Introduction
Congenital mental retardation is a
neurodevelopmental disease that affects 2–3% of the population
worldwide (1,2). Genetic factors may explain >50% of
congenital mental retardation cases (3). Chromosomal anomalies, including
chromosomal deletion, duplication and trisomy contribute to 4–28%
of cases and known monogenetic diseases explain 3–9% of cases
(4). Various methods may be used
to detect genetic abnormalities, including chromosomal microarray
(CMA) and next generation sequencing (NGS) (5–10).
However, the absence of any accompanying deformities in
non-syndromic mental retardation makes it difficult to select the
detection method. Using numerous methods increases cost and reduces
efficiency. To the best of our knowledge, no method to detect
genetic abnormalities in one step exists at present.
Typically, custom-made capture array-based targeted
NGS is used to detect disease-causing mutations in monogenic
disease (8). A custom-made capture
array that is able to detect various types of mutations, including
microdeletion, was previously designed (11). In the present study, this improved
targeted NGS method was applied to diagnose a case of non-syndromic
mental retardation of unknown cause. This may aid in the detection
of genetic abnormalities in one step.
The microdeletion 17p11.2 was successfully detected
by improved targeted NGS and no single gene mutations were
identified. Subsequently, the same microdeletion was validated
using low coverage whole-genome sequencing (LCS). Fertility
guidance was additionally given to the parents of the patient.
In the present study, the patient was diagnosed with
Smith-Magenis syndrome (SMS). SMS is a rare, congenital syndrome
that affects ~1 in every 25,000 individuals. Features include
mental retardation, behavioral abnormalities and distinctive facial
features, difficulty sleeping, delayed speech and development,
resulting from a 17p11.2 deletion encompassing the retinoic
acid-induced protein 1 (RAI1) (12–14).
It is estimated that 70% of patients with SMS have a common 3.7 Mb
deletion; the remaining 30% have larger or smaller deletions.
Approximately 90% of SMS patients have a 17p11.2 deletion; the
remaining 30% have mutations in the RAI1 gene (15).
The present study applied an improved targeted NGS
method to diagnose a patient with non-syndromic mental retardation
of an unknown cause in an effective and a fast way, without using
other methods. The result was validated by LCS. This improved
method has the potential to be developed into a screening panel to
effectively identify genetic abnormalities in non-syndromic mental
retardation and other congenital anomalies.
Case report
Patient information
An 8-year-old Chinese Han female and her parents
attended the Wuhan Medical & Health Center for Women and
Children hospital due to developmental delay and signs of mental
retardation that were present from infancy. The research was
prospectively reviewed and approved by the ethics committee of
BGI-Shenzhen (approval no. BGI-IRB 15083; Shenzen, China). Written
informed consent for participation in the present study was
obtained from the patient's parents.
The patient was the first child of unrelated parents
who had no family history of inherited diseases. The patient was
born by cesarean section with a birth weight of 3.35 kg and a body
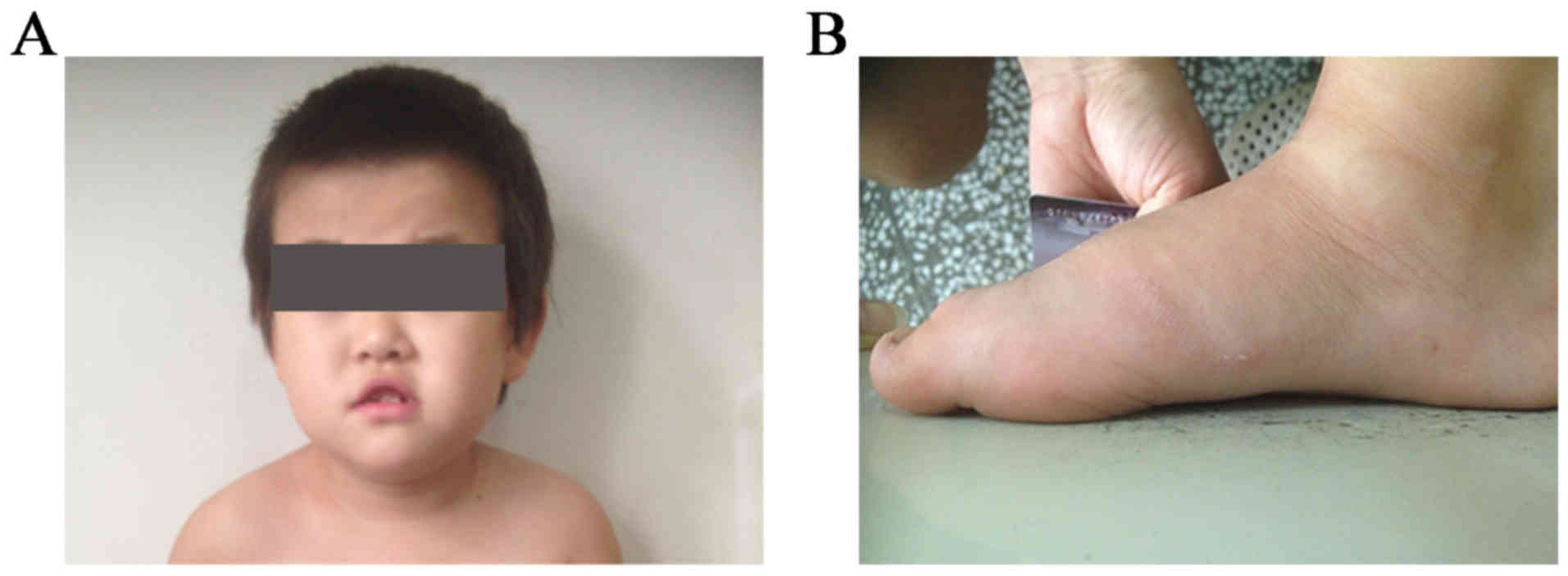
length of 50 cm. A distinct facial appearance was observed, with
short palpebral fissures, a depressed nasal bridge, hypertelorism,
and an upper lip with tented morphology and a V-like shape
(Fig. 1A and B).
By age 2 years, the patient had sleep difficulties
with nocturnal awakenings which gradually increased in frequency.
The patient exhibited signs of mental retardation and decreased
motor development compared with other children of the same age. The
patient was talkative, although failed to convey information
effectively.
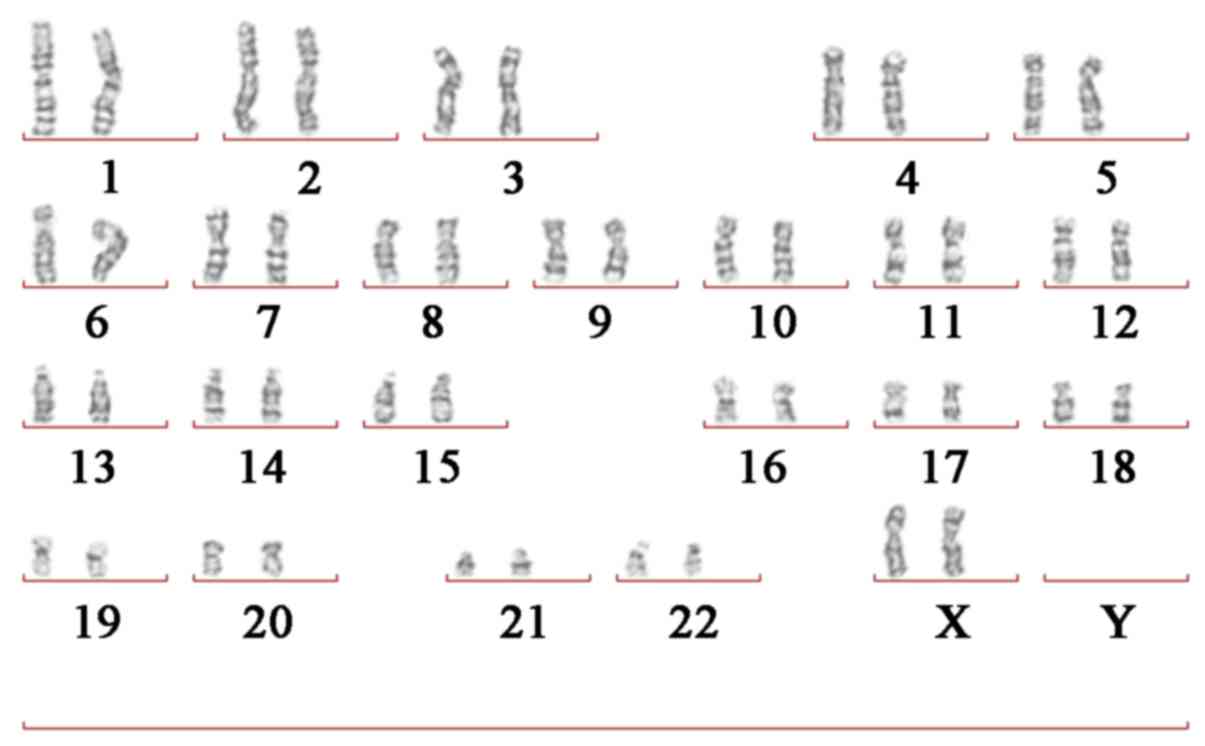
Giemsa (G)-banded cytogenetic
analysis
Routine G-banding chromosome analysis was performed
as described (16); a 5 ml
peripheral blood sample was collected from the patient and her
parents. Chromosomes from cultured lymphocytic cells were treated
with 0.25% trypsin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
for 2 min at 37°C and stained with 6% Giemsa (Sigma-Aldrich; Merck
KGaA) for 10 min at 37°C. The results revealed that the patient
carried the normal karyotype (46 chromosomes; XX; Fig. 3). In addition, the parents of the
patient carried the normal karyotype.
Improved targeted NGS
Improved targeted NGS was performed on peripheral
blood samples of the patient. The designed capture array
(NimbleGen; Roche Molecular Diagnostics, Pleasanton, CA, USA)
focuses on known associated single gene-coding regions and flanking
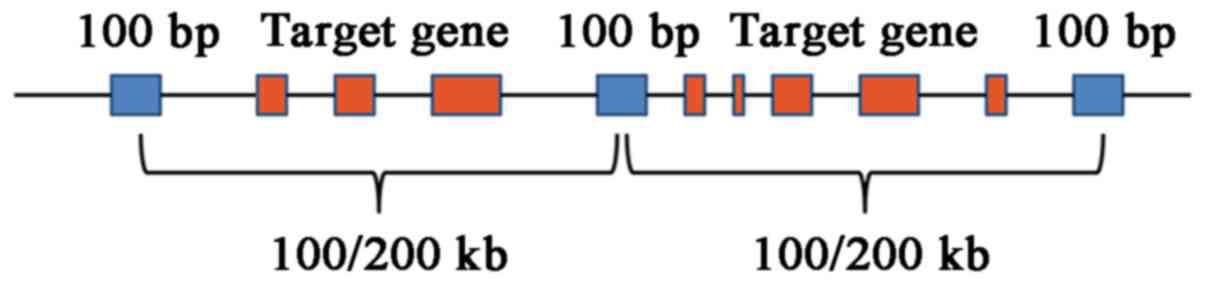
intronic boundaries 10 bp. The region that contained known
microdeletions and microduplications was covered and a target spot
at an interval of 0.1 Mb (length, 100 bp) was selected; for the
remaining genome region, a target spot at an interval of 0.2 Mb
(length, 100 bp) was selected (Fig.
2). Targeted NGS analysis with normalization was performed for
the patient (17). The
microdeletion and microduplication regions designed on this array
were selected from the database of DECIPHER (18). The 45 known microdeletion and
microduplication diseases can be identified at present listed in
Table I.
 | Table I.Known chromosomal microdeletion or
microduplication diseases. |
Table I.
Known chromosomal microdeletion or
microduplication diseases.
| Syndrome | Chromosomal
region |
|---|
| 12q14 microdeletion
syndrome | chr12:
65071919-68645525 |
| 15q13.3 microdeletion
syndrome | chr15:
30910306-32445407 |
| 15q24 recurrent
microdeletion syndrome | chr15:
74412643-75972911 |
| 15q26 overgrowth
syndrome | chr15:
99357970-102521392 |
| 16p11.2
microduplication syndrome | chr16:
29606852-30199855 |
| 16p11.2-p12.2
microdeletion syndrome | chr16:
21512062-30199854 |
| 16p11.2-p12.2
microduplication syndrome | chr16:
21475060-29284077 |
| 16p13.11 recurrent
microdeletion or microduplication | chr16:
14986684-16486684 |
| 17q21.31 recurrent
microdeletion syndrome | chr17:
43705166-44294406 |
| 1p36 microdeletion
syndrome | chr1:
10001-12840259 |
| 1q21.1 recurrent
microdeletion or microduplication | chr1:
146533376-147883376 |
| 1q21.1 susceptibility
locus for Thrombocytopenia Absent Radius syndrome | chr1:
145386506-145748067 |
| 22q11 deletion or
duplication syndrome | chr22:
19009792-21452445 |
| 22q13 deletion
syndrome | chr22:
51045516-51187844 |
| 2p15-16.1
microdeletion syndrome | chr2:
59285696-61819815 |
| 2p21 microdeletion
syndrome | chr2:
44410451-44589584 |
| 2q33.1 deletion
syndrome | chr2:
196925121-205206939 |
| 2q37 monosomy | chr2:
239969863-240322643 |
| 3q29 microdeletion or
microduplication syndrome | chr3:
195726835-197344663 |
| 7q11.23 duplication
syndrome | chr7:
72744455-74142672 |
| 8p23.1 deletion or
duplication syndrome | chr8:
8100055-11764629 |
| 9q subtelomeric
deletion syndrome | chr9:
140513443-140730578 |
| Angelman syndrome
type 1 | chr15:
22749354-28438266 |
| Angelman syndrome
type 2 | chr15:
23619912-28438266 |
|
α-thalassemia-intellectual deficit
syndrome | chr16:
60001-834372 |
| Azoospermia factor
microdeletion | Chr Y:
14352761-15154862 |
| Charcot-Marie-Tooth
syndrome type 1A | chr17:
14097915-15470903 |
| Cri du Chat
syndrome | chr5:
10001-12533304 |
| Early-onset Alzheimer
disease with cerebral amyloid angiopathy | chr21:
27252860-27543446 |
| Familial Adenomatous
Polyposis | chr5:
112043201-112181936 |
| Miller-Dieker
syndrome | chr17: 1-2588909 |
| NF1-microdeletion
syndrome | chr17:
29107097-30263321 |
| Pelizaeus-Merzbacher
disease | chrX:
103031438-103047547 |
| Smith-Magenis
syndrome | chr17:
16773072-20222149 |
| Potocki-Shaffer
syndrome | chr11:
43994800-46052450 |
| Renal cysts and
diabetes | chr17:
34815072-36215917 |
| Rubinstein-Taybi
syndrome | chr16:
3775055-3930121 |
| Sotos syndrome | chr5:
175724636-177052116 |
| Steroid sulphatase
deficiency | chrX:
6455812-8133195 |
| 11p13 deletion
syndrome | chr11:
31806339-32457087 |
| Wolf-Hirschhorn
syndrome | chr4:
1569197-2110236 |
| Xp11.22-linked
intellectual disability | chrX:
53401070-53683275 |
| Xp11.22-p11.23
microduplication | chrX:
48334549-52117661 |
| Xq28 duplication | chrX:
153287263-153363188 |
| Xq28
microduplication | chrX:
153624563-153881853 |
Sequence capture, enrichment and elution was
performed according to the standard protocol, as previously
mentioned (11). Following
enrichment, high-throughput sequencing was performed using the
Illumina Hiseq2000 analyzer (Illumina, Inc., San Diego, CA, USA).
Image analysis and base-calling were performed using the Illumina
Pipeline version 1.3.4 (Illumina, Inc.).
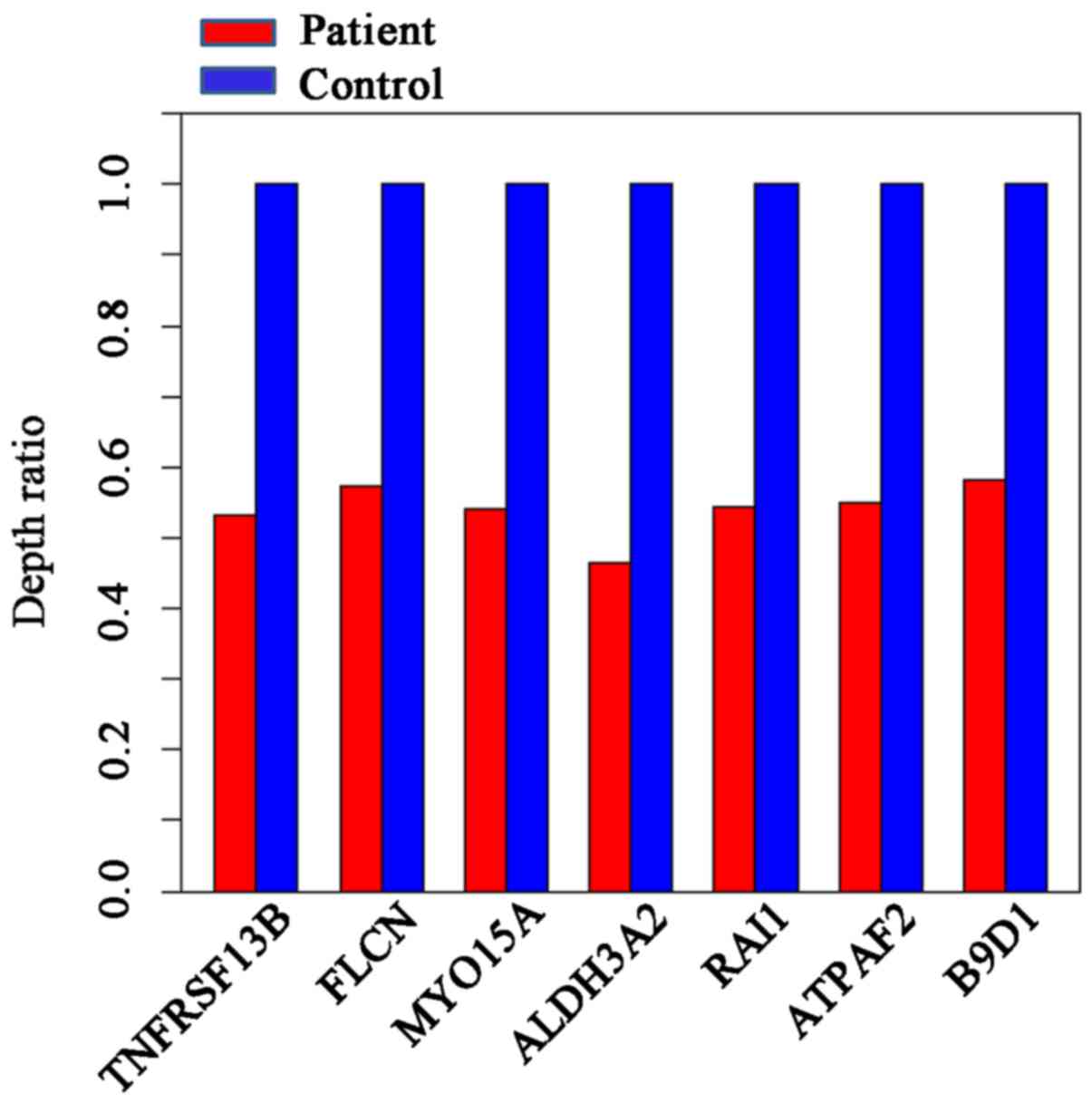
The depth ratio (patient:control) was used to
evaluate the microdeletions and microduplications; the control
referred to healthy subjects unrelated to the patient (samples
obtained from BGI). If the ratio is 1, this indicates that no
deletion has been detected. A ratio of ~0.5 indicates the detection
of a heterozygous deletion. A ratio of zero indicates a potential
homozygous deletion.
The designed capture array identified a 17p11.2
deletion. No single gene mutations were identified. The depth ratio
was obtained for seven genes located on 17p11.2; the average depth
ratio of the patient was ~0.55, indicating a heterozygous deletion
(Fig. 4). As chromosome 6 was not
designed in the capture array, the specific deletion on chromosome
6 was not identified.
LCS
To validate the results obtained from the improved
NGS method, LCS was additionally performed on the peripheral blood
samples of the patient and parents using the Illumina Hiseq2000
(Illumina, Inc.) platform with the population-scale microdeletions
and microduplications calling (PSCC) method, as described
previously (19). PSCC is a stable
and sensitive method for the detection of copy number variation
(CNV). PSCC is able to identify deletions with a resolution in the
100 kb range. It has three modules which include a two-step
correction procedure to remove the local GC content bias, a binary
segmentation method to locate the candidate microdeletions and
microduplications regions and a combined statistics test to
estimate the signal reliability. Subsequently, the microdeletions
and microduplications are determined.
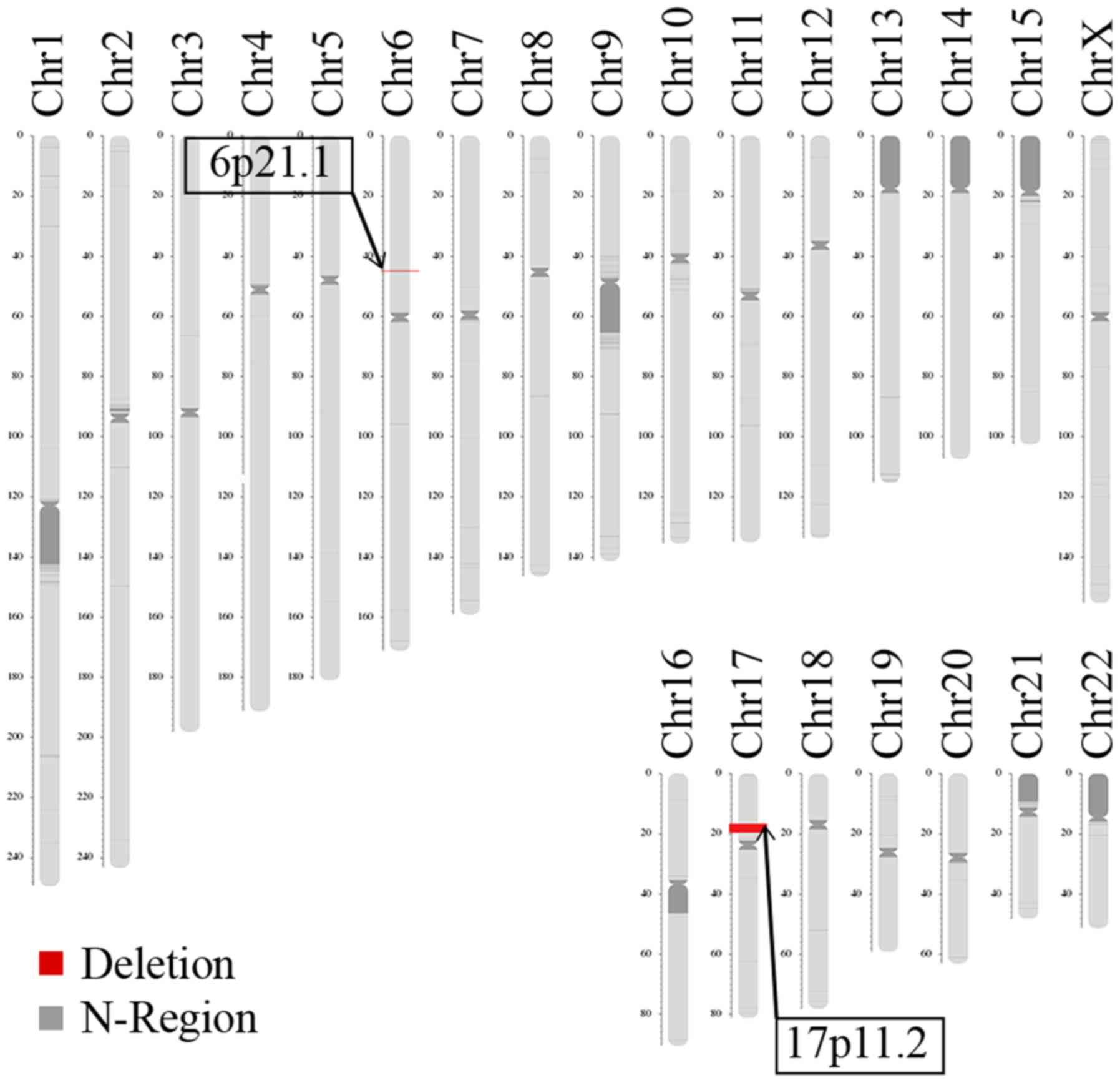
LCS identified two deletions in the patient, located
on 6p21.1 and 17p11.2 (Fig. 5).
The deletion size on 6p21.1 was approximately 172.71 kb (chromosome
6, 44895251-45077965). The Database of Genomic Variants
(dgv.tcag.ca/dgv/app/home) indicated that the identified deletion
was not causative. The deletion size on 17p11.2 was approximately
3.66 Mb (chromosome 17, 16572714-20229256), which is typically
observed in patients with SMS. The LCS results revealed the parents
were normal, with no disease-causing microdeletions and
microduplications or balanced translocation (data not shown).
Discussion
Congenital mental retardation is a
neurodevelopmental disease with a variety of causes. Genetic
factors have significant involvement in the etiology of congenital
mental retardation (3). Numerous
methods, including CMA and NGS, may be used to detect genetic
abnormalities. However, it is difficult to initially select a
method to be performed. Using a number of methods increases cost
and reduces efficiency. At present, a method to detect genetic
abnormalities in one step does not exist. Herein, an improved
targeted NGS method was developed to diagnose chromosomal anomalies
and single gene mutations in one step at low cost. In the present
study, an unknown cause of non-syndromic mental retardation was
successfully diagnosed with the improved targeted NGS method. SMS
has similar phenotypic features to other syndromes, including 9q34
deletion syndrome, Prader-Willi syndrome, 22q11.2 syndrome, Sotos
syndrome and Williams syndrome. Therefore, it may be easily
misdiagnosed (14). In the present
study, the improved targeted NGS method detected a deletion located
in 17p11.2. Thus, the patient was accurately diagnosed with
SMS.
The methods of the present study had certain
limitations. Chromosomal microdeletions and microduplications were
only detected in the targeted captured region, and the whole genome
region was not covered. Therefore, breakpoints of chromosome
aberration were unable to be detected. Additionally, there was the
potential for false negative errors. A future study will improve
the scheme to minimize the false negative rate, by increasing the
targeted captured region and optimizing the information analysis
algorithm.
Additionally, fertility guidance was given to the
parents of the patient. The origin of the majority of deletions is
de novo, and are more rarely attributed to an unbalanced
segregation of a parental balanced translocation (20). In the present case, the chromosomes
of the asymptomatic parents were normal; no disease-causing
microdeletions, microduplications or balanced translocations were
detected, indicating that the deletion 17p11.2 was de novo. The
incidence of de novo deletions may primarily occur as random
chance events during gamete formation or early embryo development.
Therefore, the risk of having another affected child may be very
low. However, the deletion may be caused by germline mosaicism, a
well reported explanation of the possible origin of autosomal
dominant and X-linked disorders (21–24).
As for de novo deletions, limited information is available
in the literature. Rothlisberger et al (20) reported that germline mosaicism is
rare, although it may never be excluded as the origin of de
novo structural aberrations. Sanchez et al (25) reported that a 15q11.2-q13 deletion
in dizygotic twins with Angelman syndrome originated from somatic
and germline mosaicism of the mother. The present study did not
assess the possibility of germline mosaicism, as it is difficult to
obtain gametes. However, prenatal testing should be proposed to
this family during genetic counselling to minimize recurrence risks
in subsequent gestation.
In summary, targeted NGS is typically used to detect
variants of monogenic diseases. In the present study, it was
investigated if the currently available targeted NGS was already
suitable for the molecular diagnosis of microdeletion.
Subsequently, the 17p11.2 deletion was identified by the designed
capture array and no single gene mutation was identified. It was
demonstrated that this method could successfully identify
microdeletion and duplication. Therefore, this improved method has
the potential to be developed into a screening panel for effective
diagnosis of genetic abnormalities in non-syndromic mental
retardation and other congenital anomalies.
Acknowledgements
Not applicable.
Funding
This work was supported by Shenzhen Technological
Innovation Plan-Technology Development Project (grant no. CXZZ
20130517144604091).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Author's contributions
WW, BM and YY designed the research and produced the
first draft of the article. LM and XG performed the experimental
studies. XW and DY contributed to drafting and revising the
manuscript. HL, YS, XW and DY performed the data analysis.
Ethics approval and consent to
participate
The research was prospectively reviewed and approved
by the ethics committee of BGI-Shenzhen (approval no. BGI-IRB
15083; Shenzen, China). Written informed consent for participation
in the present study was obtained from the patient's parents.
Consent for publication
Written informed consent for participation in the
present study was obtained from the patient's parents.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Daily DK, Ardinger HH and Holmes GE:
Identification and evaluation of mental retardation. Am Fam
Physician. 61(1059): 1067–1070. 2000.
|
|
2
|
Kaur A, Mahajan S and Singh JR:
Cytogenetic Profile of Individuals with Mental Retardation. Int J
Hum Genet. 3:13–16. 2003. View Article : Google Scholar
|
|
3
|
Chelly J, Khelfaoui M, Francis F, Chérif B
and Bienvenu T: Genetics and pathophysiology of mental retardation.
Eur J Hum Genet. 14:701–713. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Curry CJ, Stevenson RE, Aughton D, Byrne
J, Carey JC, Cassidy S, Cunniff C, Graham JM Jr, Jones MC, Kaback
MM, et al: Evaluation of mental retardation: Recommendations of a
Consensus Conference: American College of Medical Genetics. Am J
Med Genet. 72:468–477. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
de Vries BB, Pfundt R, Leisink M, Koolen
DA, Vissers LE, Janssen IM, Reijmersdal Sv, Nillesen WM, Huys EH,
Leeuw Nd, et al: Diagnostic genome profiling in mental retardation.
Am J Hum Genet. 77:606–616. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pinto IP, Minasi LB, da Cruz AS, de Melo
AV, da Cruz E, Cunha DM, Pereira RR, Ribeiro CL, da Silva CC, de
Melo E Silva D and da Cruz AD: A non-syndromic intellectual
disability associated with a de novo microdeletion at 7q and 18p,
microduplication at Xp, and 18q partial trisomy detected using
chromosomal microarray analysis approach. Mol Cytogenet. 7:442014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tucker T, Zahir FR, Griffith M, Delaney A,
Chai D, Tsang E, Lemyre E, Dobrzeniecka S, Marra M, Eydoux P, et
al: Single exon-resolution targeted chromosomal microarray analysis
of known and candidate intellectual disability genes. Eur J Hum
Genet. 22:792–800. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Martinez F, Caro-Llopis A, Roselló M,
Oltra S, Mayo S, Monfort S and Orellana C: High diagnostic yield of
syndromic intellectual disability by targeted next-generation
sequencing. J Med Genet. 54:87–92. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morgan A, Gandin I, Belcaro C, Palumbo P,
Palumbo O, Biamino E, Dal Col V, Laurini E, Pricl S, Bosco P, et
al: Target sequencing approach intended to discover new mutations
in non-syndromic intellectual disability. Mutat Res. 781:32–36.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ehmke N, Karge S, Buchmann J, Korinth D,
Horn D, Reis O and Häßler F: A de novo nonsense mutation in ZBTB18
plus a de novo 15q13.3 microdeletion in a 6-year-old female. Am J
Med Genet A. 173:1251–1256. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Y, Wei X, Kong X, Guo X, Sun Y, Man J,
Du L, Zhu H, Qu Z, Tian P, et al: Targeted next-generation
sequencing for clinical diagnosis of 561 mendelian diseases. PLoS
One. 10:e01336362015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Smith AC, McGavran L, Robinson J,
Waldstein G, Macfarlane J, Zonona J, Reiss J, Lahr M, Allen L and
Magenis E: Interstitial deletion of (17)(p11.2p11.2) in nine
patients. Am J Med Genet. 24:393–414. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Girirajan S, Elsas LJ II, Devriendt K and
Elsea SH: RAI1 variations in Smith-Magenis syndrome patients
without 17p11.2 deletions. J Med Genet. 42:820–828. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Elsea SH and Girirajan S: Smith-Magenis
syndrome. Eur J Hum Genet. 16:412–421. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Slager RE, Newton TL, Vlangos CN, Finucane
B and Elsea SH: Mutations in RAI1 associated with Smith-Magenis
syndrome. Nat Genet. 33:466–468. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Seabright M: A rapid banding technique for
human chromosomes. Lancet. 2:971–972. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Magi A, Tattini L, Pippucci T, Torricelli
F and Benelli M: Read count approach for DNA copy number variants
detection. Bioinformatics. 28:470–478. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bragin E, Chatzimichali EA, Wright CF,
Hurles ME, Firth HV, Bevan AP and Swaminathan GJ: DECIPHER:
Database for the interpretation of phenotype-linked plausibly
pathogenic sequence and copy-number variation. Nucleic Acids Res.
42:(Database issue). D993–D1000. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li X, Chen S, Xie W, Vogel I, Choy KW,
Chen F, Christensen R, Zhang C, Ge H, Jiang H, et al: PSCC:
Sensitive and reliable population-scale copy number variation
detection method based on low coverage sequencing. PLoS One.
9:e850962014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Röthlisberger B and Kotzot D: Recurrence
risk in de novo structural chromosomal rearrangements. Am J Med
Genet A. 143A:1708–1714. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wilton SD, Chandler DC, Kakulas BA and
Laing NG: Identification of a point mutation and germinal mosaicism
in a Duchenne muscular dystrophy family. Hum Mutat. 3:133–140.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Slavin TP, Lazebnik N, Clark DM,
Vengoechea J, Cohen L, Kaur M, Konczal L, Crowe CA, Corteville JE,
Nowaczyk MJ, et al: Germline mosaicism in Cornelia de Lange
syndrome. Am J Med Genet A. 158A:1481–1485. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bermúdez-López C, García-de Teresa B,
González-del Angel A and Alcántara-Ortigoza MA: Germinal mosaicism
in a sample of families with Duchenne/Becker muscular dystrophy
with partial deletions in the DMD gene. Genet Test Mol Biomarkers.
18:93–97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miyagawa M, Nishio SY, Hattori M, Takumi Y
and Usami S: Germinal mosaicism in a family with BO syndrome. Ann
Otol Rhinol Laryngol. 124 Suppl 1:118S–122S. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sánchez J, Fernández R, Madruga M,
Bernabeu-Wittel J, Antiñolo G and Borrego S: Somatic and germ-line
mosaicism of deletion 15q11.2-q13 in a mother of dyzigotic twins
with Angelman syndrome. Am J Med Genet A. 164A:370–376. 2014.
View Article : Google Scholar : PubMed/NCBI
|