ischemic-associated release of
L-glutamate may be inhibited by the activation of γ-aminobutyric
acid (GABA) receptors (4,5). GABA- and glutamate-associated
transporters serve roles in the maintenance of extracellular GABA
and glutamate levels (4). However,
in abundance, these transporters have been associated with numerous
pathological brain conditions, in particular ischemia and
epilepsy.
Glutamate transporters serve crucial functions in
the prevention of neuronal cell death by reducing
glutamate-associated toxicity. Five associated glutamate
transporters have been reported: Glutamate-aspartate
transporter/excitatory amino acid transporter (EAAT)1, glial
glutamate transporter (GLT)-1/EAAT2, EAA carrier (EAAC)1/EAAT3,
EAAT4 and EAAT5 (6,7). The functional relevance of GLT-1 was
clearly demonstrated in a study using GLT1 knockout mice, which
developed severe epilepsy (8).
However, the roles of EAAC1 in neuronal death have not been
resolved compared with glial glutamate transporters, including
GLT-1. In addition to maintaining extracellular glutamate, a
previous study investigating EAAC1-deficient mice revealed that
EAAC1 can function as a cysteine transporter and maintain neuronal
glutathione homeostasis (9).
Additionally, EAAC1 has been reported to protect injured motor
neurons via interactions with holocytochrome c synthetase (10). EAAC1 may therefore perform
additional functions in addition to glutamate homeostasis.
Sodium- and chloride-dependent GABA transporter 1
(GAT1) is primarily responsible for the removal of GABA from the
synaptic cleft and termination of GABA-mediated neurotransmission
(11). Chronic neurological
abnormalities that develop following hypoxia at an early age may be
associated with alterations of GAT functions (12–15).
A previous study reported that the protein expression levels of
GAT1 were reduced within the brains of thrombotic infarct rat
models (16). The interplay
between these transporters and ischemia require further
investigation. The present study aimed to investigate the
expression and associated functions of GAT1 and EAAC1 under hypoxic
conditions in vivo and in vitro. However, the
interplay between these transporters in ischemia remains unclear.
The present study aimed to investigate the expression and
associated functions of GAT1 and EAAC1 under hypoxia in vivo
and in vitro.
Materials and methods
Middle cerebral artery occlusion
(MCAO) animal models
14 male Sprague-Dawley rats (250–300 g; Experimental
Animal Center, Shanghai Jiao Tong University School of Medicine,
Shanghai, China) were housed in a climate-controlled room (25°C,
50% humidity), 5 rats per cage; food and water was provided ad
libitum under a 12 h-light/dark cycle. The animal study protocol
was approved by the ethics committee of Shanghai Pudong Hospital
(Shanghai, China) and was conducted according to guidelines of the
Animal Experimentation group of Shanghai Jiao Tong University
School of Medicine.
Rats were randomly allocated into the following
groups (n=6 in sham group, n=8 in MACO group): A sham-operated
control group, which underwent the operation with no occlusion; and
a 24 h post-MCAO/reperfusion group. Briefly, rats were anesthetized
with an intraperitoneal (i.p.) injection of 50 mg/kg pentobarbital
and placed in a stereotaxic instrument (Advanced Scientifics Inc.;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). A 28-gauge
stainless steel injection cannula was inserted into the right
lateral ventricle as previously described (17). MCAO rat models were generated as
previously described (18).
Briefly, a 3-0 monofilament nylon suture (Beijing Shandong
Technology Co., Ltd., Beijing, China) with a heat-treated rounded
tip was introduced into the right internal carotid artery via the
external carotid artery until slight resistance was achieved. The
suture was maintained in place for 90 min and was then withdrawn to
facilitate reperfusion.
24 h following the onset of reperfusion, the rats
were deeply anesthetized using sodium pentobarbital (100 mg/kg,
i.p.). For mRNA and protein expression analysis, rats were
transcardially perfused with 4°C normal saline; regions
corresponding to the ischemic core and penumbra were dissected on
ice using previously described methods (18). For immunostaining, the rats were
transcardially perfused with 4°C normal saline and fixed with 4%
paraformaldehyde (PFA; pH=7.4). Brains were extracted and
post-fixed in 4% PFA, the tissues were subsequently cryoprotected
in 20% sucrose, followed by 30% sucrose at 4°C for 48 h. Serial
coronal sections (25 µm) were collected between −1.80 and −4.80 mm
bregma levels. Every fifth section from a total of 24 sections was
selected for staining.
Cells and cell culture
Primary neurons derived from rat were cultured in
Dulbecco's modified Eagle's medium (Thermo Fisher Scientific)
supplemented with 10% fetal bovine serum (PAA Laboratories; GE
Healthcare, Chicago, IL, USA). Briefly, brain tissue was removed
from freshly euthanized rats into a cold, buffered salt solution.
Using a dissecting microscope, cerebral cortex can be carefully
isolated for further processing. The dissected tissue was first
minced using a scalpel or scissors. The resulting tissue pieces
were then transferred to a new container. A proteolytic enzyme
solution including trypsin and papain were then added to digest the
extracellular matrix proteins that bind cells together. Following a
short incubation of 15 min in a warm incubator of 37°C, tissue
pieces were gently washed with buffer to remove the enzymes. The
softened tissue pieces were dissociated by trituration, which
involved passing the tissue through a pipet, multiple times so that
cells become a single cell suspension. At this point, cells were
counted and checked for viability by Trypan blue. Cells were
cultured at 37°C in a humidified atmosphere containing 5%
CO2. To simulate a hypoxic environment, dissociated
cells were seeded in a 10-cm dish with or without 250 µM
CoCl2 treatment for 24 or 48 h at 37°C prior to
analysis.
Immunofluorescence
Slides were fixed with 4% PFA at 4°C for 15 min and
incubated with PBS containing 0.1% saponin (Beyotime Institute of
Biotechnology, Himen, China) and 1% normal goat serum or 2% normal
donkey serum (Beyotime Institute of Biotechnology) at room
temperature for 30 min. Slides were then incubated with primary
mouse polyclonal anti-EAAC1 (1:500; cat. no. orb149931; Biorbyt,
Ltd., Cambridge, UK), or GAT1 (1:500; cat. no. ab426; Abcam,
Cambridge, UK) at 4°C overnight. Slides were subsequently washed
and incubated with secondary fluorescent Alexa-Fluor-488-conjugated
goat anti-mouse or rabbit immunoglobulin G (cat. no. a24920/a24922;
1:100; Thermo Fisher Scientific Inc.) in a dark room for 2 h at
room temperature. DAPI staining (Beyotime Institute of
Biotechnology) was used for nuclear staining. Slides were observed
with a laser-scanning confocal microscope (TCS SP5; Leica
Microsystems, Inc., Buffalo Grove, IL, USA).
Lentiviral-mediated short hairpin RNA
(shRNA) gene knockdown or overexpression
Primary neurons exhibiting stable knockdown of EAAC1
or GAT1 were generated via transduction with a lentiviral-mediated
expression-specific target shRNA (109 TU/ml; HanYin
Biotech, Shanghai, China). Lentivirus containing an empty vector
was used as a negative control (NC, HanYin Biotech). The targeted
knockdown sequence for EAAC1 was 5′-caa caa tgt ctg aga aca a-3′
and for GAT1 was 5′-cca aat gac aga tgg gct a-3′. Cells were seeded
in six-cm dishes at a density of 5×105. Cells were then
infected with the same titer virus with 8 µg/ml
Polybrene® (HanYin Biotech) on the following day.
Overexpression of EAAC1 or GAT1 was generated following infection
of cells with EAAC1- or GAT1-expressing lentiviruses
(108 TU/ml, HanYin Biotech) with 8 µg/ml
Polybrene® (HanYin Biotech) on the following day. After
48 h, cells were harvested, and the knockdown or overexpression
efficiency was determined using reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and western blotting.
RT-qPCR
Cellular RNA was isolated using TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Subsequently, DNA was removed from the samples via DNase
treatment (DNA-free kit; Ambion; Thermo Fisher Scientific, Inc.)
and cDNA was synthesized from the purified RNA using a Moloney
murine leukemia virus RT kit (Promega Corporation, Madison, WI,
USA). GAPDH primer sets were used as a normalization control.
Primer sequences were as follows: GAT1 forward,
5′-GCAATCGCCGTGAACTCTTC-3′ and reverse,
5′-AGGAAATGGAGACACACTCAAAGA-3′; EAAC1 forward,
5′-CTCCACCACCGTCATTGCT-3′ and reverse, 5′-TGGCAGGCTTCACTTCTTCAC-3′;
GAPDH forward, 5′-GTATGTCGTGGAGTCTACTG-3′ and reverse,
5′-CTTGAGGGAGTTGTCATATTTC-3′. RT-qPCR cycling conditions were:
Initial denaturation for 3 min at 95°C followed by 45 cycles of
95°C (10 sec) and 58°C (45 sec), and data were acquired at the end
of the annealing/extension phase. RT-qPCR was performed in
triplicate using the SYBR-Green PCR Master Mix (Applied Biosystems)
on a 7900HT Fast Real-Time PCR machine according to the
manufacturer's protocol (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The relative quantification method
(2−ΔΔCq) (19) was used
to analyze quantitative RT-PCR data using GAPDH as normalizer. NC
were served as a reference.
Western blot analysis
Radioimmunoprecipitation assay buffer, a protease
inhibitor cocktail and a phosphorylation inhibitor cocktail
(Beyotime Institute of Biotechnology) were used to extract total
protein from cells. Protein concentration was determined by BCA
method. A total of 30 µg protein was separated by 10–15% SDS-PAGE
and was transferred onto a nitrocellulose membrane (EMD Millipore,
Billerica, MA, USA). The membrane was blocked with 5% non-fat milk
30 min at room temperature. The membrane was further incubated with
primary antibodies [EAAC1, (1:1,000; cat. no. orb149931; Biobyt,
Ltd.) GAT1 (1:1,000) and Actin (1:3,000; both Abcam)] overnight at
4°C and subsequently incubated with HRP conjugated anti-mouse or
rabbit immunoglobulin G (1:10,000) for 1 h at room temperature. The
signal was observed and developed with a Kodak film (Kodak,
Rochester, NY, USA) via enhanced chemiluminescence plus western
blotting detection reagent (Amersham; GE Healthcare). β-actin was
used as a control.
Apoptosis assay
An Annexin V-phycoerythrin (PE) Apoptosis Detection
kit (BD Biosciences, Franklin Lakes, NJ, USA) was employed to
assess apoptosis according to the manufacturer's protocol. Briefly,
cells treated with or without CoCl2 (control, CK) were resuspended
in 1X Binding Buffer at a concentration of 1×106
cells/ml, and 100 µl of this suspension was added to each of the
following tubes: i) An empty tube, ii) a tube containing Annexin
V-PE reagent (5 µl); iii) a tube containing 7-aminoactinomycin D
(AAD) reagent (5 µl); and iv) a tube containing Annexin V-PE
reagent (5 µl) and 7-AAD reagent (5 µl). The tubes were gently
vortexed and were incubated for 15 min at room temperature in the
dark. 1X Binding Buffer (400 µl) was then added to each tube and
the cells were analyzed by flow cytometry (Beckman gallios,
flowjo10.07).
Statistical analysis
Statistical analysis was performed using the
Student's t-test for comparison between two groups and one-way
analysis of variance followed by and a Tukey test for multiple
comparisons. P≤0.05 was considered to indicate a statistically
significant difference.
Results
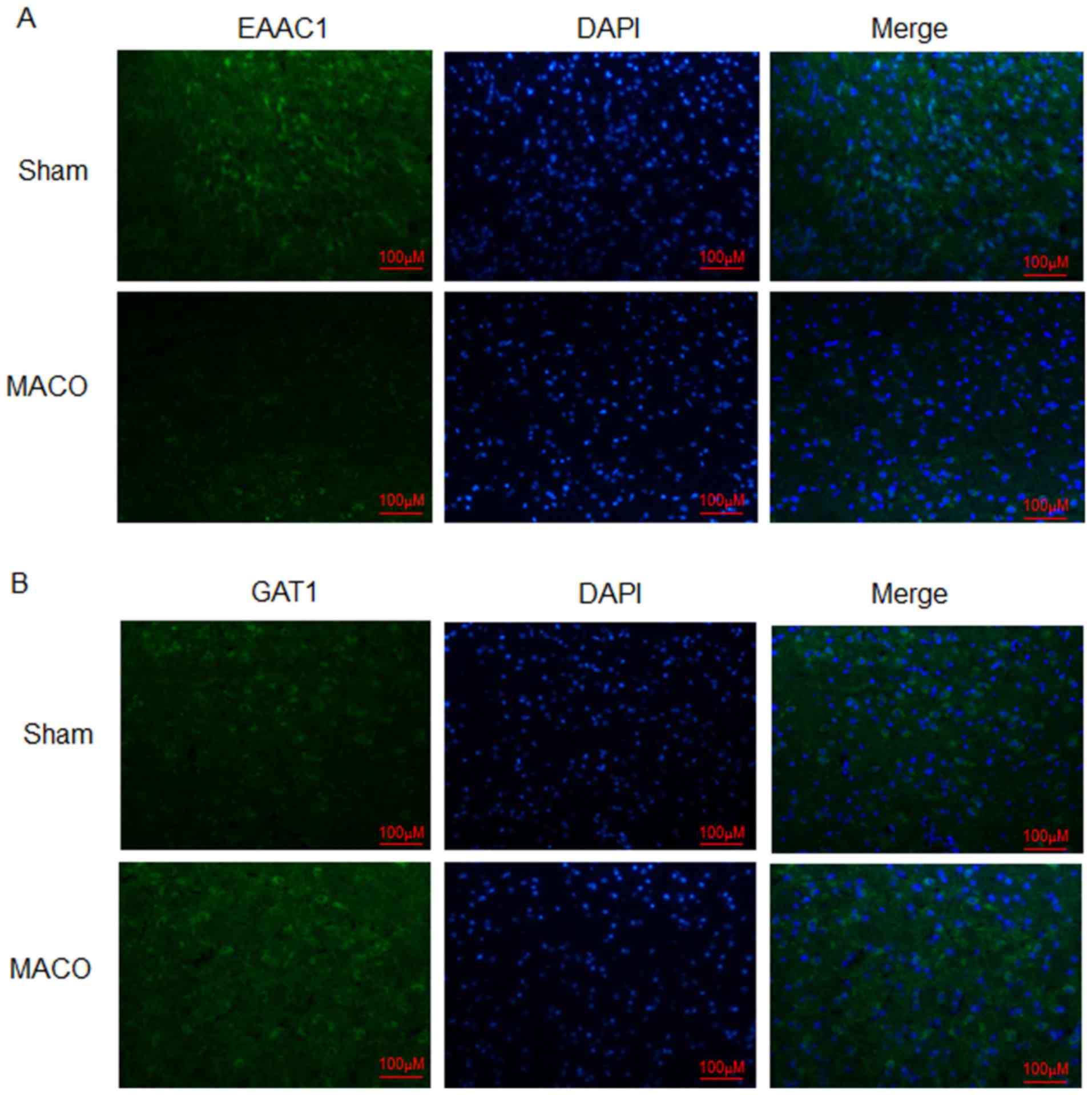
Expression levels of EAAC1 and GAT1
within the penumbra in a rat model of MCAO/reperfusion
Expression levels of EAAC1 and GAT1 within the
penumbra were analyzed via immunofluorescence. The results of the
present study revealed that the protein expression levels of EAAC1
were markedly reduced at 24 h post-MCAO/reperfusion compared with
the sham group (Fig. 1A). Protein
expression levels of GAT1 were increased compared with the sham
group (Fig. 1B). These results
indicated that EAAC1 and GAT1 may serve different functions in
hypoxia-induced ischemia.
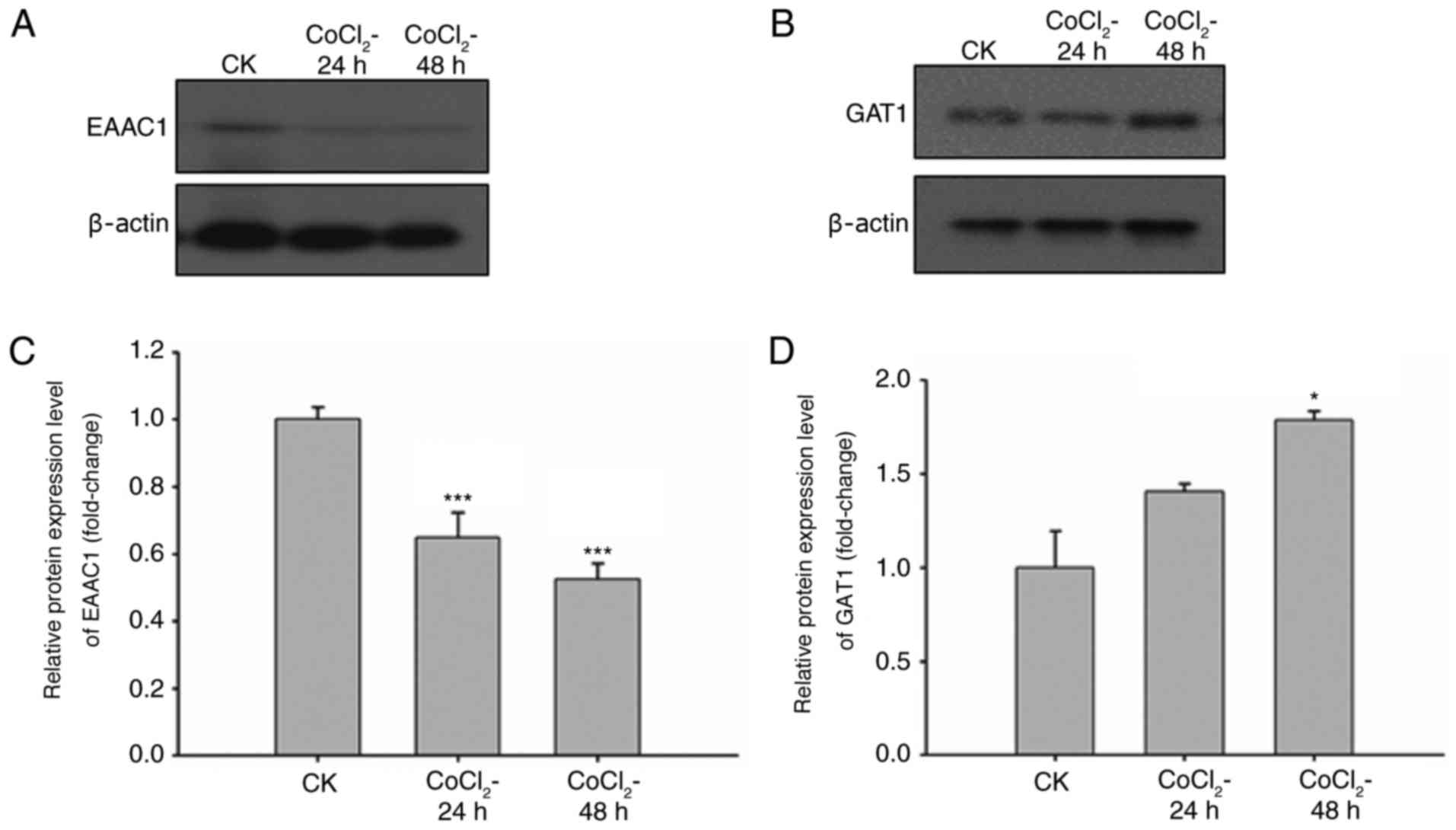
Expression levels of EAAC1 and GAT1
are influenced by hypoxic stimuli within neuronal cells
The pathogenesis of ischemia-induced brain injury is
mainly caused by neuronal death in oxygen- and energy-deficient
environments. Therefore, the expression levels of EAAC1 and GAT1
under hypoxic conditions were analyzed. CoCl2 is a
commonly used agent to induce oxygen failure. Neuronal cells were
treated with CoCl2 for 24 or 48 h, and the expression
levels of EAAC1 and GAT1 were detected through western blot
analysis and RT-qPCR. The results revealed that the protein
expression levels of EAAC1 were reduced in a time-dependent manner
in response to CoCl2 (Fig.
2A). GAT1 protein expression levels were slightly increased
following CoCl2 treatment (Fig. 2B). The mRNA expression levels of
EAAC1 and GAT1 were detected via RT-qPCR following CoCl2
treatment for 24 and 48 h. Compared with the untreated cells, mRNA
levels of EAAC1 were significantly reduced and those of GAT1 were
increased in response to CoCl2 treatment (Fig. 2C and D). The expression levels of
EAAC1 and GAT1 within primary neuronal cells were altered following
exposure to hypoxia; therefore, EAAC1 and GAT1 may serve important
roles in the hypoxia-induced ischemia.
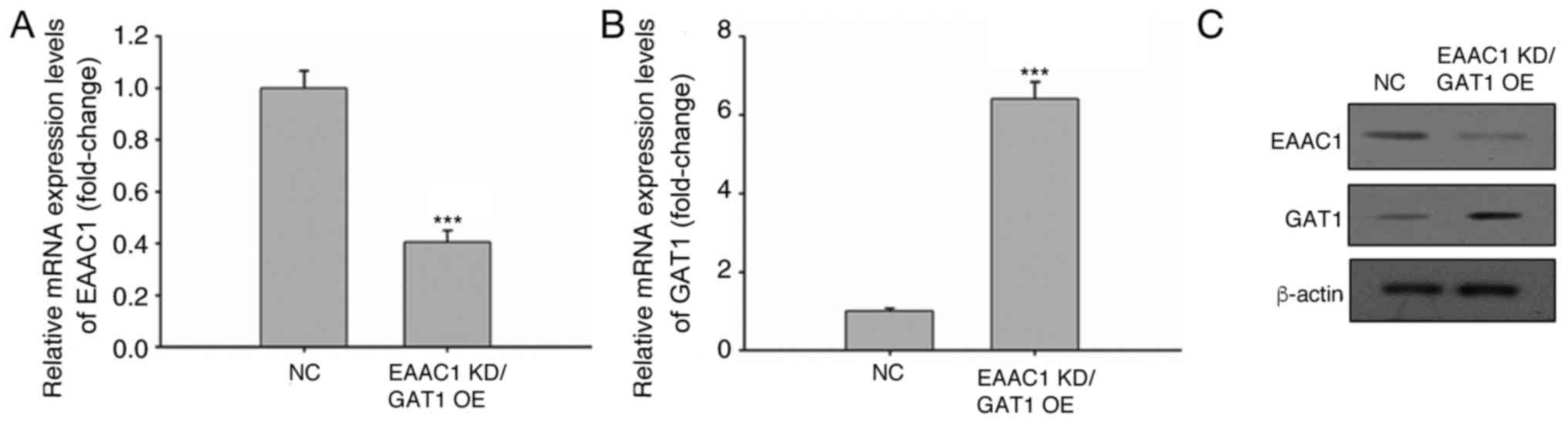
EAAC1 suppression and GAT1
overexpression increases apoptosis under hypoxic conditions
Following CoCl2 treatment, the mRNA and
protein expression levels of EAAC1 and GAT1 were reduced and
elevated, respectively. The effects of EAAC1 knockdown and
increased GAT1 expression on neuronal cell apoptosis were
subsequently investigated. EAAC1 expression was targeted within
neuronal cells via a lentiviral shRNA gene knockdown system, which
demonstrated a >60% decrease in EAAC1 expression (Fig. 3A). Lentivirus-mediated gene
overexpression significantly increased GAT1 expression (Fig. 3B). The protein expression levels of
EAAC1 and GAT1 were also confirmed via western blotting (Fig. 3C).
Subsequently, cells were treated with
CoCl2 and analyzed with a fluorescence-activated cell
sorting (FACS) apoptosis assay. Analysis indicated that EAAC1
suppression with GAT1 overexpression elevated neuronal death under
hypoxic conditions compared with the negative control group. As
presented in Fig. 4, the
percentage of apoptotic cells was significantly increased compared
with control cells following CoCl2 treatment.
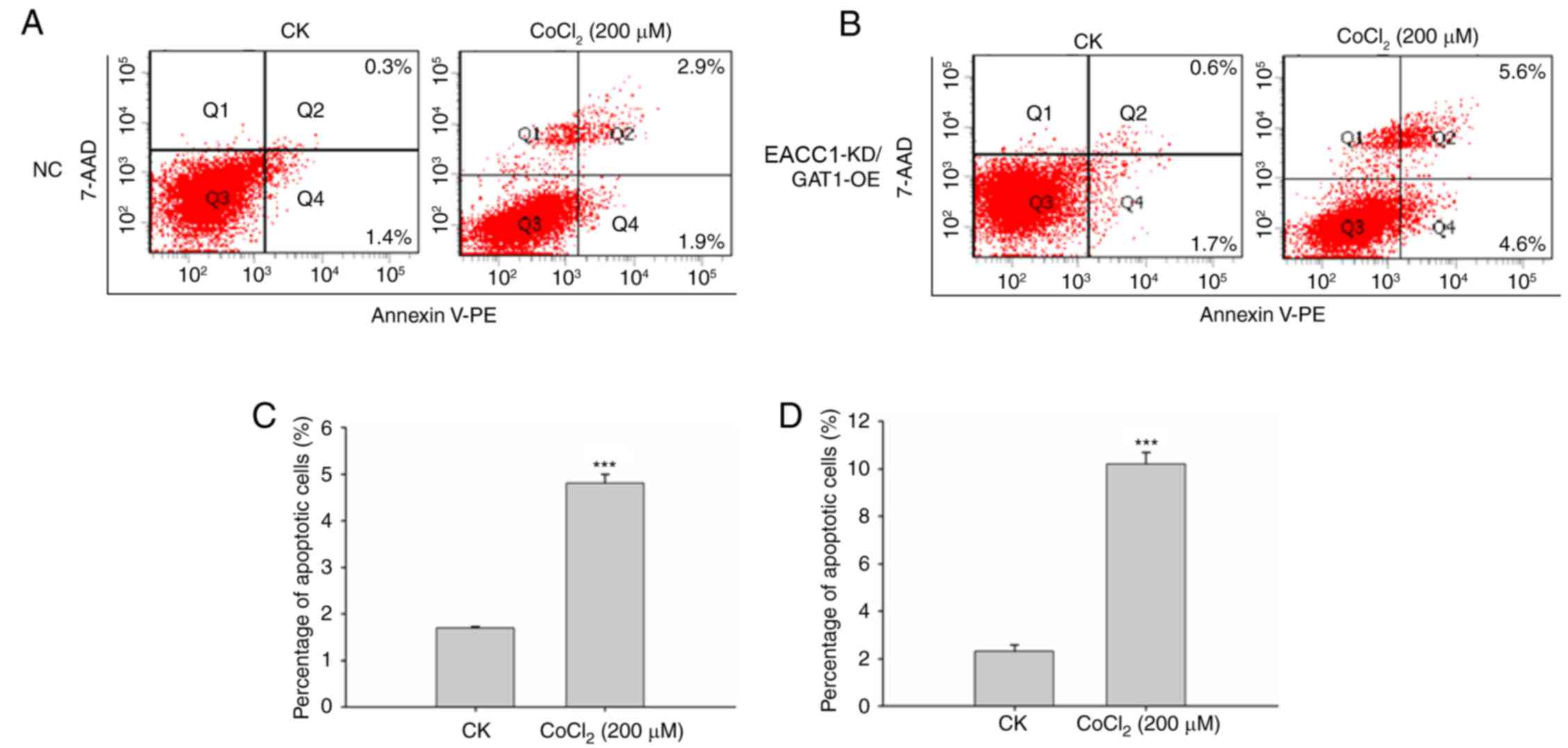 | Figure 4.EAAC1 suppression with overexpression
of GAT1 elevates neuronal death under hypoxia. Primary neuronal
cells transduced with the (A) NC lentiviral vector or (B) EAAC1
shRNA/GAT1 RNA were treated with or without CoCl2 for 48
h, apoptosis was detected by a fluorescence-activated cell sorting
assay. Number of apoptotic cells following transduction with (C)
negative control lentiviral vector or (D) EAAC1 shRNA/GAT1.
Apoptotic rate is expressed as the mean percentage of apoptotic
cells ± standard deviation. 7-AAD, 7-aminoactinomycin D; EAAC1,
excitatory amino acid carrier 1; GAT1, γ-aminobutyric acid
transporter 1; KD, knockdown; NC, negative control; OE,
overexpression; CK, control: No CoCl2 treatment; PE,
phycoerythrin; shRNA, short hairpin RNA. ***P<0.001. |
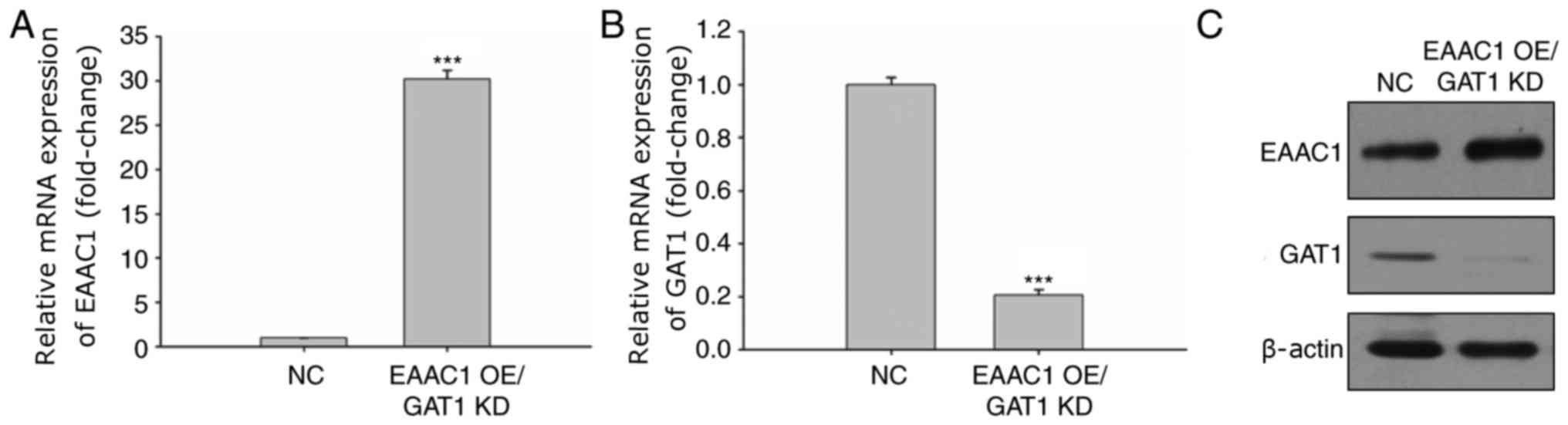
Overexpression of EAAC1 with GAT1
knockdown reduces neuronal cell apoptosis
To further investigate the effects of EAAC1 and GAT1
expression on neuronal cell apoptosis, EAAC1 overexpression and
GAT1 knockdown was applied. The mRNA expression levels of EAAC1
were markedly upregulated within neuronal cells (Fig. 5A), whereas >80% of GAT1 mRNA
expression was reduced (Fig. 5B).
The protein expression levels of EAAC1 and GAT1 were also confirmed
via western blotting (Fig.
5C).
The FACS apoptosis assay suggested that EAAC1
overexpression with GAT1 knockdown reduced neuronal cell apoptosis
under hypoxic conditions. As presented in Fig. 6, the percentage of apoptotic cells
was significantly reduced compared with control cells following
CoCl2 treatment compared with the negative control
group. These results demonstrated the importance of EAAC1 with GAT1
within hypoxic ischemia-induced brain injury and suggested that
upregulation of EAAC1 with GAT1 suppression may provide benefits
for the therapy of ischemia-associated diseases.
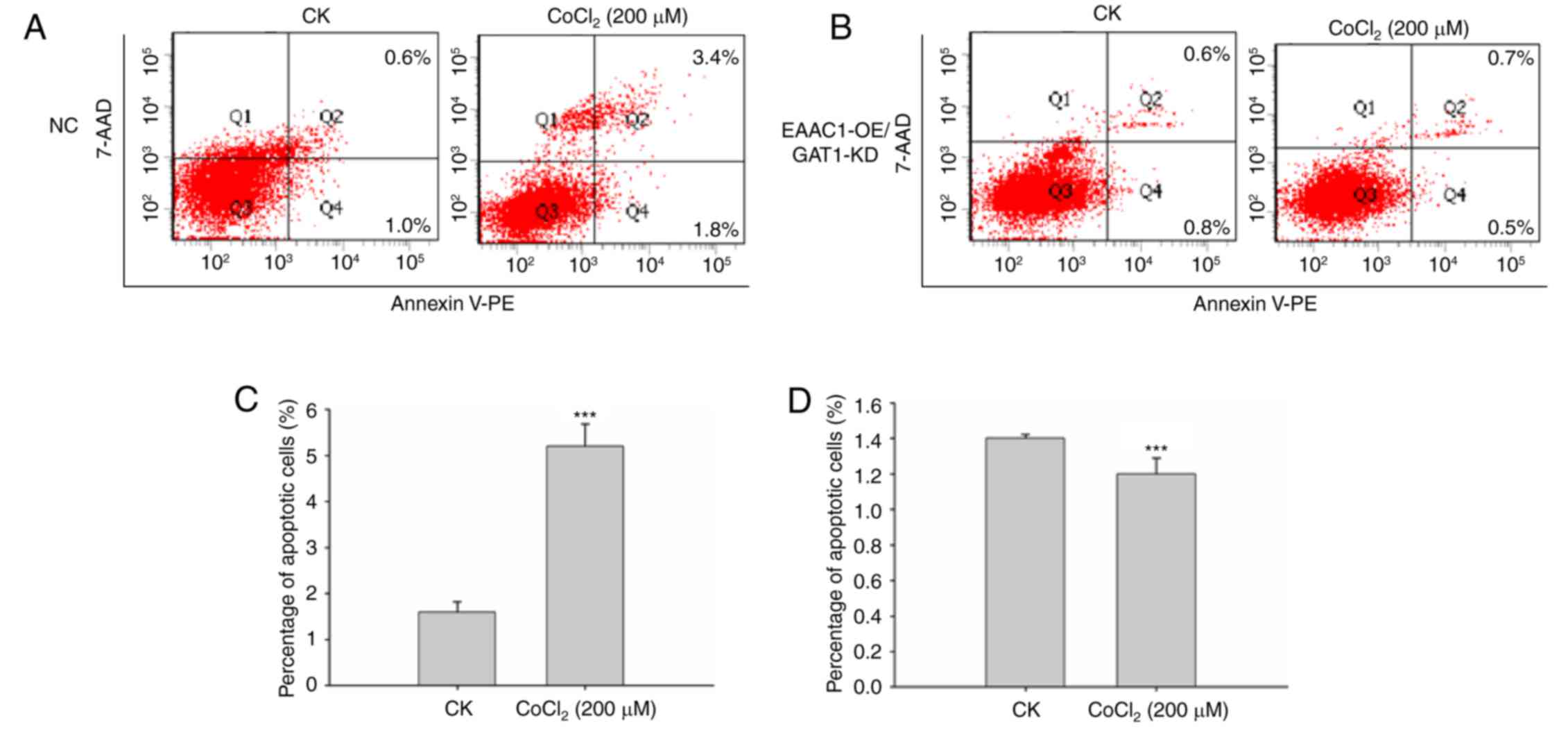 | Figure 6.Overexpression of EAAC1 with GAT1
knockdown reduces neuronal death. (A) Primary neuronal cells
transduced with (A) NC lentiviral vector or (B) EAAC1
RNA/GAT1-short hairpin RNA were treated with CoCl2 for
48 h, apoptosis was detected by a FACS assay. (C and D) Apoptotic
rate is expressed as the mean percentage of apoptotic cells ±
standard deviation. 7-AAD, 7-aminoactinomycin D; EAAC1, excitatory
amino acid carrier 1; FACS, fluorescence-activated cell sorting;
GAT1, γ-aminobutyric acid transporter 1; GLT1, glutamate
transporter 1; KD, knockdown; NC, negative control; OE,
overexpression; CK, control: No CoCl2 treatment; PE,
phycoerythrin. ***P<0.001. |
Discussion
Hypoxic ischemia-induced brain injury is a major
cause of morbidity and mortality in infants and children (3,20–22);
at present, effective treatments for these diseases are
unavailable. Glutamate and GABA are important neurotransmitters in
the human nervous system, however, whether glutamate/GABA
transporters serve important functions in hypoxic ischemia remains
unclear (23). In the present
study, it was reported that EAAC1 expression was reduced within the
cerebrum of focal cerebral ischemic MCAO rat models, as well as in
primary neurons cultured under hypoxia. Conversely, the expression
levels of GAT1 were slightly elevated under ischemic conditions.
Additionally, analysis of apoptosis revealed that EAAC1 suppression
with co-overexpression of GAT1 elevated neuronal cell apoptosis
under hypoxia; however, EAAC1 overexpression with GAT1 knockdown
reduced neuronal cell death. The present study indicated the
expression of glutamate and GABA transporters may be associated
with ischemia. Increasing the expression levels of EAAC1 and
suppressing GAT1 expression may provide beneficial effects in the
treatment of epilepsy or ischemia treatment.
Previous studies have demonstrated that the
pathogenesis of ischemic injury may be due to cellular apoptosis in
oxygen- and energy-deficient environments (24,25).
Neuronal cell apoptosis increased following CoCl2
treatment; however, apoptosis induced by CoCl2 may be
inhibited by increased expression levels of EAAC1 and suppressed
GAT1.
In conclusion, the results of the present study
demonstrated the importance of glutamate and GABA transporters in
hypoxic-ischemic brain injury; therefore, targeting the functions
of EAAC1 and GAT1 may provide advantages for the development of
hypoxic ischemia therapies.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Shanghai Municipal Health and Family Planning Commission of Chinese
Medicine Research Grants (grant no. 2014JP025A), the Shanghai
Pudong New Area Science and Technology Commission Science and
Technology Development Grants (grant no. PKJ2016-Y45) and the
Training Plan for Scientific Research of Renji Hospital (grant no.
RJZZ13-021).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
ZQ contributed to acquisition, analysis and
interpretation of data and wrote the main manuscript. YL, JX and YQ
contributed to data analysis, and LR designed the study and
contributed to revision of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Choi DW and Rothman SM: The role of
glutamate neurotoxicity in hypoxic-ischemic neuronal death. Ann Rev
Neurosci. 13:171–182. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mwaniki MK, Atieno M, Lawn JE and Newton
CR: Long-term neurodevelopmental outcomes after intrauterine and
neonatal insults: A systematic review. Lancet. 379:445–452. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Douglas-Escobar M and Weiss MD:
Hypoxic-ischemic encephalopathy: A review for the clinician. JAMA
Pediatr. 169:397–403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matsumoto N, Kumamoto E, Furue H and
Yoshimura M: GABA-mediated inhibition of glutamate release during
ischemia in substantia gelatinosa of the adult rat. J Neurophysiol.
89:257–264. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Voytenko LP, Lushnikova IV, Savotchenko
AV, Isaeva EV, Skok MV, Lykhmus OY, Patseva MA and Skibo GG:
Hippocampal GABAergic interneurons coexpressing alpha7-nicotinic
receptors and connexin-36 are able to improve neuronal viability
under oxygen-glucose deprivation. Brain Res. 1616:134–145. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou Y and Danbolt NC: GABA and glutamate
transporters in brain. Front Endocrinol (Lausanne).
4:1652013.PubMed/NCBI
|
|
7
|
Danbolt NC: Glutamate uptake. Prog
Neurobiol. 65:1–105. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tanaka K, Watase K, Manabe T, Yamada K,
Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi
T, et al: Epilepsy and exacerbation of brain injury in mice lacking
the glutamate transporter GLT-1. Science. 276:1699–1702. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aoyama K, Suh SW, Hamby AM, Liu J, Chan
WY, Chen Y and Swanson RA: Neuronal glutathione deficiency and
age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat
Neurosci. 9:119–126. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kiryu-Seo S, Gamo K, Tachibana T, Tanaka K
and Kiyama H: Unique anti-apoptotic activity of EAAC1 in injured
motor neurons. EMBO J. 25:3411–3421. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Conti F, Minelli A and Melone M: GABA
transporters in the mammalian cerebral cortex: Localization,
development and pathological implications. Brain Res Brain Res Rev.
45:196–212. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pozdnyakova N, Yatsenko L, Parkhomenko N
and Himmelreich N: Perinatal hypoxia induces a long-lasting
increase in unstimulated gaba release in rat brain cortex and
hippocampus. The protective effect of pyruvate. Neurochem Int.
58:14–21. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Richards DA and Bowery NG: Comparative
effects of the GABA uptake inhibitors, tiagabine and NNC-711, on
extracellular GABA levels in the rat ventrolateral thalamus.
Neurochem Res. 21:135–140. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Richerson GB and Wu Y: Role of the GABA
transporter in epilepsy. Adv Exp Med Biol. 548:76–91. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Smith MD, Saunders GW, Clausen RP, Frølund
B, Krogsgaard-Larsen P, Larsson OM, Schousboe A, Wilcox KS and
White HS: Inhibition of the betaine-GABA transporter (mGAT2/BGT-1)
modulates spontaneous electrographic bursting in the medial
entorhinal cortex (mEC). Epilepsy Res. 79:6–13. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Frahm C, Siegel G, Grass S and Witte OW:
Stable expression of the vesicular GABA transporter following
photothrombotic infarct in rat brain. Neuroscience. 140:865–877.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakajima H, Kubo T, Semi Y, Itakura M,
Kuwamura M, Izawa T, Azuma YT and Takeuchi T: A rapid, targeted,
neuron-selective, in vivo knockdown following a single
intracerebroventricular injection of a novel chemically modified
siRNA in the adult rat brain. J Biotechnol. 157:326–333. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo L, Lan J, Lin Y, Guo P, Nie Q, Mao Q,
Ren L and Qiu Y: Hypoxia/ischemia up-regulates Id2 expression in
neuronal cells in vivo and in vitro. Neurosci Lett. 554:88–93.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Graham EM, Ruis KA, Hartman AL,
Northington FJ and Fox HE: A systematic review of the role of
intrapartum hypoxia-ischemia in the causation of neonatal
encephalopathy. Am J Obstet Gynecol. 199:587–595. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Strasser K, Lueckemann L, Kluever V,
Thavaneetharajah S, Hoeber D, Bendix I, Fandrey J, Bertsche A and
Felderhoff-Mueser U: Dose-dependent effects of levetiracetam after
hypoxia and hypothermia in the neonatal mouse brain. Brain Res.
1646:116–124. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Murray DM, Bala P, O'Connor CM, Ryan CA,
Connolly S and Boylan GB: The predictive value of early
neurological examination in neonatal hypoxic-ischaemic
encephalopathy and neurodevelopmental outcome at 24 months. Dev Med
Child Neurol. 52:e55–e59. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pettmann B and Henderson CE: Neuronal cell
death. Neuron. 20:633–647. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Broughton BR, Reutens DC and Sobey CG:
Apoptotic mechanisms after cerebral ischemia. Stroke. 40:e331–e339.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fu F, Wu D and Qian C: The MicroRNA-224
inhibitor prevents neuronal apoptosis via targeting spastic
paraplegia 7 after cerebral ischemia. J Mol Neurosci. 59:421–429.
2016. View Article : Google Scholar : PubMed/NCBI
|