Introduction
Diabetic nephropathy (DN) is a serious complication
of diabetes and may result in end-stage renal failure (1,2). In
total, ~30% of patients with diabetes mellitus (DM) developed DN
following a disease duration of 15–30 years (3,4). The
morbidity of DN is markedly rising with the increasing incidence
and prevalence of diabetes. Albuminuria is regarded as the
principal feature of DN and an independent risk factor for renal
failure, in addition, hyperglycemia invariably acts as an
initiating and maintaining factor during the development of end
stage renal disease (5,6); however, the pathogenesis of DN has
not been fully elucidated. A previous study has demonstrated that
the damage of renal hemodynamics and metabolism caused by chronic
hyperglycemia may lead to the secretion of inflammatory factors,
followed by infiltration of immune cells (7). Therefore, the inflammatory response
has been postulated to serve a key role in the pathogenesis of DN
(8). A previous study conducted by
Wang et al (9) demonstrated
that inflammation is associated with DN, and overexpression of
renal inflammasome components NLR family pyrin domain containing 3
(NLRP3), apoptosis-associated speck-like protein containing a CARD
(ASC) and caspase-1, resulting in elevation of interleukin (IL)-1β
and IL-18, subsequently contribute to renal injury. These
observations suggest that the NLRP3 inflammasome may be a
therapeutic target for diabetes with kidney injury.
Keller et al (10) demonstrated that NLRP3 is involved
in the regulation of the activity of caspase-1, which in turn lead
to the maturation and secretion of pro-inflammatory cytokines,
including IL-1β against pathogen infection, and may additionally
drive pyroptosis (3). The c-Jun
N-terminal kinase (JNK) signaling pathway is activated through
lysosome rupture, which subsequently leads to the complete
activation of the NLRP3 inflammasome in macrophages (11). It was hypothesized that high
glucose may induce activation of the JNK signaling pathway. In the
present study, it was demonstrated that JNK, a stress-responsive
mitogen-activated protein kinase, was activated following high
glucose stimulation and a JNK inhibitor suppressed NLRP3
inflammasome activation.
Spleen tyrosine kinase (Syk) is a non-receptor
protein tyrosine kinase, which transmits B-cell antigen receptor or
Fc-receptor signaling of hematopoietic cells, and Syk may result in
gene transcriptions of C-C motif chemokine ligand 2 and
transforming growth factor β-1, which may be involved in the
development of DN (12). It was
additionally observed that a tyrosine phosphorylation site is on
ASC acts as a molecular switch controlling inflammasome assembly
(3,7). In the present study, it was
demonstrated that Syk was involved in JNK-dependent NLRP3
inflammasome activation in high glucose-induced HK2 cells and rat
glomerular mesangial cells (RGMCs).
Materials and methods
Animals
Sixty Male Sprague-Dawley rats (age, 5-week-old;
weight, 180–200 g) were purchased from The Laboratory Animal Center
of the Academy of Military Medical Sciences (Beijing, China). They
were maintained under standard conditions of temperature (23±5°C)
and humidity (60±5%) with an alternating 12 h light/dark cycles.
All the animals had access to clean drinking water and a standard
pellet diet. The rats in the experimental group (n=36) were given a
single intraperitoneal injection of fresh streptozotocin (STZ; 65
mg/kg; Sigma-Aldrich; Merck KGaA) in 0.1 M sodium citrate buffer
(pH 4.3); whereas, the control group rats (n=24) received the same
dosage of sodium citrate buffer only. At 72 h following injection,
blood glucose ≥16.7 mM was considered as diabetes. The body and
kidney weight, blood and urine glucose and albumin were determined
at week 12 and 16. Seven DN and six control rats were sacrificed at
12, 16, 20 and 33 weeks, respectively, and kidneys were analyzed
for mRNA and protein expression at 12, 16, 20 and 33 weeks. All the
experimental procedures in the present study were approved by the
Animal Care and Welfare Committee of Tianjin Medical University
(Tianjin, China).
Reagents and antibodies
NLRP3 (cat. no. 13158, 1:1,500), phosphorylated
(p)-Syk (cat. no. 2710, 1:1,500), p-JNK (cat. no. 9255, 1:1,500),
Syk (cat. no. 13198, 1:1,500), JNK (cat. no. 9252, 1:1,500), and
cleaved caspase-1 (cat. no. 4199, 1:1,500) antibodies were obtained
from Cell Signaling Technology, Inc. (Danvers, MA, USA); caspase-1
(cat. no. ab179515, 1:1,500), pro-IL-1β (cat. no. ab2105, 1:2,000)
and mature (m)IL-1β (cat. no. ab9722, 1:2,000) antibodies from
Abcam (Cambridge, UK); apoptosis regulator Bax (cat. no. 200958,
1:1,000) and Bcl-2 (cat. no. 230004, 1:500) apoptosis regulator
(Bcl-2; BH3 Domain Specific) antibodies from Zen BioScience
(Chengdu, China); ASC (cat. no. sc-271054, 1:500); β-actin (cat.
no. sc-47778, 1:1,000) and mouse anti-rabbit IgG-HRP (cat. no.
sc-2357, 1:4,000) antibodies from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA); anti-mouse IgG HRP Conjugate (cat. no. W4021,
1:5,000) from Promega (Madison, WI, USA). All cell culture reagents
were obtained from Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). JNK inhibitor II was purchased from Merck KGaA (Darmstadt,
Germany), and Syk inhibitor IV, BAY61-3606 from Santa Cruz
Biotechnology, Inc. The radioimmunoprecipitation assay lysis buffer
[50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium
deoxycholate, 0.1% SDS] was purchased from Beyotime Institute of
Biotechnology (Haimen, China). TRIzol® reagent was
obtained from Thermo Fisher Scientific, Inc. The fluorescein
iosthiocyanate (FITC)-Annexin V Apoptosis Detection kit was
obtained from BioLegend, Inc. (London, UK).
Lipofectamine® 3000 was purchased from Invitrogen
(Thermo Fisher Scientific, Inc.) Small interfering (si)RNA specific
to Syk was purchased from Santa Cruz Biotechnology, Inc.
Cell culture
The HK2 cell line was derived from a normal adult
human kidney (13) and RGMC was
derived from rat renal glomeruli (14). In the present study, the HK2 cells
(cat. no. ZQ0313) and RGMCs (HBZY-1; cat. no. ZQ0540) were
purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd.
(Shanghai, China). HK2 cells were maintained in Dulbecco's modified
Eagle's medium (DMEM)/F-12 (1:1) basic (1X) medium (Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS,
Gibco; Thermo Fisher Scientific, Inc.), 1% streptomycin/penicillin.
RGMCs were cultured in DMEM containing 5 mM glucose and 10% FBS at
37°C and 5% CO2. The cells were added to the 6-well
plate at a density of 1×106 cells/well and treated with
5 or 25 mM glucose or high mannitol (Mtol) concentration (5 mM
glucose + 20 mM Mtol), then pretreated with Syk inhibitor (1 µM) or
Syk-siRNA for 12, 24, 36 and 48 h to detect the expression of the
NLRP3 inflammasome or for 10, 20, 30 and 40 min to detect the
protein level of p-JNK.
Transient transfection
siRNA (50 nM) specific to Syk (Syk-siRNA) was used
to knockdown Syk and a scramble siRNA, termed negative control
(NC)-siRNA, was used as a control in the experiment. HK2 cells and
RGMCs were transfected using Lipofectamine® 3000 reagent
following the manufacturer's protocol. Sequences for Syk-siRNA were
as follows: Sense, 5′-GCAUGAGUGAUGGGCUUUATT-3′; antisense,
5′-UAAAGCCCAUCACUCAUGCTT-3′. Sequences for NC-siRNA as follows:
Sense, 5′-UUCUCCGAACGUGUCACGUTT-3′; antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. Following 48 h of transfection, the
cells were treated with the high glucose and harvested for western
blotting.
Histological examination
For histological assessment, the renal cortex was
fixed in 4% neutral buffered paraformaldehyde for 24 h at 4°C,
embedded in paraffin and cut to 5-µm sections. The sections were
dewaxed using standard sequential techniques at room temperature.
Some sections were stained with hematoxylin and eosin (H&E),
the slides were dipped consecutively in 100, 90, 70 and 50% alcohol
for 2 min each, and placed over running tap water for 10 min before
and after dipping in haematoxylin for 10–15 min. The slides were
dipped twice in 1% acid alcohol and again placed over running tap
water for 10 min before and after dipping twice in 1% ammonia
solution. Finally, the slides were dipped in 2% eosin solution for
2–3 min and washed with absolute alcohol twice. The slides were
mounted in mounting medium (Solarbio, China) and observed under a
light microscope.
For periodic acid-Shiff (PAS) staining, the
formaldehyde sections were dewaxed, hydrated, stained with schiff's
reagent for 10–15 min at room temperature, then washed with running
water for 5 min. The sections were re-dyed with hematoxylin for 1–2
min, then washed and soaked in 1% acetic acid aqueous solution for
3–5 sec, differentiated with 1% acidified ethanol for 3–5 sec at
room temperature to remove the excessive binding dyes, stained with
aniline blue for 5 min at room temperature, immersed in 0.2% acetic
acid aqueous solution for 3–5 sec, treated with 95% ethanol and
absolute ethanol, cleared with xylene, and mounted with neutral
gum. A total of 10 fields were randomly observed using a light
microscope (magnification, ×200 and ×400).
Immunohistochemistry
Sections were permeabilized with 1% Triton X-100 for
2 h and blocked with normal goat serum (Beyotime Institute of
Biotechnology, Haiman, China) for 30 min at room temperature, the
sections were incubated sequentially with NLRP3 (1:500; cat. no.
ab223687; Abcam), caspase-1 (1:500; cat. no. ab108362; Abcam) and
mIL-1β (1 µg/ml; cat. no. ab9722; Abcam) antibodies at 4°C
overnight. The next day, after rewarming for 1 h, sections were
washed with PBS and then incubated with mouse anti-rabbit IgG-HRP
(cat. no. sc-2357, 1:100) antibody for 2 h at room temperature. To
visualize the signals, sections were treated with peroxidase
substrate 3,3′-diaminobenzidine (DAB, 0.05%, ZSGB-Bio, China) and
counterstained with hematoxylin for 1 min at room temperature.
Sections were viewed and imaged under a light microscope (Ni-U;
Nikon Corporation, Tokyo, Japan). Images were analyzed
quantitatively using Image-Pro Plus 6.0 (Media Cybernetics, Inc.,
Rockville, MD, USA).
Western blot analysis
The renal cortex was excised and homogenized in
protein extraction buffer and centrifuged at 13,000 × g, 4°C for 20
min. Protein concentration of the supernatants of tissue
homogenate, HK2 cells and RGMCs were measured using a bicinchoninic
acid protein assay kit. 25 µg protein was loaded per lane and
separated on 10 or 12% SDS-PAGE and transferred to polyvinylidene
difluoride membranes. Following blocking with 5% fat-free dry milk
or BSA for 2 h at room temperature, the membranes were incubated
with the primary antibody (mentioned above) overnight at 4°C.
Following three washes with Tris-buffered saline/Tween 20, the
membranes were probed with secondary antibodies [anti-mouse
immunoglobulin G (IgG) or anti-rabbit IgG] at room temperature for
1 h. The protein bands were visualized with a Horseradish
Peroxidase Substrate Peroxide Solution (EMD Millipore, Billerica,
MA, USA) and quantified using ImageJ software 6.0 (National
Institutes of Health, Bethesda, MD, USA).
Reverse transcription-polymerase chain
reaction (RT-PCR)
To measure specific gene expression, the primer
sequences for NLRP3, caspase-1 and IL-1β were synthesized (Table I). Total RNA of rat renal cortex
from the control group and DM group was isolated using
TRIzol® reagent, according to the manufacturer's
protocol. RT (42°C for 1 h; 70°C for 5 min) was conducted using the
TIANGEN RNA PCR kit (Tiangen Biotech Co., Ltd., Beijing, China).
The DNA polymerase was purchased from Invitrogen (Thermo Fisher
Scientific, Inc.). PCR reactions were performed at an initial
denaturation at 94°C for 3 min, followed by 35 cycles at 94°C for
30 sec, 55/59/60°C for 30 sec, 72°C for 1 min and final extension
step at 72°C for 5 min. The amplified products were detected by
1.5% agarose gel electrophoresis, stained with ethidium bromide
(0.5 µg/ml) for 40 min at room temperature. Gene expression was
normalized to β-actin by ImageJ software 6.0 (National Institutes
of Health).
 | Table I.Primers for reverse
transcription-polymerase chain reaction. |
Table I.
Primers for reverse
transcription-polymerase chain reaction.
| Gene | Forward sequence
(5′-3′) | Reverse sequence
(5′-3′) |
|---|
| NLR family pyrin
domain containing 3 |
AGGGCTCTGTTCATTG |
CTTCCACGTCTCGGTTC |
| Caspase-1 |
TGCCTGGTCTTGTGACTTGGAG |
ATGTCCTGGGAAGAGGTAGAAACG |
| Interleukin-1β |
TGGGATGATGACGACCTGC |
GGAGAATACCACTTGTTGGCTTA |
| β-actin |
GTTGACATCCGTAAAGACC |
GACTCATCGTACTCCTGCTC |
Flow cytometry
The cells were treated with BAY61-3606 for 2 h,
followed by high glucose treatment for 36 h, and washed twice with
cold Cell Staining Buffer (BioLegend, Inc.). Subsequently, cells
were resuspended in annexin V binding buffer at a density of
0.25–1.00×107 cells/ml and added 5 µl FITC-annexin V and
10 µl propidium iodide solution, then placed at room temperature
for 15 min in the dark. Annexin V binding buffer (400 µl) was added
to each tube and analyzed using a flow cytometer. All data were
analyzed using FlowJo software 7.6 (FlowJo LLC, Ashland, OR, USA),
according to the manufacturers' protocol.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. At least three independent experiments and differences
between groups were analyzed by GraphPad Prism 5 software (GraphPad
Software, Inc., La Jolla, CA, USA). Student's t-test was used for
comparison between two groups. One-way analysis of variance was
used followed by Dunnett's post hoc test for comparing between all
columns and control column, or Tukey's post hoc test for comparing
all pairs of columns. P<0.05 was considered to indicate a
statistically significant difference.
Results
Rat model of DN
The body weight, blood glucose and urine glucose of
the diabetic rats were significantly increased compared with the
corresponding control rats (Table
II; P<0.001). Additionally, urine amount and albumin
excretion were significantly increased compared with the control
groups (Table II; P<0.01),
which indicated the dysfunction of kidneys of diabetic rats.
Furthermore, PAS and H&E staining for the kidneys demonstrated
glomerular hypertrophy and mesangial expansion in the diabetic rats
(Fig. 1). In contrast, these
alterations were not observed in the rats of the control group,
suggesting that the renal structure of diabetic rats was
disorganized.
| Table II.The renal function parameters of
streptozotocin-induced diabetic rats. |
Table II.
The renal function parameters of
streptozotocin-induced diabetic rats.
|
| 12 weeks | 16 weeks |
|---|
|
|
|
|
|---|
| Parameters | Control | Diabetes | Control | Diabetes |
|---|
| Body weight
(g) |
516.3±11.20 |
236.7±17.32c |
624.3±1.202 |
329.0±24.06c |
| Kidney/body weight
ratio (%) |
0.27±0.030 |
0.59±0.037b |
0.30±0.0053 |
0.56±0.00a |
| Blood glucose
(mmol/l) |
6.26±0.27 |
32.27±0.61c |
6.07±0.43 |
32.70±0.30c |
| Urine amount
(ml) |
10.67±3.18 |
198.3±22.28b |
11.20±2.88 |
250.8±17.15c |
| Urine glucose
(mmol/l) |
2.45±0.13 |
357.2±28.89c |
3.03±0.91 |
425.1±32.20c |
| Albumin excretion
(mg/24 h) |
21.63±5.17 |
140.3±17.45b |
14.16±3.721 |
100.3±6.86c |
NLRP3 inflammasome is activated in
rats with DN
Previous studies reported that inflammasome
activation participates in the development of DN; therefore, the
renal injury was estimated by immunohistochemical staining of
NLRP3, caspase-1 and mIL-1β. As demonstrated in Fig. 2A and B, the expression of NLRP3,
caspase-1 and mIL-1β in the DN group was significantly higher
compared with the control group (P<0.01). Protein expression
levels of NLRP3 inflammasome was examined in DN rat kidneys at
different weeks, but its protein expression levels at 16 weeks was
upregulated more. Furthermore, there was no difference in the
expression levels of NLRP3 inflammasome in control rats at
different weeks. Therefore, the 16-week-old DN and control rats was
chosen as the experimental time point. Three different rats from
the control group at 16 weeks (termed C1, C2 and C3) and three
different rats from the DN group at 16 weeks (termed DN1, DN2 and
DN3) was selected to assess the mRNA expression levels of NLRP3 in
kidney tissues (Fig. 2C-E). There
was an ~2 fold increase in the expression level of NLRP3 in the
diabetic rats group compared with the control group (Fig. 2E; P<0.01). Furthermore, the mRNA
expression levels of caspase-1 and IL-1β were also significantly
increased (Fig. 2E;
P<0.05).
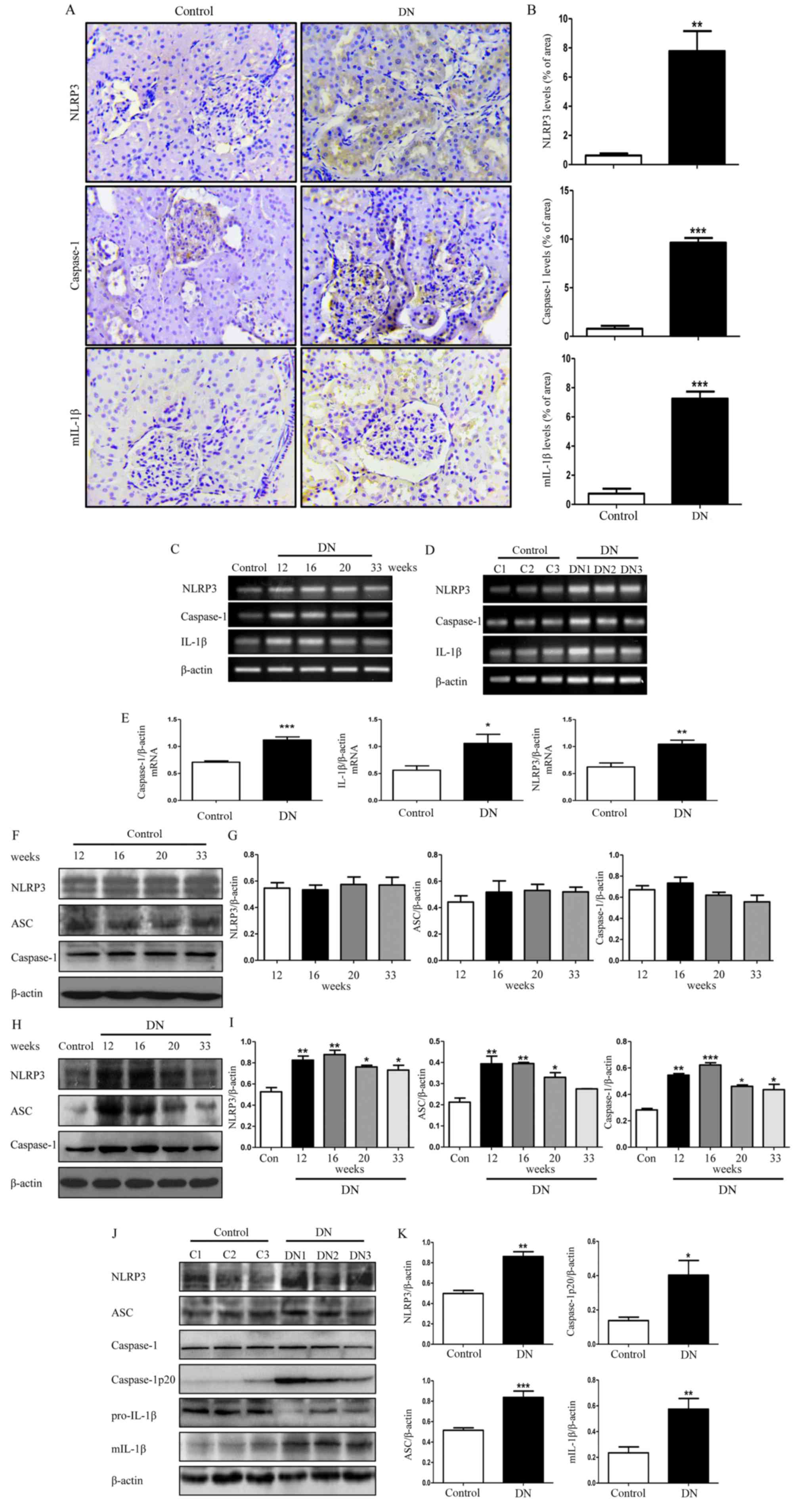 | Figure 2.NLRP3 inflammasome activity in DN
rats. (A) Representative images of paraformaldehyde-fixed kidney
sections from the control group and DN group rats at 16 weeks were
stained with anti-NLRP3, anti-caspase-1 and anti-mIL-1β antibodies
and representative images (×400× magnification). (B) Staining of
NLRP3, caspase-1, mIL-1β in rat kidney was quantified using
Image-Pro Plus 6.0. (C) Total RNA from the kidneys of the
16-week-old control rats and DN rats was extracted and subjected to
RT-PCR for NLRP3, caspase-1 and IL-1β at 12, 16, 20 and 33 weeks.
(D) RNA was extracted from kidneys of three different control rats
and three different DN rats, and then subjected to RT-PCR at week
16. (E) Densitometry analysis of the mRNA levels of NLRP3,
caspase-1 and IL-1β in 16-week-old control rat kidneys and DN rat
kidneys. (F) Total lysates from kidneys in control rats at
different weeks were extracted and subjected to western blot
analysis for NLRP3, ASC and caspase-1. (G) Densitometry analysis of
NLRP3, ASC and caspase-1 in. (H) The protein expression levels of
NLRP3, ASC and caspase-1 in DN rats at 12, 16, 20 and 33 weeks and
control rats at 16 weeks. (I) Densitometry analysis of NLRP3, ASC
and caspase-1. (J) Protein was extracted from kidneys of three
different DN rats and three different control rats, and then
subjected to western blot to detect the protein levels of these
molecules mentioned above in 16-week-old DN and control rats. (K)
Densitometry analysis of NLRP3, ASC, caspase-1p20 and mIL-1β. Data
are presented as the mean ± standard error of the mean from three
independent experiments. Student's t-test was used for comparison
between control group and DN group (for A-E, J and K). One-way
ANOVA followed by Dunnett's post hoc (for H and I) or Tukey's post
hoc test (F and G). *P<0.05, **P<0.01, ***P<0.001 vs.
control group. β-actin was used as the internal loading control.
RT-PCR, reverse transcription-polymerase chain reaction; ANOVA,
analysis of variance; DN, diabetic nephropathy; NLRP3, NLR family
pyrin domain containing 3; mIL-1β, mature interluekin-1β; C,
control; ASC, apoptosis-associated speck-like protein containing a
CARD. |
The protein expression levels of NLRP3
inflammasome-associated molecules in the control group were
detected at 12, 16, 20 and 33 weeks, and normalized to β-actin as a
control. It was identified that there was no alteration in the
expressions of these molecules in the control group at different
weeks (Fig. 2F and G), thus we
randomly selected the 16-week-old control rats as the control group
for our further examination. We detected the protein level of NLRP3
inflammasome in the DN rat kidneys and the result revealed that the
protein expression levels of NLRP3, ASC and caspase-1 in the DN
rats at 12, 16, 20 and 33 weeks were significantly increased
compared with the control group rats at 16 weeks (Fig. 2H and I; P<0.05) with the
exception of ASC at week 33. Among them, the most marked difference
in expression was observed in 16-week-old rats (Fig. 2H and I); therefore, we selected the
16-week-old DN rats as our experimental group for further
examination. Three different rats from the control group at 16
weeks (C1, C2 and C3) and three different rats from the DN group at
16 weeks (DN1, DN2 and DN3) were selected for further evaluation.
As demonstrated in Fig. 2J and K,
in addition to upregulation of NLRP3, the expression of
caspase-1p20, an active form of caspase-1, was significantly higher
compared with the control group (Fig.
2K; P<0.05). Simultaneously, the protein expression level of
mIL-1β was significantly increased (Fig. 2K; P<0.01).
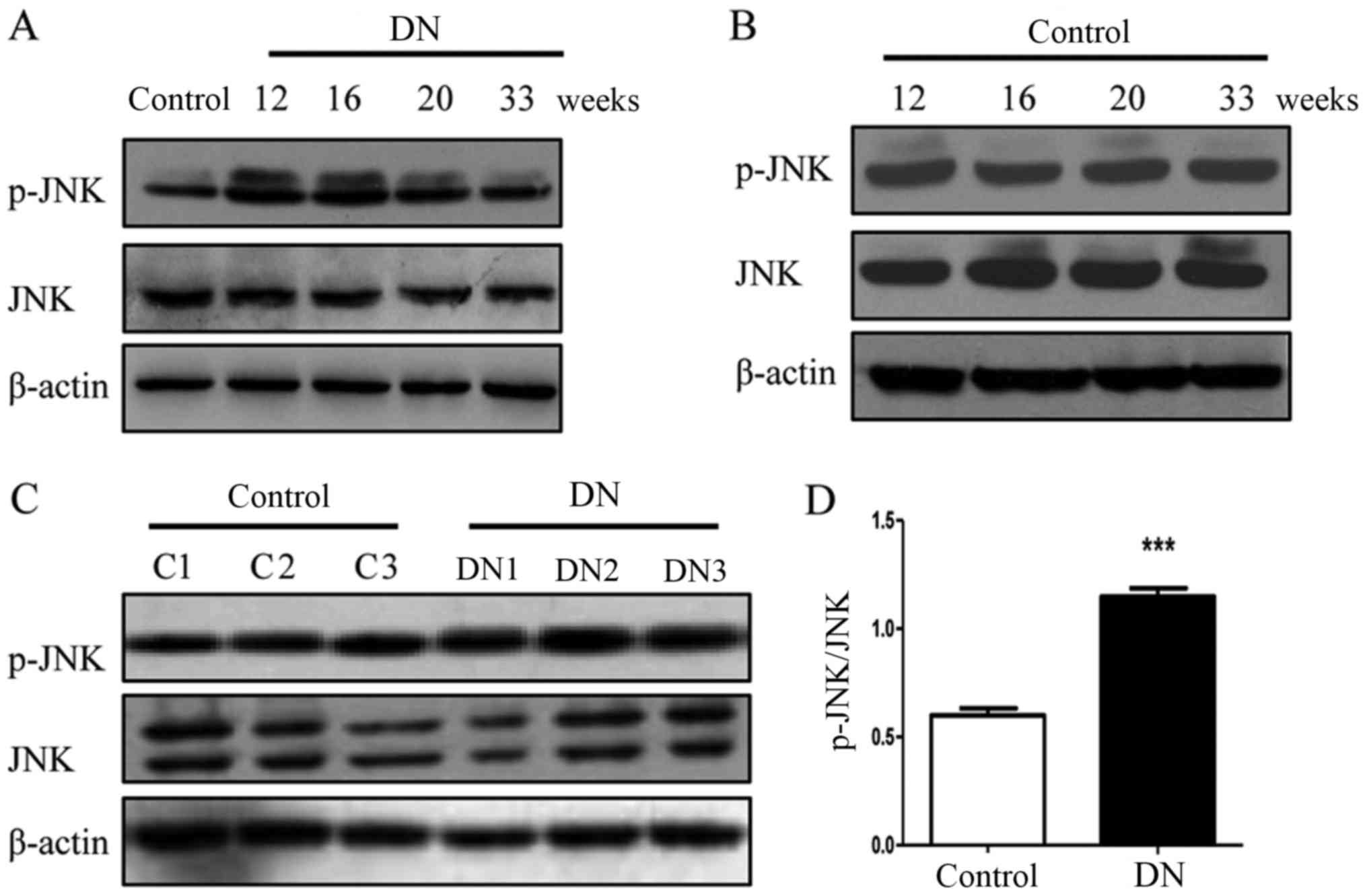
Phosphorylation of JNK is increased in
rats with DN
In a similar trend to the NLRP3
inflammasome-associated molecules, the phosphorylation of JNK was
increased in the diabetic rats at 12, 16, 20 and 33 weeks (Fig. 3A and B) and the phosphorylation
levels of JNK in the three 16-week-old DN rats (DN1, DN2 and DN3)
were significantly increased compared with the three 16-week-old
rats (C1, C2 and C3) in the control group (P<0.001; Fig. 3C and D). These results suggest that
JNK is involved in the pathogenesis of DN.
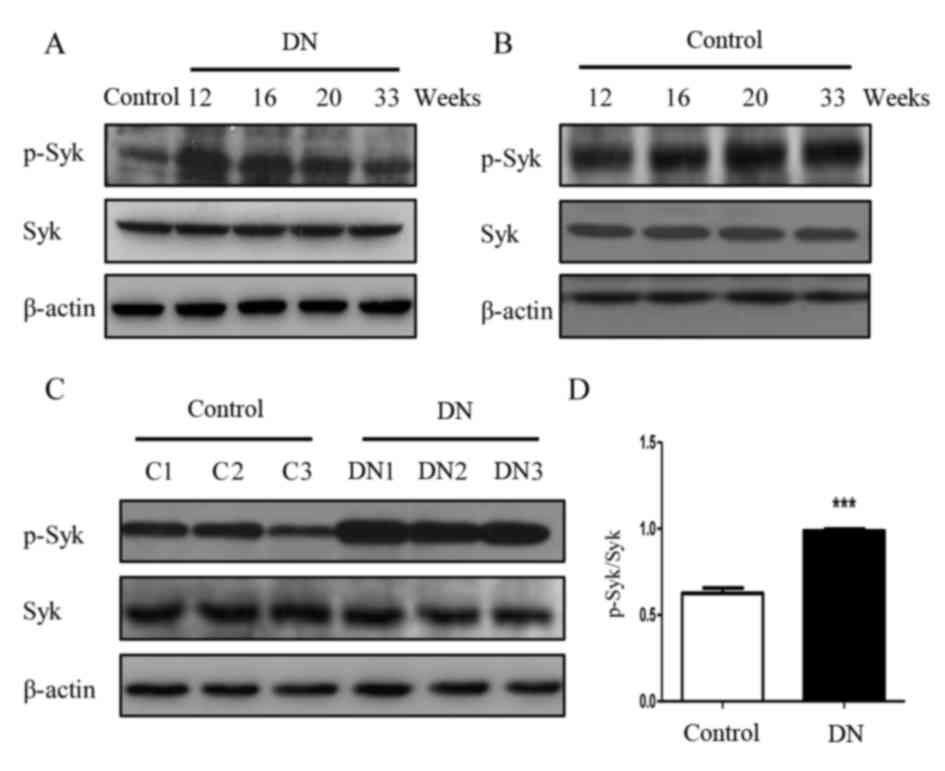
Syk is activated in rats with DN
To determine whether Syk is involved in the
pathogenesis of DN, the protein expression of Syk was detected in
renal tissue. Western blot analysis demonstrated that level of
p-Syk in the diabetic group rats appeared increased compared with
the control group at the corresponding 12, 16, 20 and 33 weeks
(Fig. 4 and B). Further confirming
these observations, phosphorylation of Syk in the three different
16-week-old DN rats (DN1, DN2 and DN3) kidney was significantly
increased compared with the three 16-week-old rats (C1, C2 and C3)
in the control group (P<0.001; Fig.
4C and D).
Inhibition of JNK attenuates high
glucose-induced NLRP3 inflammasome activation in HK2 cells
To confirm the role of JNK, HK2 cells were treated
with or without the JNK inhibitor (10 µM) for 2 h, and subsequently
exposed to 5 or 25 mM glucose for 24 h. As demonstrated in Fig. 5A and B, high glucose (25 mM)
induced a significant increase in the phosphorylation level of JNK,
particularly at 10 and 20 min, compared with normal glucose
(P<0.01). Furthermore, the protein expression levels of NLRP3
and ASC were also increased subsequent to treatment with high
glucose, particularly at 12, 24 and 36 h. The JNK inhibitor
downregulated the high glucose-induced increased expression of
NLRP3, caspase-1p20 and ASC expression and along with a decrease in
the maturation of IL-1β (Fig. 5C and
D), confirming that JNK may have a critical role in
NLRP3-dependent maturation of IL-1β during the development of
DN.
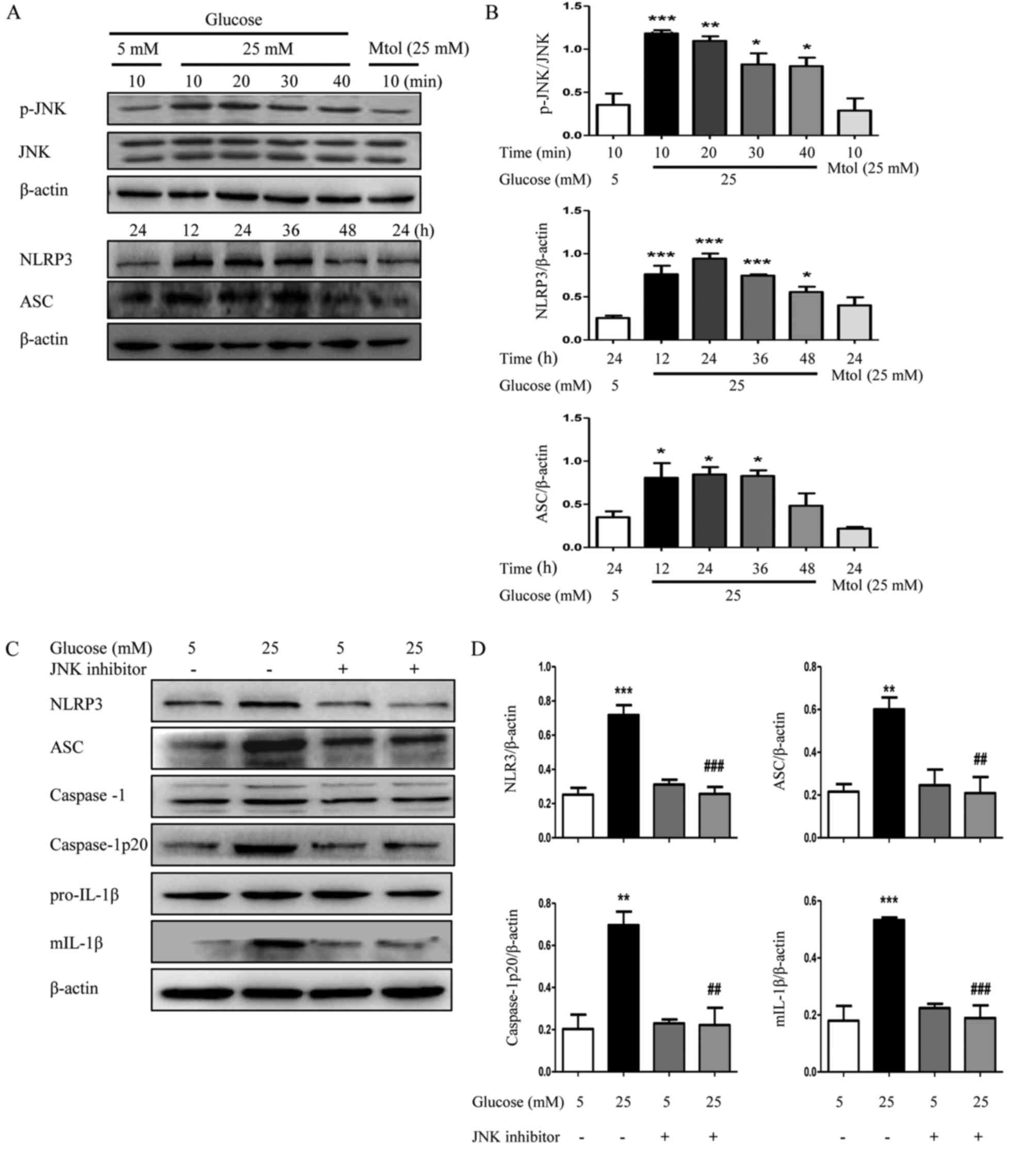 | Figure 5.Inhibition of JNK suppresses NLRP3
inflammasome activation in HK2 cells. (A) HK2 cells were cultured
in Dulbecco's modified Eagle's medium/F12 (1:1) containing 5 mM
glucose or 25 mM glucose for different times. The protein
expression levels of p-JNK, NLRP3 and ASC were detected by western
blotting. (B) Densitometry analysis of p-JNK, NLRP3 and ASC. (C)
The protein expression levels of NLRP3, ASC, caspase-1,
caspase-1p20, pro-IL-1β and mIL-1β of HK2 cells were detected by
western blotting following treatment with JNK inhibitor (10 µM) and
(D) densitometry analysis was performed. The data are presented as
the mean ± standard error of the mean of three independent
experiments. Statistical significance was determined by one-way
analysis of variance followed by Dunnett's post hoc test (for A and
B) or Tukey's post hoc test (for C and D). *P<0.05, **P<0.01,
***P<0.001 vs. 5 mM glucose. ##P<0.01,
###P<0.001 vs. 25 mM glucose. Mtol, mannitol; p,
phosphorylated; JNK, c-Jun N-terminal kinase; NLRP3, NLR family
pyrin domain containing 3; ASC, apoptosis-associated speck-like
protein containing a CARD; mIL-1β, mature interleukin 1β. |
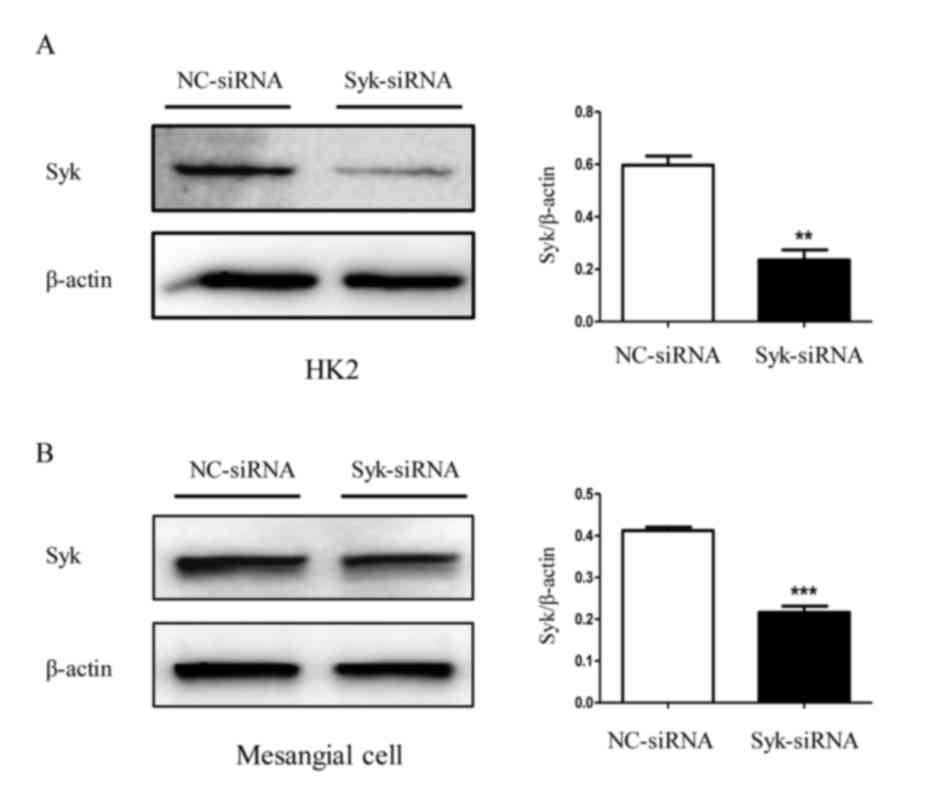
Syk is involved in JNK-dependent NLRP3
inflammasome activation in high glucose-induced HK2 cells and
RGMCs
The role of Syk in JNK-dependent NLRP3 inflammasome
activation induced by high glucose was further investigated. The
protein level of Syk in HK2 cells and RGMCs was evaluated following
transfection with Syk-siRNA. As expected, Syk protein levels were
significantly decreased by Syk-siRNA transfection compared with
control (Fig. 6A and B). As
demonstrated in Fig. 7A,
phosphorylation of Syk was increased following treatment with high
glucose, significantly at 10 and 20 min compared with normal
glucose levels (P<0.05). By contrast, the hyperosmotic Mtol
control group exhibited no effect on the production of p-Syk in HK2
cells. In Fig. 7B, high glucose
increased the phosphorylation of JNK and the addition of BAY61-3606
decreased high glucose-induced phosphorylation of JNK. The
increased protein expression of downstream molecules, including
NLRP3, ASC, caspase-1p20 and mIL-1β induced by high glucose was
significantly reduced following Syk inhibition (Fig. 7C and D; P<0.05). The addition of
Syk-siRNA demonstrated a similar effect. The expression of p-JNK
and NLRP3 inflammasome was markedly decreased in Syk-siRNA-treated
HK2 cells compared with high glucose-treated HK2 cells (Fig. 7E-G), which suggests an important
role of Syk in regulating JNK-dependent activation of NLRP3
inflammasome and subsequent maturation of IL-1β upon stimulation
with high glucose.
 | Figure 7.Syk is involved in JNK-dependent
NLRP3 inflammasome activation in high glucose-induced HK2 cells.
(A) Level of p-Syk was determined in HK2 cells. (B) Effects of
BAY61-3606 on high glucose-induced phosphorylation of JNK
determined by western blotting. (C) Protein expression level of
NLRP3, ASC, caspase-1, caspase-1p20, pro-IL-1β and mIL-1β were
examined by western blotting in HK2 cells and (D) analyzed using
densitometry. (E) HK2 cells transfected with siRNA against Syk were
exposed to high glucose condition for 48 h and the expression of
p-JNK was measured and quantified. (F) HK2 cells were additionally
examined to determine the protein expression levels of NLRP3, ASC,
caspase-1, caspase-1p20, pro-IL-1β and mIL-1β by western blotting
and (G) densitometry was performed using ImageJ software. The data
are presented as the mean ± standard error of the mean of three
independent experiments. *P<0.05, **P<0.01 vs. 5 mM glucose
by one-way ANOVA followed by Dunnett's post hoc test (for A).
#P<0.05, ##P<0.01,
###P<0.001 vs. 25 mM glucose by one-way ANOVA
followed by Tukey's post hoc test (for B-G). ANOVA, analysis of
variance; NC, negative control; siRNA, small interfering RNA; Syk,
spleen tyrosine kinase; p, phosphorylated; JNK, c-Jun N-terminal
kinase; NLRP3, NLR family pyrin domain containing 3; ASC,
apoptosis-associated speck-like protein containing a CARD; mIL-1β,
mature interleukin 1β. |
It was additionally demonstrated that Syk had a
critical role in the JNK/NLRP3/IL-1β pathway in high glucose
induced RGMCs. As demonstrated in Fig.
8A-C, the Syk inhibitor BAY61-3606 significantly decreased the
level of p-JNK and the downstream molecules, including NLRP3, ASC,
caspase-1p20 and mIL-1β, induced by high glucose (P<0.05).
Similar effects were also observed in Syk-siRNA-treated cells
(Fig. 8D-F). Taken together, these
data demonstrated that Syk acts upstream of JNK and NLRP3
inflammasome in RGMCs.
 | Figure 8.Syk is involved in JNK-dependent
NLRP3 inflammasome activation in high glucose-induced RGMCs. (A)
Level of p-JNK was examined by western blotting in RGMCs following
treatment with BAY61-3606 (1 µM) and analyzed by densitometry. (B)
Protein expression level of NLRP3, ASC, caspase-1, caspase-1p20,
pro-IL-1β and mIL-1β were examined by western blotting in RGMCs,
and (C) analyzed by densitometry. (D) RGMCs transfected with siRNA
against Syk were exposed to high glucose condition for 48 h and the
phosphorylation of JNK was examined. (E) RGMCs were additionally
examined to determine the protein expression levels of NLRP3, ASC,
caspase-1, caspase-1p20, pro-IL-1β and mIL-1β by western blotting
and (F) analyzed using ImageJ. The data are presented as the mean ±
standard error of the mean of three independent experiments.
*P<0.05, **P<0.01, ***P<0.001 vs. 5 mM glucose and
NC-siRNA. #P<0.05, ##P<0.01 vs. 25 mM
glucose and NC-siRNA by one-way analysis of variance followed by
Tukey's post hoc test. RGMCs, rat glomerular mesangial cells; p,
phosphorylated; JNK, c-Jun N-terminal kinase; NLRP3, NLR family
pyrin domain containing 3; ASC, apoptosis-associated speck-like
protein containing a CARD; IL-1β, interleukin 1β; mIL-1β, mature
IL-1β; NC, negative control; siRNA, small interfering RNA; Syk,
spleen tyrosine kinase. |
Syk is involved in high
glucose-induced apoptosis of HK2 cells
The aforementioned results collectively support that
Syk is involved in the process of apoptosis; to verify the
implication of Syk in the pathomechanism of apoptosis in DN, a
series of experiments were performed. HK2 cells were pretreated
with high glucose for 36 h and the protein expression levels of Bax
and Bcl-2 were subsequently determined by western blotting. As
demonstrated in Fig. 9A,
expression of Bax was significantly increased and Bcl-2
significantly decreased by high glucose (P<0.001). Furthermore,
high glucose-induced activation of Bax was decreased and Bcl-2 was
increased in HK2 cells upon incubation with the Syk inhibitor
BAY61-3606 (Fig. 9B).
Additionally, the flow cytometry analysis demonstrated that the
inhibition of Syk significantly reduced apoptosis of HK2 cells in
high glucose (Fig. 9C). Although
the apoptotic rate of HK2 cells in 5 mM glucose+BAY61-3606 treated
group was higher than that in 5 mM glucose treated group, that was
not statistically significant (data not shown). Taken together,
these results indicated that Syk was involved in high
glucose-induced apoptosis of HK2 cells; however, the specific
mechanism requires further investigation.
Discussion
DN is a serious complication of DM, with 25–40% of
patients with type 1 DM developing DN within 20–25 years of
diabetes and leads to a high mortality rate worldwide (15). Therefore, finding novel therapeutic
strategies against DN is an important unmet medical requirement at
present (16). Previous studies
reported that the immune-mediated inflammatory response
participates in the development of DN. Numerous inflammatory
cytokines, including IL-1β, IL-18, tumor necrosis factor-α, C-C
motif chemokine 2 and intercellular adhesion molecule 1 are
significantly increased in renal tissues during DN and attenuating
the expression of these cytokines may protect against diabetic
renal injury (17–19). In the present study, it was
demonstrated that the expression of NLRP3 was upregulated in
high-glucose induced HK2 cells, which also led to upregulation of
ASC expression, cleavage of caspase-1 and maturation of IL-1β.
Furthermore, it was identified that the phosphorylation levels of
Syk and JNK were significantly increased in the DN kidneys compared
with control animals. While, the increase in phosphorylation of Syk
and JNK appears to have been diminished over time. Kanellis et
al (20) demonstrated that in
I/R rat kidneys, delaying JNK inhibitor treatment until 1 h
following reperfusion conferred no benefit, combined with the
present results, it may suggest that the early peak of JNK
activation is the main pathologic event during kidney injury. To
examine the effects of Syk on the inflammasome pathway during the
pathogenesis of DN, the core inflammatory molecular expression was
investigated in HK2 cells and RGMCs. BAY61-3606 inhibited
JNK-mediated expression of inflammasome genes, including NLRP3,
ASC, caspase-1 and mIL-1β. Similarly, Syk-siRNA reduced the high
glucose-induced upregulation of p-JNK and decreased the expression
of NLRP3, ASC, caspase-1p20 and mIL-1β in HK2 cells and RGMCs.
These results suggest that Syk is a pivotal protein in regulating
the pathophysiology of HK2 cells and RGMCs under high glucose
condition and Syk inactivation is crucial for protective effects on
high glucose-treated HK2 cells and RGMCs.
A previous study demonstrated that high glucose may
induce the expression of NLRP3 and pro-caspase-1 in mesangial
cells, which leads to the maturation of inflammatory cytokines
through proteolysis and tissue inflammation (21). Okada et al (11) reported that JNK regulates the NLRP3
inflammasome through the oligomerization of ASC in THP-1 cells. In
addition, Syk served a crucial role in mediating NLRP3
stimuli-induced processing of pro-caspase-1 and the consequent
activation of caspase-1 in 293T cells, and Syk may directly
associate with NLRP3 and ASC, and, interact indirectly with
pro-caspase-1 (3). Furthermore,
substantial amount evidence supports that Syk is required for
activation of JNK signaling, acute neutrophil-mediated glomerular
injury and cell death (22,23).
Therefore, it was hypothesized that Syk serves a key role in
activating JNK signaling, and subsequently induces activation of
the NLRP3 inflammasome and mIL-1β during the development of DN. The
present study suggested that the Syk/JNK/NLRP3 signaling pathway is
a novel signaling pathway involved in DN. Similarly, it was also
identified that the Syk/JNK/NLRP3 pathway served an important role
in diabetic cardiomyopathy, Syk-siRNA and JNK-siRNA attenuated high
glucose-induced upregulation of NLRP3 (data not shown). Thus, this
signaling pathway may serve a pivotal role in renal and cardiac
function.
Short-term application of inhibitors in vivo
was usually selected (24,25), and the long-term application of
inhibitors (>20 days) is mainly focused on diseases that are
accompanied with few complications and little influence on basic
metabolism (26,27). The present study was conducted over
4–5 months and it is noteworthy that diabetes deteriorates with
time, thus the same dose may exert different effects on diabetic
rats at different time points. Therefore, a specific dose of
inhibitor, which may be effective in an early stage, may not exert
an effect in a later stage. In addition, the internal factors are
complex and the application of inhibitors in vivo may not be
targeted to the kidney. Thus, the effects of Syk and JNK inhibitors
in the STZ-induced diabetic rats was not assessed and only the
association of Syk, JNK, NLRP3 inflammasome and IL-1β in two types
of cells in vitro was clarified. Based on the histological
examination, diffuse lesions were observed in the diabetic rat
kidneys. HK2 cells are frequently used in studies associated with
renal inflammatory process (28,29),
thus HK2 cells were selected for examination in the present study.
Mesangial cells are additionally frequently examined in studies
concerning kidney diseases (30,31);
therefore, the effect of Syk on RGMCs under high glucose condition
was additionally examined. Taken together, it was demonstrated that
the Syk signaling pathway was involved in renal tubular injury and
glomerular injury by high glucose in the present study.
Previous studies have demonstrated that high glucose
rapidly activates Syk, which leads to tyrosine phosphorylation of
nuclear factor (NF)-κB inhibitor α and thus activates NF-κB in
proximal tubular epithelial cells and glomerular mesangial cells;
while deficiency of Syk reverses the effect (32–34),
which indicates NF-κB may be involved in the Syk signaling pathway
under high glucose conditions. Excessive production of reactive
oxygen species (ROS) may promote the generation of various
cytokines and stimulate the activation of signaling pathways to
affect the bioactivity of renal cells, which may ultimately
initiate and participate in the pathogenesis of DN (35). Wei et al (36) demonstrated that ROS production
leads to activation of mitogen-activated protein kinase 3/1, JNK
and NF-κB transcription factor in podocyte. Furthermore, high
glucose was able to induce mesangial cell proliferation and
fibronectin expression through the JNK/NF-κB/NADPH oxidase/ROS
signaling pathways (37) and
activate the pathway of ROS/thioredoxin-interacting protein
(TXNIP)/NLRP3 inflammasome signaling and results in the release of
IL-1β in GMCs (24). TXNIP is
implicated in the activation of ROS in rats and humans with DN and
closely associated with renal fibrosis (38,39).
Thus, the specific roles of inflammatory molecules, including ROS,
NF-κB and TXNIP in the Syk/JNK/NLRP3 signaling pathway require
further examination in DN. Gasdermin-D (Gsdmd) is a generic
substrate for caspase-1 and caspase-4/5/11 and is additionally
associated with NF-κB (40,41).
The function of Gsdmd in DN requires clarification.
Previous studies suggest that the mechanisms of
apoptosis involved in the pathogenesis of DN primarily includes
hyperglycemia-mediated oxidative stress-induced apoptosis (42), endoplasmic reticulum stress-induced
apoptosis (43), and pro-apoptotic
(including Bax and Bcl-2-associated agonist of cell death) and
anti-apoptotic (including Bcl-2 and Bcl-xl) Bcl-2 family
proteins-mediated apoptosis. However, it was observed that Syk
serves an essential role in numerous types of cells, including
T-cell non-Hodgkin lymphoma cell lines, human retinoblastoma cells,
breast cancer cells, immunocytes and neurons (44–48);
therefore, it was investigated whether Syk is involved in the
mechanism of apoptosis in high glucose-induced HK2 cells. It was
demonstrated that high glucose indeed increased the apoptosis of
HK2 cells, and the expression of pro-apoptotic protein Bax was
markedly increased; whereas, anti-apoptotic protein Bcl-2 was
decreased. The Syk inhibitor eliminated these alterations. All the
present data demonstrated that Syk was involved in high
glucose-induced apoptosis in HK2 cells; however, the specific
mechanism requires further investigation.
In conclusion, the present study demonstrated that
the NLRP3 inflammasome acts as a sensor and a regulator of the
inflammatory response in DN, resulting in cleavage of pro-caspase-1
and maturation of cytokine IL-1β. The phosphorylation of Syk may
predominantly increase the phosphorylation level of JNK and the
expression of its downstream molecules, including NLRP3,
caspase-1p20, ASC and mIL-1β in high glucose-induced HK2 cells and
RGMCs, which may be inhibited by the Syk inhibitor BAY61-3606 or
Syk-siRNA. Furthermore, Syk was involved in high glucose-induced
apoptosis of HK2 cells. However, the effect of Syk and JNK
inhibitors on the STZ-induced diabetic rats was not detected,
therefore, the specific mechanism requires further examination. The
present results may help to clarify the cellular and molecular
basis of the pathogenesis in DN, providing a novel potential target
for the treatment of DN.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Basic Research Program of China (973 Program; grant no.
2015CB553605), National Natural Science Foundation of China (grant
nos. 81772252, 31400762 and 81200116), the Natural Science
Foundation of Tianjin (grant no. 15JCYBJC49700), the Natural
Science Foundation of Tianjin Medical University (grant no.
2014KYQ12), the Key Laboratory of Myocardial Ischemia, Harbin
Medical University, Chinese Ministry of Education (grant no.
KF201303).
Availability of data and materials
The datasets used during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
YaS and YCu conceived the present study and edited
the manuscript. YQ and XT performed the experiments and wrote the
manuscript. LM was responsible for the detection of renal function
of all rats. SL and MX performed the HE and PAS staining, and
analyzed data. YCh, YH, PZ and GL participated in the construction
of the DN model. YuS and RL were responsible for rat blood glucose
monitoring and management of rats. YL and ZQ provided advice and
guidance for the implementation of the experiments. All authors
discussed the results and implications and commented on the
manuscript at all stages.
Ethics approval and consent to
participate
All the experimental procedures in the present study
were approved by the Animal Care and Welfare Committee of Tianjin
Medical University (Hexi, China). The animal use protocol was
reviewed and approved by the Animal Ethical and Welfare Committee
on 10th January 2017.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang YW, Wang YG, Luo MY, Wu H, Kong LL,
Xin Y, Cui WP, Zhao YJ, Wang JY, Liang G, et al: Novel curcumin
analog C66 prevents diabetic nephropathy via JNK pathway with the
involvement of p300/CBP-mediated histone acetylation. Biochim
Biophys Acta. 1852:34–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sun XY, Qin HJ, Zhang Z, Xu Y, Yang XC,
Zhao DM, Li XN and Sun LK: Valproate attenuates diabetic
nephropathy through inhibition of endoplasmic reticulum
stress-induced apoptosis. Mol Med Rep. 13:661–668. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gao C, Huang W, Kanasaki K and Xu Y: The
role of ubiquitination and sumoylation in diabetic nephropathy.
Biomed Res Int. 2014:1606922014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Prasad N, Gupta P, Jain M, Bhadauria D,
Gupta A, Sharma RK and Kaul A: Outcomes of De Novo allograft
diabetic nephropathy in renal allograft recipients. Exp Clin
Transplant. 11:215–221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Samra YA, Said HS, Elsherbiny NM, Liou GI,
El-Shishtawy MM and Eissa LA: Cepharanthine and Piperine ameliorate
diabetic nephropathy inrats:role of NF-κB and NLRP3 inflammasome.
Life Sciences. 157:187–199. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stryker LS: Modifying risk factors:
Strategies that work diabetes mellitus. J Arthroplasty.
31:1625–1627. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hara H, Kohsuke Tsuchiya, Kawamura I, Fang
RD, Cuellar EH, Shen YN, Mizuguchi J, Schweighoffer E, Tybulewicz V
and Masao Mitsuyama: Phosphorylation of ASC acts as a molecular
switch controlling the formation of speck-like aggregates and
inflammasome activity. Nat Immunol. 14:1247–1255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wada J and Makino H: Inflammation and the
pathogenesis of diabetic nephropathy. Clin Sci (Lond). 124:139–152.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang C, Pan Y, Zhang QY, Wang FM and Kong
LD: Quercetin and allopurinol ameliorate kidney injury in
STZ-treated rats with regulation of renal NLRP3 inflammasome
activation and lipid accumulation. PLoS One. 7:e382852012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Keller M, Rüegg A, Werner S and Beer HD:
Active caspase-1 is a regulator of unconventional protein
secretion. Cell. 132:818–831. 2007. View Article : Google Scholar
|
|
11
|
Okada M, Matsuzawa A, Yoshimura A and
Ichijo H: The lysosome rupture-activated TAK1-JNK pathway regulates
NLRP3 inflammasome activation. J Biol Chem. 289:32926–32936. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang WS, Chang JW, Han NJ, Lee SK and Park
SK: Spleen tyrosine kinase mediates high glucose-induced
transforming growth factor-β1 up-regulation in proximal tubular
epithelial cells. Exp Cell Res. 318:1867–1876. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ryan MJ, Johnson G, Kirk J, Fuerstenberg
SM, Zager RA and Torok-Seorb B: HK-2: An immortalized proximal
tubule epithelial cell line from normal adult human kidney. Kidney
Int. 45:48–57. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gennero I, Fauvel J, Nieto M, Cariven C,
Gaits F, Briand-Mésange F, Chap H and Salles JP: Apoptotic effect
of sphingosine 1-phosphate and increased sphingosine 1-phosphate
hydrolysis on mesangial cells cultured at low cell density. J Biol
Chem. 277:12724–12734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Elsherbiny NM and Al-Gayyar MM: The role
of IL-18 in type 1 diabetic nephropathy: The problem and future
treatment. Cytokine. 81:15–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qian X, Li XH, Ma FF, Luo SS, Ge RW and
Zhu YZ: Novel hydrogen sulfide-releasing compound,
S-propargyl-cysteine, prevents STZ-induced diabetic nephropathy.
Biochem Biophys Res Commun. 473:931–938. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pan Y, Zhang XH, Wang Y, Cai L, Ren LQ,
Tang LG, Wang JY, Zhao YJ, Wang YG, Liu Q, et al: Targeting JNK by
a new curcumin analog to inhibit NF-κB-mediated expression of cell
adhesion molecules attenuates renal macrophage infiltration and
injury in diabetic mice. PLoS ONE. 8:e790842013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jun W and Hirofumi M: Inflammation and the
pathogenesis of diabetic Nephropathy. Clin Sci (Lond). 124:139–152.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim SM, Lee SH, Kim YG, Kim SY, Seo JW,
Choi YW, Kim DJ, Jeong KH, Lee TW, Ihm CG, et al:
Hyperuricemia-induced NLRP3 activation of macrophages contributes
to the progression of diabetic nephropathy. Am J Physiol Renal
Physiol. 308:F993–F1003. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kanellis J, Ma FY, Kandane-Rathnayake R,
Dowling JP, Polkinghorne KR, Bennett BL, Friedman GC and
Nikolic-Paterson DJ: JNK signaling in human and experimental renal
ischaemia/reperfusion injury. Nephrol Dial Transplant.
25:2898–2908. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Feng H, Gu JL, Gou F, Huang W, Gao CL,
Chen G, Long Y, Zhou XQ, Yang MJ, Liu S, et al: High glucose and
lipopolysaccharide prime NLRP3 inflammasome via ROS/TXNIP pathway
in mesangial cells. J Diabetes Res. 2016:69731752016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ryan J, Ma FY, Kanellis J, Delgado M,
Blease K and Nikolic-Paterson DJ: Spleen tyrosine kinase promotes
acute neutrophil-mediated glomerular injury via activation of JNK
and p38 MAPK in rat nephrotoxic serum nephritis. Lab Invest.
91:1727–1738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee CK, Yang Y, Chen C and Liu J:
Syk-mediated tyrosine phosphorylation of Mule promotes TNF-induced
JNK activation and cell death. Oncogene. 35:1988–1995. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu HM, Fang L, Shen QY and Liu RY:
SP600125 promotes resolution of allergic airway inflammation via
TLR9 in an OVA-induced murine acute asthma model. Mol Immunol.
67:311–316. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shen H, Wu N, Wang Y, Han X, Zheng Q, Cai
X, Zhang H and Zhao M: JNK inhibitor SP600125 attenuates
paraquat-induced acute lung injury: An in vivo and in vitro study.
Inflammation. 40:1319–1330. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Long AJ, Sampson E, McCarthy RW, Harris
CM, Barnard M, Shi D, Conlon D, Caldwell R, Honor D, Wishart N, et
al: Syk Inhibition induces platelet dependent peri-islet hemorrhage
in the rat pancreas. Toxicol Pathol. 44:998–1012. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Llop-Guevara A, Porras M, Cendón C, Di
Ceglie I, Siracusa F, Madarena F, Rinotas V, Gómez L, van Lent PL,
Douni E, et al: Simultaneous inhibition of JAK and SYK kinases
ameliorates chronic and destructive arthritis in mice. Arthritis
Res Ther. 17:3562015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fu Y, Wang C, Zhang D, Xin Y, Li J, Zhang
Y and Chu X: Increased TRPC6 expression is associated with tubular
epithelial cell proliferation and inflammation in diabetic
nephropathy. Mol Immunol. 94:75–81. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen P, Yuan Y, Zhang Ty, Xu B, Gao Q and
Guan TJ: Pentosan polysulfate ameliorates apoptosis and
inflammation by suppressing activation of the p38 MAPK pathway in
high glucose-treated HK2 cells. Int J Mol Med. 41:908–914.
2018.PubMed/NCBI
|
|
30
|
Li J, Bao L, Zha D, Zhang L, Gao P, Zhang
J and Wu X: Oridonin protects against the inflammatory response in
diabetic nephropathy by inhibiting the TLR4/p38-MAPK and TLR4/NF-κB
signaling pathways. Int Immunopharmacol. 55:9–19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang J, Kan M and Wu GY: Bergenin
ameliorates diabetic nephropathy in rats via suppressing renal
inflammation and TGF-β1-Smads pathway. Immunopharmacol
Immunotoxicol. 38:145–152. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang WS, Kim JS, Han NJ, Lee MJ and Park
SK: Toll-like receptor 4/spleen tyrosine kinase complex in high
glucose signal transduction of proximal tubular epithelial cells.
Cell Physiol Biochem. 35:2309–2319. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang S, Yang Z, Xiong F, Chen C, Chao X,
Huang J and Huang H: Betulinic acid ameliorates experimental
diabetic-induced renal inflammation and fibrosis via inhibiting the
activation of NF-κB signaling pathway. Mol Cell Endocrinol.
434:135–143. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang WS, Seo JW, Han NJ, Choi J, Lee KU,
Ahn H, Lee SK and Park SK: High glucose-induced NF-kappaB
activation occurs via tyrosine phosphorylation of IkappaBaplha in
human glomerular endothelial cells: Involvement of Syk tyrosine
kinase. Am J Physiol Renal Physio. 1294:F1065–F1075. 2008.
View Article : Google Scholar
|
|
35
|
Qi W, Niu J, Qin Q, Qiao Z and Gu Y:
Glycated albumin triggers fibrosis and apoptosis via an NADPH
oxidase/Nox4-MAPK pathway-dependent mechanism in renal proximal
tubular cells. Mol Cell Endocrinol. 405:74–83. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wei MM, Li ZG, Xiao L and Yang Z: Effects
of ROS-relative NF-κB signaling on high glucose-induced TLR4 and
MCP-1 expression in podocyte injury. Mol Immunol. 68:261–271. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang LY, Pang SS, Deng B, Qian LH, Chen
J, Zou JJ, Zheng JY, Yang LH, Zhang CY, Chen XF, et al: High
glucose induces renal mesangial cell proliferation and fibronectin
expression through JNK/NF-NF-κB/NADPH oxidase/ROS pathway, which is
inhibited by resveratrol. Int J Biochem Cell Biol. 44:629–638.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Devi TS, Lee I, Hüttemann M, Kumar A,
Nantwi KD and Singh LP: TXNIP links innate host defense mechanisms
to oxidative stress and inflammation in retinal muller glia under
chronic hyperglycemia: Implications for diabetic retinopathy. Exp
Diabetes Res. 2012:4382382012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tan SM, Zhang Y, Cox AJ, Kelly DJ and Qi
WE: Tranilast attenuates the up-regulation of
thioredoxin-interacting protein and oxidative stress in an
experimental model of diabetic nephropathy. Nephrol Dial Transpl.
26:100–110. 2011. View Article : Google Scholar
|
|
40
|
Shi JJ, Zhao Y, Wang K, Shi XY, Wang Y,
Huang HW, Zhuang YH, Cai T, Wang FC and Shao F: Cleavage of GSDMD
by inflammatory caspases determines pyroptotic cell death. Nature.
526:660–676. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu ZJ, Lu Gan, Xu YT, Luo D, Ren Q, Song
Wu S and Sun C: Melatonin alleviates inflammasome-induced
pyroptosis through inhibiting NF-κB/GSDMD signal in mice adipose
tissue. J Pineal Res. 63:2017. View Article : Google Scholar
|
|
42
|
Pal PB, Sinha K and Sil PC: Mangiferin
attenuates diabetic nephropathy by inhibiting oxidative stress
mediated signaling cascade, TNFα related and mitochondrial
dependent apoptotic pathways in streptozotocin-induced diabetic
rats. PLoS One. 9:e1072202014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yao F, Li Z, Ehara T, Yang L, Wang D, Feng
L, Zhang Y, Wang K, Shi Y, Duan H and Zhang L: Fatty acid-binding
protein 4 mediates apoptosis via endoplasmic reticulum stress in
mesangial cells of diabetic nephropathy. Mol Cell Endocrinol.
411:232–242. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wilcox RA, Sun DX, Novak A, Dogan A,
Ansell SM and Feldman AL: Inhibition of Syk protein tyrosine kinase
induces apoptosis and blocks proliferation in T-cell non-Hodgkin
lymphoma cell lines. Leukemia. 24:229–232. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qiu Q, Yang C, Xiong W, Tahiri H, Payeur
M, Superstein R, Carret AS, Hamel P, Ellezam B, Martin B, et al:
SYK is a target of lymphocyte-derived microparticles in the
induction of apoptosis of human retinoblastoma cells. Apoptosis.
20:1613–1622. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang WH, Childress MO and Geahlen RL: Syk
interacts with and phosphorylates nucleolin to stabilize Bcl-x(L)
mRNA and promote cell survival. Mol Cell Biol. 34:3788–3799. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gobessi S, Laurenti L, Longo PG, Carsetti
L, Berno V, Sica S, Leone G and Efremov DG: Inhibition of
constitutive and BCR-induced Syk activation downregulates Mcl-1 and
induces apoptosis in chronic lymphocytic leukemia B cells.
Leukemia. 23:686–697. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Scheib JL, Sullivan CS and Carter BD:
Jedi-1 and MEGF10 signal engulfment of apoptotic neurons through
the tyrosine kinase Syk. J Neurosci. 32:13022–13031. 2012.
View Article : Google Scholar : PubMed/NCBI
|