Introduction
MYCN proto-oncogene bHLH transcription factor
(MYCN) amplification and 1p36 deletion are important factors
associated with poor prognosis in neuroblastoma (1–3), one
of the most types of infant malignancy (4). MYCN amplification, which leads
to MYCN overexpression, has been reported in 18–38% of cases
of neuroblastoma and in a panel of neuroblastoma cell lines
(3,5–9). As
a developmentally-regulated gene, MYCN is highly expressed
in dorsal root ganglia, sympathetic chain ganglia and the spinal
cord in the human fetus during the development of the sympathetic
nervous system at 8.5 weeks of gestation (9). In addition, the chromosome 1p36 locus
is frequently deleted in neuroblastoma cell lines (10). Chromodomain helicase DNA binding
protein 5, calmodulin binding transcription activator 1, kinesin
family member 1B β, castor zinc finger 1 and microRNA
(miR)−34a have been analyzed as the strongest
candidate tumor suppressor genes at the 1p36 locus in neuroblastoma
(11,12).
c-MYB proto-oncogene transcription factor (c-Myb)
has been reported to be associated with cell growth and
proliferation in neuroblastoma (13). On induction by retinoic acid,
c-myb and MYCN expression levels decrease during the
differentiation stage of neuroblastoma cells (14–18).
In humans, c-myb, MYB proto-oncogene like 2 (B-myb)
and MYB proto-oncogene like 1 (A-myb) belong to the
myb gene family, and contain highly-conserved N-terminal
domains (19). The functional
orthologs B-myb and Drosophila melanogaster-myb
(Dm-myb) share essential conserved functions required for
cell proliferation, whereas the paralogous genes, A-myb and
c-myb, have acquired novel functions (20). The Dm-Myb protein complex is
directly involved in DNA replication and binds to
amplification-control-element-on-3 (ACE3) and replication
origin-β in a site-specific manner (21), necessary for chorion gene
amplification on the third chromosome in Drosophila ovarian
follicle cells (22).
The Dm-myb complex regulates the expression of
developmentally-regulated genes (23). Additionally, it has been proposed
that this complex may be involved in the activation or repression
of transcription and DNA replication, depending on the presence of
E2F transcription factor 1 (E2F1) or E2F transcription factor 2
(E2F2) with other particular cofactors, respectively.
Drosophila lethal (3)
malignant brain tumor [D-L(3)mbt] protein has also been associated
with the Myb-MuvB repressor complex (23).
The human homolog of D-l(3)mbt, L3MBTL1 histone
methyl-lysine binding protein (L3MBTL1), is expressed in
cancer cell lines and a variety of normal human tissues (24). L3MBTL1, a candidate tumor
suppressor in del(20q12) myeloid disorders, is required for normal
progression of the replication fork, and interacts with the
minichromosome maintenance complex component 2–7, cell division
cycle 45 and proliferating cell nuclear antigen components of the
DNA replication machinery (25).
Chromatin licensing and DNA replication factor 1
(hCdt1) is an essential factor of the pre-replication complex and
is inhibited by geminin (GMNN) protein to prevent re-replication
during the S, G2 and M phases of the cell cycle
(26). GMNN and
hCdt1 are overexpressed in tumors and a variety of
cancer-derived cell lines (27–29).
MYCN transcriptionally activates the p53
tumor suppressor gene to induce apoptosis (30); however, MYCN suppresses the
cyclin dependent kinase inhibitor 1A (p21) gene,
resulting in anti-apoptotic activity of neuroblastoma (31). MYCN overexpression
sensitizes MYCN-amplified neuroblastoma cells to apoptosis
via the induction of p53 (32). In
addition, E2F-regulated B-myb and c-myb expression
are elevated by apoptotic stimuli, causing neuronal death (33).
In the present study, potential c-Myb target genes,
and the effect of c-myb RNA interference (RNAi) on
MYCN expression and amplification in neuroblastoma were
investigated. For this, a plasmid vector-mediated RNAi method with
a short hairpin RNA (shRNA) directed against c-myb mRNA was
used in MYCN-amplified neuroblastoma cell lines. The present
study demonstrated that c-myb may induce the expression of
E2F1 and L3MBTL1 and that E2F1 may be
associated with the induction of MYCN, B-myb, p21 and
hCdt1 expression, in addition to the repression of
GMNN. In addition, the results demonstrated that MYCN
gene copy number was increased following treatment with
c-myb RNAi. These findings revealed that c-myb is
involved in controlling MYCN expression and amplification in
MYCN-amplified neuroblastoma cell lines. Following
c-myb RNAi treatment, L3MBTL1 expression was
completely silenced, whereas GMNN was upregulated; the
results indicate G2/M arrest. Consequently, the present
study demonstrated that the MYCN gene may be amplified
during S phase, which may occur via a replication-based
mechanism.
Materials and methods
Sequence comparison
The DNA sequences encompassing the D.
melanogaster-ACE3 element and that upstream of human
MYCN were compared using the LFASTAn alignment program
(version 2;
bioinfo.hku.hk/services/analyseq/cgi-bin/lfastan_in.pl). The DNA
sequences of D. melanogaster-chorion gene cluster (GenBank
accession no. X02497.1) and Homo sapiens-chromosome 2
genomic contig, including the MYCN gene (NCBI reference
sequence NT_005334.16; region, 8493966-11135164) were downloaded
from the NCBI website (ncbi.nlm.nih.gov).
Transcription factor binding site
search
Transcription factor binding sites upstream (−1,021
to −143), including the enhancer and proximal promoter of
MYCN gene were investigated using the TFSEARCH program
(version 1.3; cbrc.jp/research/db/TFSEARCH.html). In addition, the
location information of the regulatory transcription factor binding
sites in the promoters of all genes investigated in the present
study was obtained from Qiagen, Inc. (Valencia, CA, USA) as
predicted by Text Mining Application (SABioscience Corporation;
Qiagen, Inc.) and the University of California Santa Cruz (UCSC)
Genome Browser (sabiosciences.com/chipqpcrsearch.php?app=TFBS).
Cell culture
Kelly (no. ACC 355), IMR32 (no. ACC 165), SIMA (no.
ACC 164), MHH-NB-11 (no. ACC 157) and SH-SY5Y (no. ACC 209) cell
lines were purchased from the Leibniz Institute DSMZ-German
Collection of Microorganisms and Cell Cultures GmbH (Braunschweig,
Germany). Kelly, SIMA and MHH-NB-11 cells were cultured in
RPMI-1640 (cat. no. FG1215; Biochrom AG; Merck KGaA, Darmstadt,
Germany) supplemented with 10% fetal bovine serum (cat. no. S0113;
FBS; Biochrom AG; Merck KGaA), 2 mM L-glutamine, 100 U/ml
penicillin and 100 µg/ml streptomycin. In addition, the culture
medium of MHH-NB-11 cells included 1X non-essential amino acids.
IMR32 cells were cultured in RPMI-1640 (Biochrom AG; Merck KGaA)
supplemented with 20% FBS, 2 mM L-glutamine, 100 U/ml penicillin,
100 µg/ml streptomycin and 1X non-essential amino acids. SH-SY5Y
cells were cultured in Dulbecco's modified Eagle's medium (cat. no.
FG0415; DMEM; Biochrom AG; Merck KGaA) supplemented with 20% FBS, 2
mM L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin.
Cells were incubated at 37°C in a humidified atmosphere containing
5% CO2.
Fluorescence in situ hybridization
(FISH)
FISH was performed as previously described (3,34).
In FISH experiments, MYCN gene (2p24)/Chromosome 2
α-Satellite (red/green; cat. no. PONC0224; Qbiogene, Inc.; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), 1p36/Chr 1 SE (Poseidon
probe red/green, cat. no. KB-10705; Kreatech Diagnostics Corp.,
Amsterdam, LG, Netherlands) and 1p36/1q25 (Vysis probe
orange/green, cat. no. 32-231004; Abbott Pharmaceutical Co. Ltd.,
Lake Bluff, IL, USA) probes were used.
Neuroblastoma cells were seeded into
25-cm2 tissue culture flasks and grown in culture medium
specific for each cell line (please see ‘Cell culture’
section for the characteristics of each growth medium) in a
humidified atmosphere containing 5% CO2 at 37°C. The
cells were detached with trypsin-EDTA (Biochrom AG; Merck KGaA) and
incubated with 80 µl colcemid (Biological Industries, Kibbutz Beit
Haemek, Israel) in a tube containing 5 ml culture medium in a 37°C
water bath for 30 min. Following centrifugation at 300 × g for 10
min at room temperature, the cell pellets were incubated with 8 ml
hypotonic solution (0.075 M) (Biochrom AG; Merck KGaA) in a 37°C
water bath for 30 min. For the fixation of the cell pellets, 5 ml
fresh Carnoy's solution (3:1 Methanol:Glacial Acetic Acid; Merck
KGaA) was used for 2–3 min at room temperature followed by
centrifugation at 300 × g for 10 min (repeated five times).
The homogenized cells were dropped onto slides, and
a 2X SSC (AppliChem GmbH, Darmstadt, Germany)/0.5% NP-40
(AppliChem) mixture was used for washing the slides in a 37°C water
bath for 30 min. The slides were dehydrated in an ethanol
(AppliChem) series of 70, 85 and 96%, respectively. The
double-stranded DNAs on the slides were denatured in 70% formamide
(AppliChem)/2X SSC (AppliChem) at 70°C for 2 min (5 min for
Abbott/Vysis 1p36 probe). The denaturation of probes was performed
in a 96°C water bath for 5 min (5 and 10 min at 75°C for Vysis and
Poseidon 1p36 probes, respectively). The DNAs were hybridized with
the probes via overnight incubation at 37°C in a hybridization box.
The 0.5X SSC (AppliChem)/0.1% SDS (Honeywell Riedel-de Haën AG,
Seelze, Germany), 1X PBD including NP40 (AppliChem) and Tween 20
(Santacruz Biotechnology, Inc., Dallas, Texas, USA) and 70% ethanol
(AppliChem), respectively, were used for washing the slides after
hybridization overnight. Cell nuclei were counterstained with
4′,6-diamidino-2-phenylindole II (DAPI II; cat. no. 30-804841;
Abbott Pharmaceutical Co. Ltd.) suspended in an antifade
solution.
FISH slides were analyzed under an epifluorescence
microscope (magnification, ×100; Nikon Eclipse E600, Nikon
Corporation, Tokyo, Japan) equipped with DAPI, FITC, rhodamine and
triple band-pass filter sets. FISH images from interphase nuclei
and metaphase spreads were captured using a high-sensitivity
monochrome charge-coupled device camera, which was integrated with
a Macintosh computer and processed with MacProbe imaging software
(version 4.0, PSI Scientific Systems, League City, TX, USA).
In FISH analyses, 2 red and 2 green signals for
MYCN and internal control per diploid genome were considered
normal; 3–5 red signals vs. 2 green signals per diploid genome were
scored as a low copy number of the MYCN gene; 6–10 red
signals vs. 2 green signals per diploid genome were scored as an
intermediate copy number of MYCN gene, whereas >10 red
signals vs. 2 green signals per diploid genome indicate high copy
number of MYCN.
For 1p36 and the internal control, 2 orange/red and
2 green signals per diploid genome were considered normal. Only 1
orange/red signal vs. ≥2 green signals per diploid genome was
classified as a 1p36 deletion. A total of 2 orange/red signals vs.
3 green signals per diploid genome and so forth (representing at
least one more signal number of control than that of the 1p36
probe) were considered to indicate an imbalance in 1p36 copy
number; 3 orange/red and 3 green signals per diploid genome were
classified as a 1p36-3/3 balanced alteration.
DNA extraction
Kelly, SIMA, IMR32, MHH-NB-11 and SH-SY5Y cells were
seeded into 75-cm2 tissue culture flasks and grown in a
culture medium until cells reached near-confluence in a humidified
atmosphere containing 5% CO2 at 37°C. Genomic DNA was
extracted using phenol:chloroform:isoamyl alcohol (25:24:1,
respectively), pH 8.0 (cat. no. A0889.0500; AppliChem GmbH,
Darmstadt, Germany) as previously described (35).
cDNA synthesis
Kelly, SIMA, IMR32, MHH-NB-11 and SH-SY5Y cells were
seeded into 24-well plates at a density of 4×104 cells
in 0.75 ml culture medium per well and grown in a culture medium
until cells reached near-confluence in a humidified atmosphere
containing 5% CO2 at 37°C. First-strand cDNAs were
synthesized directly from adherent cultured cells, without
requiring RNA purification or RNase H digestion steps, using the
FastLane Cell cDNA kit (cat. no. 215011; Qiagen, Inc.) according to
the manufacturer's protocol. The procedure of this kit involves
four steps: i) removing extracellular contaminants, ii) performing
cell lysis and RNA stabilization, iii) eliminating genomic DNA and
iv) producing the first-strand cDNA via reverse transcription. This
kit has been optimized for use particularly in real-time, two-step
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR). The genomic DNA from the FastLane lysate was eliminated
using gDNA Wipeout Buffer at 42°C for five min. The lysate was
placed immediately on ice. The reverse-transcription reaction was
performed at 42°C for 30 min; later reverse transcriptase enzyme
was inactivated for finishing reaction at 95°C for three min
according to the manufacturer's protocol.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
In neuroblastoma cell lines, the expression levels
of c-myb and potential target genes, and MYCN gene
copy number compared with hypoxanthine phosphoribosyltransferase 1
(HPRT1) and p53 reference genes, respectively, were
determined via qPCR on a LightCycler® 2.0 instrument
(Roche Diagnostics, Basel, Switzerland). Relative quantitative qPCR
experiments were performed using the LightCycler®
TaqMan® Master kit (cat. no. 04535286001; Roche Applied
Science, Penzberg, Germany) with prevalidated hydrolysis probes,
Universal ProbeLibrary Set, Human (cat. no. 04683633001; Roche
Applied Science) and primers according to the manufacturer's
protocol.
Probe and primer sets were designed using the
web-based ProbeFinder software (version 2.40) via the UPL assay
design center (www.universalprobelibrary.com). Primer pairs were
synthesized by Gene Link, Inc. (Hawthorne, NY, USA). NCBI accession
numbers, probe/primer sets and amplicon sizes are presented in
Table I.
 | Table I.Probe and primer pairs used for
quantitative polymerase chain reaction. |
Table I.
Probe and primer pairs used for
quantitative polymerase chain reaction.
| A, mRNA |
|---|
|
|---|
| NCBI accession
number | Probe catalogue
number | Right primer
(5′-3′) | Left primer
(5′-3′) | Amplicon size
(nt) |
|---|
| A-myb
BC101186.1 | 2, 04684982001 |
aagcaagtggctgggaca |
ctccttttaagaatgcgcttg | 75 |
| B-myb
X13293.1 | 26,
04687574001 |
gccagagacttccggacttt |
cccgagaagcagaagagga | 68 |
| c-myb
M15024.1 | 62,
04688619001 |
agctgcatgtgtggttctgt |
tgctcctaatgtcaaccgaga | 72 |
| MYCN
BC002712.2 | 55,
04688520001 |
cctcttcatcatcttcatcatctg |
ccacaaggccctcagtacc | 68 |
| E2F1
BC050369.2 | 5, 04685024001 |
ctgggtcaacccctcaag |
tccaagaaccacatccagtg | 75 |
| E2F2
BC053676.1 | 68,
04688678001 |
gccttgacggcaatcact |
ggacaaggccaacaagagg | 93 |
| CDK2
BC003065.2 | 50,
04688112001 |
cagaatctccagggaataggg |
cctcctgggctgcaaata | 104 |
| GMNN
AF067855.1 | 53,
04688503001 |
ccagaggttcaccattcagtc |
aactggcagaagtagcagaaca | 72 |
| hCdt1
AB053172.1 | 10,
04685091001 |
agcaggtgcttctccatttc |
gcggagcgtctttgtgtc | 117 |
| L3MBTL1
BC039820.1 | 82,
04689054001 |
ttccttcttcttgcttctcca |
agcgcagggaataccagag | 72 |
| p21
BC000275.1 | 70,
04688937001 |
agctgctcgctgtccact |
ccgaggcactcagaggag | 112 |
| p53
AB082923.1 | 12,
04685113001 |
ccctttttggacttcaggtg |
aggccttggaactcaaggat | 85 |
| p27
BC001971.1 | 1, 04684974001 |
cgggttaactcttcgtggtc |
agatgtcaaacgtgcgagtg | 130 |
| HPRT1
BC000578.2 | 73,
04688961001 |
cgagcaagacgttcagtcct |
tgaccttgatttattttgcatacc | 102 |
|
| B, DNA |
|
| NCBI accession
number | Probe catalogue
number | Right primer
(5′-3′) | Left primer
(5′-3′) | Amplicon size
(nt) |
|
| MYCN
Y00664.1 | 36,
04687949001 |
ggcctttagggtcagacaga |
tgaccagggtcatgcaacta | 60 |
| p53
U94788.1 | 23,
04686977001 |
ctctagccaagcttccatcc |
ttcagctcgggaaaatcg | 88 |
In the calculation of both mRNA expression levels
and gene copy number in neuroblastoma cell lines, the following
equations (∆Cp method) were used: Presumed as E=2,
∆Cp=(Cptarget-Cpreference) and
R=2−∆Cp (36). A heatmap for visualizing gene
expression and MYCN gene copy number within neuroblastoma
cell lines was produced using Heatmapper software (www.heatmapper.ca) (37).
In addition, MYCN copy number and gene
expression levels from MYCN-amplified neuroblastoma cell
lines prior to- and post-treatment with c-myb RNAi as
normalized to the Cp values of target and reference genes of
SH-SY5Y cells, were determined using the following equation (ΔΔCp
method): Presumed as E=2
R1=2-ΔΔCp=2-[ΔCptarget(sample - control)-ΔCpref(sample -
control)]
.
In the efficiency-corrected (dilution) method, to
determine the slope (S), the standard curves were produced
between mean Cp values and logarithms (logs) of five serial
starting template concentrations (ranging from 2–32 ng) using Excel
(v. 2; Turkish, Home and Student version/initial release date;
07/17/2007, Microsoft Corporation, Redmond, WA, USA). E and
R2 values were calculated using the following equations:
Etargetorref=10[-1/S]andR2=(Etarget)ΔCptarget(control -
sample)(Eref)ΔCpref(control - sample)
.
RNAi
In the MYCN-amplified Kelly, SIMA, IMR32 and
MHH-NB-11 cell lines, c-myb mRNA expression was dysregulated
using a specific shRNA-expressing pre-made plasmid DNA vector
(custom-made psiRNA-h7SKneo G1 kit; cat. no. ksirna3-n21;
InvivoGen, San Diego, CA, USA) according to the manufacturer's
protocols.
A shRNA insert in the double-stranded RNA
structure, which specifically targeted c-myb mRNA (NCBI
accession no. M15024.1), was designed using InvivoGen siRNA wizard
software (version 2.4; sirnawizard.com) and later ligated into the
psiRNA-h7SKneo G1 expression vector. Pre-made plasmid DNA vectors
(InvivoGen) expressing shRNAs for enhanced green fluorescent
protein (EGFP) or irrelevant genes served as shRNA controls.
Plasmid vectors containing the sequences of shRNA targeting
c-myb mRNA and control shRNAs were transformed into
E. coli LyoComp GT116 strain (InvivoGen).
For SpeI enzyme digestion and DNA
sequencing, plasmid DNA was extracted from transformed cells using
the High Pure Plasmid Isolation kit (Roche Applied Science,
Penzberg, Germany) according to the manufacturer's protocols.
Plasmid DNA vector expressing the shRNA directed against
c-myb mRNA and EGFP control vector were cut with the
SpeI restriction enzyme (cat. no. ER1252; Thermo Fisher
Scientific, Inc.) at a concentration of 10 U/µl (final
concentration 0.5 U/µl) incubated in a 37°C water bath for 14 h and
imaged following agarose gel electrophoresis (1%) stained with
ethidium bromide. DNA Marker II was used for genomic DNA analysis
(cat. no. SM0351; Fermentas; Thermo Fisher Scientific, Inc.). The
shRNA sequences of c-myb and EGFP vectors were confirmed by
plasmid DNA sequencing (Macrogen, Inc., Seoul, Korea) in both
directions using forward (OL559) and reverse (OL408) primers.
Primer sequences: OL559 primer (forward)
5′-CGATAAGTAACTTGACCTAAGTG-3′ and OL408 primer (reverse)
5′-GCGTTACTATGGGAACATAC-3′; c-myb shRNA, oligo 1 (forward)
5′-ACCTCGGTTATCTGCAGGAGTCTTCATCAAGAGTGAAGACTCCTGCAGATAACCTT-3′ and
oligo 2 (reverse)
5′-CAAAAAGGTTATCTGCAGGAGTCTTCACTCTTGATGAAGACTCCTGCAGATAACCG-3′;
EGFP shRNA, oligo 1 (forward)
5′-ACCTCGCAAGCTGACCCTGAAGTTCACCACCTGAACTTCAGGGTCAGCTTGCTT-3′ and
oligo 2 (reverse)
5′-CAAAAAGCAAGCTGACCCTGAAGTTCAGGTGGTGAACTTCAGGGTCAGCTTGCG-3′.
For stable transfection, purified plasmid DNA was
isolated in intermediate quantities using the Genopure Plasmid Midi
kit (Roche Applied Science). LyoVec reagent (InvivoGen) was used
for the transfection of plasmid DNAs (5–6 µg) into
MYCN-amplified neuroblastoma cell lines [cell number per
well (6-well plate): 8×105]. Selection and maintenance
of transfected cells expressing the neomycin-resistance
(neo) gene were performed with G418 treatment (InvivoGen) at
a concentration of 500–800 µg/ml for 4 weeks.
Statistical analysis
Statistical differences were determined using a
one-tailed Wilcoxon signed-rank test. For two-tailed analysis,
Spearman's (rs) and Pearson's (r)
coefficients were used to investigate correlation; correlation
coefficients (r, rs) were evaluated according to
the classified criteria as low (0.00–0.24), moderate (0.25–0.49),
strong (0.50–0.74) and very strong (0.75–1.00) (39). P<0.05 was considered to indicate
a statistically significant difference. All analyses were performed
using SPSS software version 11.0 (SPSS, Inc., Chicago, IL,
USA).
Results
Sequence upstream of human MYCN gene
shares ~44% sequence identity with a region encompassing ACE3,
upstream of chorion genes in D. melanogaster
It has been determined that promoter and enhancer
regions are located ~200 and 800 bp upstream of MYCN,
respectively, and have been reported to be responsible for basal
and cell type-specific expression in a variety of murine and human
cell lines (40). In addition, the
similar organization of the origin elements and replicators,
including ACE3 and DHFR, from Drosophila,
mammals, Tetrahymena and Sciara has been reviewed
previously (41). In normal human
cells, amplified DNA has not been observed at frequencies greater
than 1/108, and MYCN expression has been reported to be
limited in normal human adult tissues (42,43).
Taken together, previous reports have suggested
that certain trans-acting and epigenetic factors may serve
important roles in the transcriptional and/or epigenetic control of
MYCN expression and amplification. It was hypothesized that
the DNA sequences of upstream cis-regulatory elements of
amplifiable genes in a variety of organisms may be evolutionarily
conserved, at least in part. The present study thus investigated
the degree of sequence similarity between sequence upstream of the
human MYCN gene and the ACE3 element controlling
chorion gene amplification in D. melanogaster (44).
Initially, the DNA sequences encompassing
MYCN upstream and ACE3 were compared using the
LFASTAn program (Fig. 1A). The
results revealed that sequences upstream of the human MYCN
gene (−1,021 to −143; bases 2,466,909 to 2,467,787), including the
enhancer and proximal promoter, share a 43.545% sequence identity
in an overlap of 914 nucleotides with a DNA fragment (bases 14 to
884) spanning the D. melanogaster ACE3 sequence. This result
suggested that the expression and/or amplification mechanisms of
developmentally-regulated genes may be conserved among various
organisms during the evolutionary process.
 | Figure 1.Sequence comparison between
MYCN upstream and ACE3, and transcription factor
binding sites in MYCN upstream. (A) A 1,020 bp DNA sequence
(top; bases 1–1,020) encompassing ACE3 from Drosophila
melanogaster chorion gene cluster and a DNA sequence of ~65 kb
(bottom) containing MYCN (bases 2,416,201 to 2,481,180) from
human chromosome 2 genomic contig were compared using the LFASTAn
program. (B) Using the TFSEARCH program, putative c-Myb and E2F1
binding sites upstream (−1,021 to −143) of human MYCN gene
were detected. MYCN, MYCN proto-oncogene bHLH transcription
factor; ACE3, amplification-control-element-on-3; c-Myb,
transcriptional activator Myb; E2F1, E2F transcription factor
1. |
Putative c-Myb and E2F1 binding sites
detected in the enhancer and proximal promoter located upstream of
MYCN
Enhancer (−980 to −860), inhibition (−860 to −797)
and promoter (−279 to +108) regions of MYCN were previously
identified in IMR32 cells (45).
Additionally, MYCN expression mediated by this enhancer is
induced at higher levels within IMR32 cells than in HeLa cells,
indicating cell type-specific enhancement of MYCN
expression. Recently, it was reported that the DNA binding motif of
MYB (c-Myb) is specifically enriched in low-complexity
transcription factor binding site (TFBS)-clustered regions
(46), which suggested that c-Myb
may contribute to cell type-specific transcriptional
regulation.
To identify the putative transcription factor
binding sites upstream of MYCN, the TFSEARCH program was
employed (Fig. 1B). A total of two
c-Myb and three E2F1 binding sites were identified in the enhancer
and proximal promoter located upstream of MYCN, which shares
partial sequence identity with a region encompassing ACE3.
The bioinformatics data analysis of the present study indicated
that c-Myb and E2F1 may be involved in the expression of the
MYCN gene in neuroblastoma cells.
MYCN amplification status, 1p36
alterations and ploidy level determined by FISH analysis in
neuroblastoma cell lines
To determine MYCN amplification status and
1p36 alterations in neuroblastoma cells (Fig. 2), interphase nuclei and metaphase
spreads were analyzed using FISH. The degree of MYCN
amplification (>10 copies per nucleus) was notably high in
Kelly, SIMA, MHH-NB-11 and IMR32 cells (Table II); however, the fluorescence
signal intensity of nuclei for each MYCN-amplified cell line
differed (Fig. 2B). The SH-SY5Y
cell line lacking MYCN amplification most commonly included
three copies (93.1%) of MYCN (Table II and Fig. 2B). Our recent study demonstrated
that three copies of MYCN in SH-SY5Y may be due to an
unbalanced translocation involving the MYCN locus at 2p24
(34). The FISH analysis of
metaphase spreads typically demonstrated homogeneously staining
regions (HSRs) in Kelly, MHH-NB-11, and IMR32 cells, and double
minutes in SIMA cells (Fig. 2C).
In addition, the ploidy levels of all cell lines were determined
(Fig. 2C).
 | Figure 2.MYCN amplification and 1p36
alterations from interphase and metaphase in neuroblastoma cell
lines. (A) Neuroblastoma cells were harvested for FISH experiments
(magnification, ×40). (B) MYCN amplification status of
interphase nuclei was determined by FISH. (C) Modal number, hsr and
dmin from metaphase in neuroblastoma cell lines are demonstrated.
(D) 1p36 alterations from metaphase and interphase in neuroblastoma
cell lines are presented. Magnification for B-D, ×100. MYCN,
MYCN proto-oncogene bHLH transcription factor; ACE3,
amplification-control-element-on-3; FISH, fluorescence in
situ hybridization; del, deletion; dmin, double minute; hsr,
homogeneously staining region; imb, imbalance; SE, satellite
enumeration. |
| Table II.MYCN amplification and 1p36
alterations in neuroblastoma cell linesa. |
Table II.
MYCN amplification and 1p36
alterations in neuroblastoma cell linesa.
|
| MYCN
amplification (%) | 1p36 alterations
(%)b |
|---|
|
|
|
|
|---|
| Cell line | Normal | Low | Intermediate | High | 2/2 | 3/3c |
Deletionc | Imbalance |
|---|
| SH-SY5Y | 3.8 | 96.2 | 0.0 |
0.0 | 67.1 |
2.8 |
0.4 | 28.4 |
| Kelly | 0.0 |
0.0 | 0.0 | 100.0 |
2.4 | 49.8 |
0.1 | 11.4 |
| SIMA | 0.0 |
1.5 | 3.4 |
95.1 |
7.4 |
0.9 |
0.4 | 91.1 |
| MHH-NB-11 | 0.0 |
0.0 | 0.0 | 100.0 |
0.4 |
3.4 |
0.0 | 95.7 |
| IMR32 | 0.0 |
0.0 | 0.0 | 100.0 |
5.0 |
0.6 | 85.9 |
8.5 |
In the present study, IMR32 exhibited the highest
degree of 1p36 deletion, whereas the percentage was markedly low in
other neuroblastoma cell lines (Table
II and Fig. 2D). The 1p36
imbalance was higher in SIMA and MHH-NB-11 compared with SH-SY5Y,
Kelly and IMR32 cells (Table II
and Fig. 2D). 1p36-3/3 balanced
alterations were observed at a higher percentage in Kelly cells
compared with other cell lines and were negatively correlated with
1p36 deletion (Table II and
Fig. 2D).
Taken together, FISH analyses revealed that all the
neuroblastoma cell lines analyzed in the present study contained
aneuploid cell clones. Additionally, MYCN-amplified cell
lines exhibited MYCN amplification and at least one 1p36
alteration as structural chromosome aberrations, whereas SH-SY5Y
cells exhibited 1p36 imbalance, but no MYCN
amplification.
c-myb expression, candidate target
gene expression and MYCN gene copy number determined by qPCR in
neuroblastoma cell lines
The candidate c-Myb target genes were selected
using published literature on neuroblastoma tumor biology and gene
amplification mechanisms, together with predicted binding site data
for transcription factors in gene promoters (SABiosciences Text
Mining Application and UCSC Genome Browser).
The present study investigated the associations
between c-myb expression, candidate target gene expression,
MYCN gene copy number and 1p36 alterations. MYCN gene
copy number and the expression levels [MYCN, c-myb,
B-myb, A-myb, E2F1, E2F2, p53, p21, cyclin-dependent
kinase inhibitor 1B (p27), hCdt1, GMNN, cyclin
dependent kinase 2 (CDK2) and L3MBTL1] were
determined in neuroblastoma cell lines compared with the reference
genes, p53 and HPRT1, via qPCR prior to c-myb
RNAi (Table III and Fig. 3A). The expression levels of
MYCN and B-myb were notably higher compared with
those of E2F1, hCdt1, GMNN, p27, CDK2, p21, L3MBTL1 and
c-myb in MYCN-amplified cell lines (Table III and Fig. 3B).
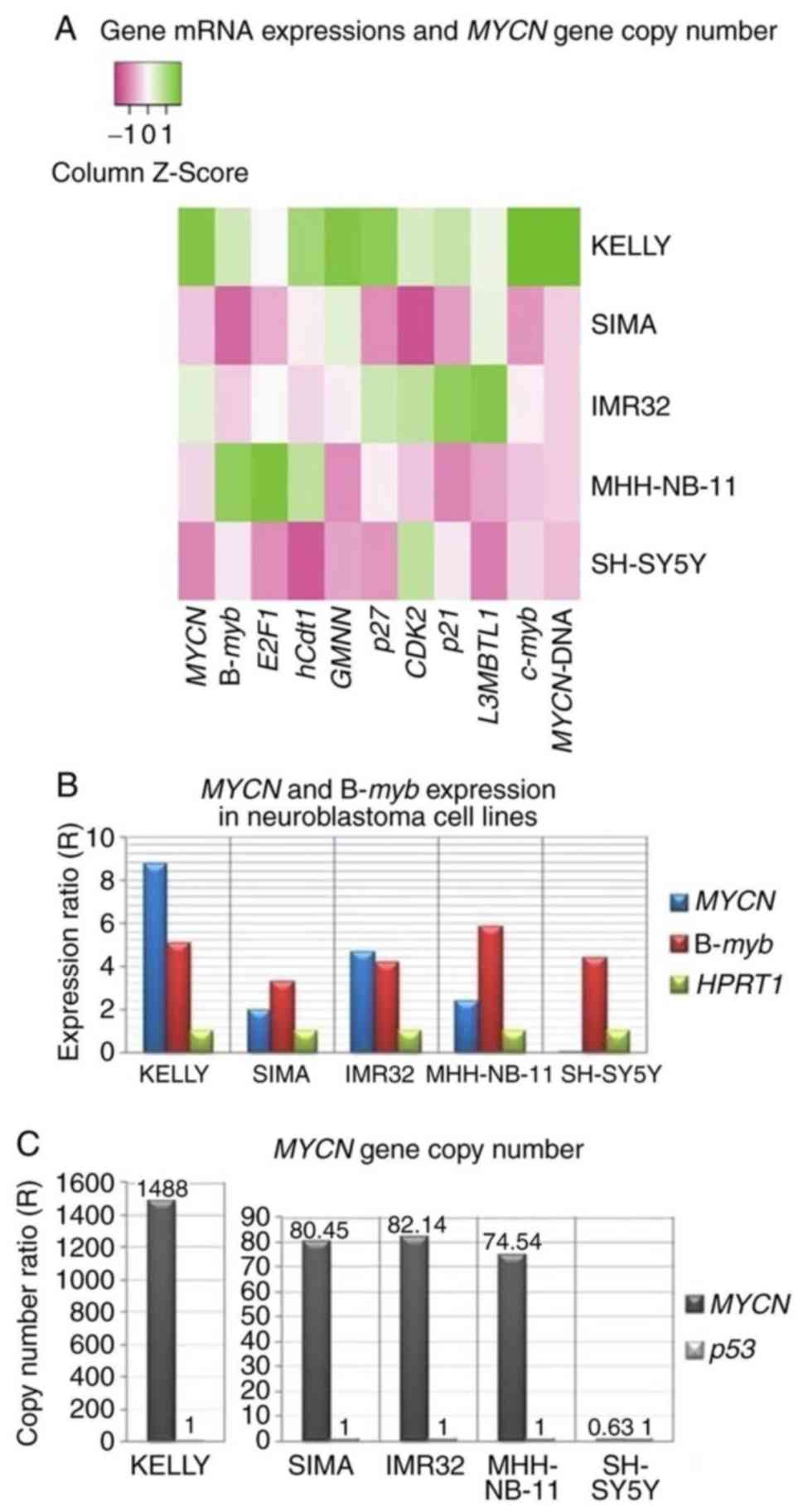 | Figure 3.Expression of potential c-Myb target
genes and MYCN gene copy number in neuroblastoma cell lines.
(A) Heat map visualizing the mRNA expression of 10 genes and
MYCN gene copy number (MYCN-DNA) from neuroblastoma
cell lines compared with reference genes, HPRT1 and
p53, respectively. The color key represents the column
Z-score of gene expression levels and copy numbers. Dark pink
indicates the lowest gene expression or copy number and dark green
indicates the highest gene expression or copy number. (B)
Expression levels of MYCN and B-myb as compared with
reference HPRT1; (C) MYCN gene copy number as
compared with reference p53 in neuroblastoma cell lines (see
also Table III). MYCN, MYCN
proto-oncogene bHLH transcription factor; B-myb, MYB
proto-oncogene like 2; E2F1, E2F transcription factor 1;
hCdt1, chromatin licensing and DNA replication factor 1;
GMNN, geminin; p27, cyclin-dependent kinase inhibitor
1B; CDK2, cyclin dependent kinase 2; p21,
cyclin-dependent kinase inhibitor 1A; L3MBTL1, L3MBTL1
histone methyl-lysine binding protein; c-Myb,
transcriptional activator Myb; HPRT1, hypoxanthine
phosphoribosyltransferase 1. |
| Table III.Expression of candidate c-Myb target
genes and MYCN gene copy numbers in neuroblastoma cell
lines. |
Table III.
Expression of candidate c-Myb target
genes and MYCN gene copy numbers in neuroblastoma cell
lines.
| A,
mRNAa |
|---|
|
|---|
|
| Cell line
(Cp/R) |
|---|
|
|
|
|---|
| Gene | SH-SY5Y | Kelly | SIMA | MHH-NB-11 | IMR32 |
|---|
|
MYCNb |
34.84±0.19/0.073 |
26.41±0.19/8.75 |
28.79±0.22/1.99 |
29.22±0.32/2.39 |
27.66±0.08/4.66 |
| B-myb |
28.94±0.27/4.38 |
27.19±0.26/5.10 |
28.07±0.91/3.27 |
27.93±0.31/5.86 |
27.82±0.05/4.17 |
| HPRT1 |
31.07±0.13/1.00 |
29.54±0.32/1.00 |
29.78±0.05/1.00 |
30.48±0.15/1.00 |
29.88±0.11/1.00 |
| E2F1 |
32.88±0.02/0.29 |
30.13±0.12/0.66 |
31.05±0.03/0.41 |
30.07±0.83/1.33 |
30.48±1.03/0.66 |
| hCdt1 |
31.97±0.14/0.54 |
29.95±0.36/0.75 |
30.40±0.36/0.65 |
30.95±0.05/0.72 |
30.54±0.06/0.63 |
|
GMNNc |
33.09±0.08/0.25 |
29.37±0.23/1.13 |
30.35±0.46/0.67 |
32.96±0.31/0.18 |
30.84±0.14/0.51 |
| p27 |
33.64±0.38/0.17 |
29.97±0.51/0.74 |
32.39±0.20/0.16 |
31.89±0.30/0.38 |
30.73±0.41/0.55 |
| CDK2 |
32.32±0.21/0.42 |
30.89±0.70/0.39 |
32.42±0.55/0.16 |
32.31±0.46/0.28 |
31.12±0.12/0.42 |
| p21 |
34.87±0.07/0.072 |
32.35±0.56/0.14 |
35.40±0.02/0.020 |
37.86±0.22/0.006 |
32.26±0.09/0.19 |
| L3MBTL1 |
38.83±1.16/0.005 |
34.99±0.71/0.023 |
35.17±1.61/0.024 |
37.17±0.81/0.010 |
34.52±1.29/0.040 |
|
c-mybb |
37.54±0.85/0.011 |
34.59±0.84/0.030 |
37.18±1.08/0.006 |
37.18±0.20/0.010 |
36.11±0.12/0.013 |
| A-myb | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| E2F2 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| p53 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
|
| B,
DNAd |
|
|
| Cell line
(Cp/R) |
|
|
|
| Gene | SH-SY5Y | Kelly | SIMA |
MHH-NB-11 | IMR32 |
|
|
MYCNc |
29.44±0.13/0.63 |
22.15±0.56/1488.87 |
24.77±0.29/80.45 |
23.89±0.66/74.54 |
24.99±0.57/82.14 |
| p53 | 28.78±0.61/1.0 | 32.69±0.60/1.0 | 31.10±0.47/1.0 | 30.11±0.80/1.0 | 31.35±0.58/1.0 |
A-myb, E2F2 and p53 were not
expressed in any of the five neuroblastoma cell lines (Table III). E2F2 has been
analyzed as a candidate tumor suppressor gene located at the 1p36
locus, which is deleted in 25% of tumors and 87% of cell lines in
neuroblastoma (2,10). According to FISH analysis, the 1p36
deletion was observed at a high percentage only in the IMR32 cell
line (Table II), suggesting that
E2F2 may be epigenetically repressed in cell lines lacking
the 1p36 deletion.
Using qPCR, MYCN gene copy number per
haploid genome compared with the reference gene, p53, was
notably high in Kelly cells (1,488.87 copies); however, 82.14,
80.45 and 74.54 copies were detected in IMR32, SIMA and MHH-NB-11
cells, respectively. The copy number for SH-SY5Y cells was 0.63
(Fig. 3C).
Pearson's correlation coefficient analysis revealed
a notably positive significant correlation between 1p36-3/3
balanced alteration and MYCN gene copy number
(r=0.99, P<0.001; Tables
II and III, and Fig. 4A), which requires further
cytogenetic examination in MYCN-amplified neuroblastoma
cells bearing the 1p36-3/3 alteration. Our recent study identified
a large interstitial deletion at 1q25-q41 in Kelly cell line
predominantly possessing the 1p36-3/3 alteration in metaphase
(34), suggesting that loss of
heterozygosity of one or more tumor suppressor genes located in
this region may contribute to a significant increase in MYCN
gene copy number.
 | Figure 4.Scatter plots for correlations of
gene expressions and genomic alterations identified in
neuroblastoma cell lines. The correlations of (A) MYCN copy
number and 1p36-3/3 alteration, (B) c-myb and MYCN
expression, (C) p21 expression and 1p36 imbalance, (D)
1p36-3/3 alteration and c-myb expression, (E) B-myb
expression and 1p36 deletion, (F) 1p36 deletion and 1p36-3/3
alteration and (G) MYCN copy number and GMNN
expression were presented. The scatter plots were produced using
Excel. MYCN, MYCN proto-oncogene bHLH transcription factor;
c-Myb, transcriptional activator Myb; B-myb, MYB
proto-oncogene like 2; GMNN, geminin; p21,
cyclin-dependent kinase inhibitor 1A. |
The mRNA expression levels of MYCN and
c-myb genes declined within 3 h of treatment with retinoic
acid of neuroblastoma cell lines (16). Statistical analysis revealed a
significant positive correlation between c-myb and
MYCN expression (Table
III and Fig. 4B). This finding
suggested that there may be an association between the expression
of c-myb and MYCN genes in neuroblastoma cells.
Correlations between 1p36 alterations
and expression of c-myb, B-myb and p21 genes
Previous reports have revealed that c-Myb and B-Myb
facilitate G2/M progression via the direct upregulation
of cyclin B1 in normal, embryonic stem and cancer cells
(47–49), whereas depletion of p21 causes
chromosome segregation and cytokinesis defects in HCT116, HeLa and
SAOS-2 cells (50). Statistical
analyses of the present study revealed a significant negative
correlation between p21 expression and 1p36 imbalance
(rs=−1.00, P<0.001; Fig. 4C); however, a significant positive
correlation between c-myb expression and 1p36-3/3 alteration
(r=0.96, P<0.02; Fig.
4D) in neuroblastoma cell lines was observed (Tables II and III). In addition, B-myb
expression and 1p36-3/3 alteration were negatively correlated with
1p36 deletion (rs=−0.82, P=0.089;
rs=−0.87, P=0.054; Tables II and III; Fig.
4E and F). All statistical data suggested that c-myb,
B-myb and p21 genes may serve an important role
against the genomic instability of chromosome 1p in neuroblastoma
cells.
c-myb and E2F1 may be involved in
controlling MYCN expression and amplification
The present study employed RNAi against
c-myb mRNA. A c-myb-specific shRNA sequence was
designed and ligated into a plasmid DNA vector. Following
transformation, plasmid DNA was transfected into
MYCN-amplified Kelly, IMR32, SIMA and MHH-NB-11
neuroblastoma cell lines. To generate stably transfected cell
lines, the cells expressing c-myb shRNA were selected using
G418. The presence of c-myb-specific and EGFP-specific
shRNAs in plasmid vectors prior to transfection was confirmed by
SpeI enzyme digestion and DNA sequencing.
RNAi reduced c-myb mRNA expression by 95.58,
77.79, 86.42 and 88.89% in Kelly, SIMA, MHH-NB-11 and IMR32 cells,
respectively (Fig. 5A).
MYCN/p53 DNA copy number ratios from
MYCN-amplified cell lines were determined using the ∆∆Cp
method (36) as normalized to the
Cp values of MYCN and p53 genes from the SH-SY5Y
control cell line in pre- and post-c-myb RNAi groups. These
results were compared with those of the efficiency corrected
(dilution) method (36,38) in Kelly, SIMA and IMR32 cells. The
results of the present study revealed that MYCN gene copy
numbers increased by 115.85, 71.70, 85.31 and 53.69% following
c-myb RNAi treatment of Kelly, SIMA, IMR32 and MHH-NB-11
cells, respectively (Fig. 5B). The
findings of the present study suggested that c-myb may be
involved in controlling MYCN amplification.
The expression levels of MYCN, c-myb,
E2F1, hCdt1, B-myb, GMNN, p21 and L3MBTL1 genes
in Kelly, SIMA, MHH-NB-11 and IMR32 cell lines as normalized to the
Cp values of target and reference (HPRT1) genes of the
control SH-SY5Y cell line were determined pre- and
post-c-myb RNAi. The mRNA expression level of each gene was
presented as the mean of related expression levels from the four
aforementioned MYCN-amplified cell lines (Fig. 6). The expression level of
MYCN was moderately decreased by 33.99±5.78% following
treatment with c-myb RNAi in MYCN-amplified cell
lines, while E2F1 expression was downregulated by
86.66±8.40%. These results suggested that c-myb may be
involved in the induction of E2F1 and MYCN
expression, potentially via putative c-Myb binding sites in their
promoters.
 | Figure 6.Alterations in potential c-Myb target
gene expression and MYCN gene copy number following
treatment with c-myb RNAi. MYCN gene copy number and
expression levels of c-myb and potential target genes
(B-myb, p21, hCdt1, E2F1, L3MBTL1, GMNN and MYCN)
were calculated in MYCN-amplified neuroblastoma cell lines
as normalized to the Cp values of target and reference genes of
SH-SY5Y control cell line using the delta-delta Cp method (36). **Data are expressed as mean
variation (%) ± standard error of the mean from Kelly, SIMA,
MHH-NB-11 and IMR32 cell lines (n=4). *P<0.05; Wilcoxon signed
rank test (one-tailed). shRNA, short hairpin RNA; RNAi, RNA
interference; B-myb, MYB proto-oncogene like 2; p21,
cyclin-dependent kinase inhibitor 1A; hCdt1, chromatin
licensing and DNA replication factor 1; c-myb,
transcriptional activator myb; E2F1, E2F transcription
factor 1; L3MBTL1, L3MBTL1 histone methyl-lysine binding
protein; GMNN, geminin; MYCN, MYCN proto-oncogene
bHLH transcription factor. |
However, the c-myb may also be indirectly
associated with MYCN expression via the upregulation of
E2F1 (Fig. 7A and B).
E2F1 may be involved in activation of the MYCN
promoter, possibly via putative E2F1 binding sites (Fig. 1B). It was previously reported that
E2F1-3 proteins activate the MYCN promoter via E2F binding
sites in Kelly and IMR32 cells (51). Taken together, the findings of the
present study indicate that c-myb and E2F1 may be
involved in controlling MYCN expression and amplification in
MYCN-amplified neuroblastoma cells.
 | Figure 7.Involvement of c-myb and
E2F1 in the expression and amplification of MYCN. (A)
Model mechanism indicates the possible contribution of c-myb
and E2F1 genes in the apoptotic elimination of
MYCN-amplified neuroblastoma (MNA NB) cells via the
upregulation of MYCN expression. MYCN overexpression
indirectly activates the E2F1-induced apoptosis signaling pathway,
potentially by inhibiting miR-20a and miR-92a, which prevent
upregulation of E2F genes. (B) Flow schema showing the
alterations in MYCN gene copy number and expression of
c-myb, E2F1 and potential target genes following treatment
with c-myb RNAi in MYCN-amplified neuroblastoma cell
lines. *Spearman's correlation coefficient, two-tailed
(rs=1.0, P<0.01). miR, microRNA; RNAi, RNA
interference. c-myb, transcriptional activator myb; MYCN,
MYCN proto-oncogene bHLH transcription factor; E2F1, E2F
transcription factor 1; p-73, tumor protein p73; p21,
cyclin-dependent kinase inhibitor 1A; L3MBTL1, L3MBTL1 histone
methyl-lysine binding protein; B-myb, MYB proto-oncogene
like 2; hCdt1, chromatin licensing and DNA replication
factor 1; Rb, RB transcriptional corepressor; GMNN,
geminin. |
E2F1 may contribute to hCdt1, p21 and
B-myb gene expression in MYCN-amplified neuroblastoma cells
In HCT116 cells, the hCdt1 promoter is
activated by E2F1 (52). In
addition, E2F1 induces p21 transcription by directly binding
to the proximal promoter of p21 via a p53-independent
mechanism in NIH3T3 cells (53),
but also transactivates the B-myb promoter in SAOS-2 cells
(54).
In the present study, the mRNA expression level of
hCdt1 declined by 37.48±12.45% following c-myb RNAi
in MYCN-amplified neuroblastoma cell lines (Fig. 6). In addition, a significant
positive correlation was identified between E2F1 and
hCdt1 expression post-c-myb RNAi. p21 was also
expressed; however, the p53 transcript was not detected in
neuroblastoma cells (Table III).
Following c-myb RNAi treatment, p21 expression was
downregulated by 92.51±4.94% in MYCN-amplified neuroblastoma
cell lines (Fig. 6). Additionally,
B-myb expression was downregulated by 82.49±6.28% following
c-myb RNAi treatment (Fig.
6).
These results suggest that E2F1 may induce
hCdt1, p21 and B-myb expression, potentially via
putative E2F1 binding sites in the promoters of these genes in
MYCN-amplified neuroblastoma cells (Fig. 7).
MYCN gene is amplified during S phase,
possibly via a replication-based mechanism: GMNN expression is
upregulated following c-myb RNAi treatment in MYCN-amplified
neuroblastoma cell lines
GMNN transcription is induced by E2F1-4
transcription factors via the RB transcriptional corepressor 1
(Rb)/E2F signaling pathway (52).
The present study reported GMMN upregulation; however,
E2F1 mRNA expression was downregulated following
c-myb RNAi treatment (Fig.
6). GMNN was also upregulated following E2F1 RNAi
treatment (data not shown) in MYCN-amplified neuroblastoma
cell lines.
The mRNA expression levels of GMNN are
higher in the S and G2/M phases compared with the
G1/S transition of the cell cycle (28,52).
In addition, Rb serves an important role in the repression of
GMNN via intragenic E2F sites during G1 (55). The promoter of Rb contains a
putative E2F1 binding site, suggesting that E2F1 may directly
regulate the Rb promoter. Therefore, E2F1 may be
associated with the repression of GMNN expression via the
induction of Rb in MYCN-amplified neuroblastoma cells
(Fig. 7B). These findings indicate
that MYCN gene copy number increased during S phase, while
neuroblastoma cells progress toward G2/M phase following
c-myb RNAi treatment (Fig.
7B).
c-myb RNAi causes silencing of L3MBTL1
expression in MYCN-amplified cell lines, indicating G2/M
arrest
Inhibition of L3MBTL1 mRNA expression leads
to G2/M arrest in a variety of cancerous and normal
human cells (25). In the present
study, L3MBTL1 expression was silenced in
MYCN-amplified cell lines following treatment with
c-myb RNAi (Fig. 6). In
addition, bioinformatics analysis demonstrated that the promoter of
L3MBTL1 harbors a putative binding site for c-Myb, but not
for E2F. These results suggest that c-myb may induce
L3MBTL1 expression, possibly via a putative c-Myb binding
site identified in the promoter of L3MBTL1 in
MYCN-amplified neuroblastoma cells (Fig. 7B).
In addition, L3MBTL1 expression was highly
downregulated by E2F1 RNAi in Kelly, SIMA and IMR32 cells
(data not shown), suggesting that E2F1 may transactivate the
promoter of c-myb in neuroblastoma cells, similar to human
glioblastoma cells (56). However,
the c-myb promoter does not include any putative E2F binding
sites.
In conclusion, the findings of the present study
suggested that L3MBTL1 may be a potential c-Myb target gene.
In addition, similar to GMNN upregulation, the silencing of
L3MBTL1 expression following c-myb RNAi treatment
indicates that MYCN gene copy number increased prior to
G2/M arrest in MYCN-amplified neuroblastoma cell
lines (Fig. 7B). Taken together,
it is concluded that MYCN gene is amplified during S phase,
potentially via a replication-based mechanism.
Discussion
Neuroblastoma is a common pediatric solid tumor
derived from the primitive cells of the sympathetic nervous system
(57). MYCN amplification
is an important factor associated with poor prognosis in
neuroblastoma (5). MYCN is
amplified in 18–38% of reported cases (3,5–7) and
in a panel of neuroblastoma cell lines (8). The mRNA expression levels of
MYCN and c-myb decline within 3 h following treatment
with retinoic acid in neuroblastoma cell lines (16). However, the expression and
amplification mechanisms of MYCN require further study.
The present study investigated the potential target
genes of c-Myb and the effects of c-myb RNAi on MYCN
expression and amplification. MYCN gene copy number and mean
expression levels of MYCN, c-myb, E2F1, hCdt1,
B-myb, GMNN, p21 and L3MBTL1 genes were determined in
MYCN-amplified neuroblastoma cell lines, which were
normalized to their counterparts from the SH-SY5Y control cell line
in pre- and post-c-myb RNAi treatment. To compare with those
of the ∆∆Cp method, MYCN gene copy numbers of Kelly, SIMA
and IMR32 cells were also determined using the dilution method.
The ∆∆Cp method revealed that MYCN gene copy
number increased by 115.85, 71.70, 85.31 and 53.69% in Kelly, SIMA,
IMR32 and MHH-NB-11 cells, respectively, following c-myb
RNAi treatment. The dilution method also revealed that MYCN
gene copy number was increased in Kelly (336.49%), SIMA (20.58%)
and IMR32 (76.83%) cells normalized to target and reference Cp
values of the control SH-SY5Y cell line. However, PCR amplification
efficiency (E) values out of range 1.95–2.05 in the dilution
method led to notable differences in the MYCN gene copy
numbers compared with those obtained from the ∆∆Cp method,
particularly for Kelly and SIMA. Using the dilution method,
E values >2.0, which are unexpected in theory, have been
practically observed (36). Taken
together, ∆∆Cp and dilution methods demonstrated an increase in
MYCN gene copy number following c-myb RNAi treatment,
suggesting that c-myb may be involved in controlling
MYCN amplification in MYCN-amplified neuroblastoma
cells.
Previously, Beall et al (21) reported that the protein product of
D. melanogaster-myb gene, which is closely associated with
the vertebrate myb gene family (including A-myb,
B-myb and c-myb) (19,20),
is required for chorion gene amplification in trans.
However, it was proposed that a Dm Myb-MuvB repressor complex,
including D-L(3)mbt protein, may be involved in the repression of
transcription and DNA replication (23). The paralogous human gene,
c-myb, has acquired novel functions not possessed by the
Dm-myb gene (20). L3MBTL1,
a human homolog of the D-l(3)mbt protein, interacts with the DNA
sliding clamp and other proteins, forming a replicative helicase
complex, and is required for progression of the replication fork
(25). Loss of L3MBTL1 mRNA
expression is an indicator of G2/M arrest (25). The present study reported that
L3MBTL1 mRNA expression was silenced, whereas MYCN
gene copy number increased following c-myb RNAi treatment of
MYCN-amplified neuroblastoma cell lines. Taken together with
published data, the findings of the present study suggest that
L3MBTL1 may act as a guard against arrest or abnormal forms
of replication forks during MYCN amplification. Furthermore,
the present study indicated that MYCN gene copy number was
increased prior to G2/M arrest.
Statistical analysis revealed a notably significant
positive correlation between c-myb and MYCN
expression levels in neuroblastoma cells (Table III). Furthermore, the expression
levels of MYCN were moderately decreased in
MYCN-amplified neuroblastoma cells following c-myb
RNAi treatment. In addition, bioinformatics analysis demonstrated
that the enhancer and promoter of MYCN include putative
c-Myb binding sites. The results of the present study suggest that
MYCN may be a potential target gene of c-Myb and that
c-myb may be associated with the induction of MYCN
expression, potentially via the promoter and/or enhancer regions of
MYCN. Upon induction by retinoic acid in human neuroblastoma
cells, the mRNA expression levels of c-myb and MYCN
decrease within 3 h (14–18).
Following treatment with c-myb RNAi, the
mRNA expression levels of E2F1 and MYCN decreased,
whereas the MYCN gene copy number increased in
MYCN-amplified neuroblastoma cells. Bioinformatics analyses
demonstrated that the MYCN promoter includes putative E2F1
binding sites, while that of E2F1 harbors c-Myb binding
sites. These results suggested that E2F1 may be a potential
target gene of c-Myb; and c-myb gene is involved in the
induction of E2F1 expression potentially via a putative
c-Myb-binding site in the E2F1 promoter. As with c-myb,
E2F1 may also be associated with the induction of MYCN
expression, potentially via E2F1 binding sites in the MYCN
promoter. Our preliminary studies demonstrated that MYCN
expression is downregulated by E2F1 RNAi in Kelly and IMR32
cells (data not shown). E2F1-3 have been reported to activate the
proximal promoter of MYCN gene in Kelly and IMR32 cells
(51). Collectively, these
findings suggested that c-myb and E2F1 may be
involved in controlling MYCN expression and amplification
via the promoter and/or enhancer regions of MYCN.
In the present study, B-myb and p21
were observed to be downregulated following treatment with
c-myb RNAi. In addition, the expression levels of
B-myb and p21 decreased following E2F1 RNAi
treatment in Kelly, SIMA, MHH-NB-11 and IMR32 cell lines (data not
shown). Bioinformatics data revealed that the p21 promoter
includes putative E2F1 binding sites without a c-Myb binding site
(Qiagen, Inc.). These results indicated that E2F1 may be
associated with the induction of B-myb and p21 mRNA
expression, possibly via putative E2F1 binding sites in their
respective promoters. A direct interaction causing S-phase arrest
between the E2F1 transcription factor and the proximal promoter of
p21 was previously demonstrated in NIH3T3 cells (53), and E2F1 transactivates the
B-myb promoter in SAOS-2 cells (54).
E2F1 transcriptionally activates the Tp73 tumor
suppressor that induces p53-responsive genes and apoptosis
(58). The expression level of p21
protein is increased in the IGR-N-91 neuroblastoma cell line
(containing mutated p53) infected with TAp73α recombinant
adenovirus; this leads to G1 arrest via a
p53-independent signaling pathway (59). In MYCN-amplified
neuroblastoma, MYCN transactivates miR-17-5p, which in turn
accelerates cell cycle progression and protects cells from
apoptosis via the downregulation of p21 and BIM, respectively
(60). MYCN overexpression
sensitizes MYCN-amplified neuroblastoma cells to apoptosis
via the induction of p53 (32).
Additionally, enforced MYCN expression causes a significant
reduction in cell viability via the apoptosis signaling pathway in
SY5Y and SK-N-AS cell lines lacking MYCN amplification
(61).
Furthermore, Guglielmi et al (18) reported that the mRNA expression
levels of pro-apoptotic E2F1 and Tp73 are increased
without modulating Tp53 mRNA expression, due to the
inhibition of miR-9, miR-20a and miR-92a by upregulated MYCN
expression in SK-N-AS cells. It was concluded that MYCN may
be required during the activation of neuroblastoma differentiation
to induce apoptosis in undifferentiated cells. In addition,
Guglielmi et al (18)
reported that cell death was not observed following treatment with
retinoic acid of MYCN-silenced LAN-5 cells (~40%) harboring
MYCN amplification, whereas control LAN-5 cells did exhibit
retinoic acid-induced apoptosis.
In the present study, p21 was identified to
be transcribed in neuroblastoma cells, whereas p53 mRNA was
not expressed under normal conditions. Taken together, the results
suggested that c-myb and E2F1 may contribute to the
sensitization of MYCN-amplified cells to apoptosis by
inducing MYCN overexpression that indirectly activates E2F1
protein, which in turn upregulates p73 and p21 in neuroblastoma
(18,32,53,58,62–64).
That is, MYCN amplification may be partially controlled via
E2F1-induced G1/S arrest and apoptosis in
MYCN-amplified neuroblastoma cells (Fig. 7A).
In a previous study, c-Myc expression was reported
to be upregulated in 5–15% of the p53−/− mice cells in
all the tissues examined, whereas <1% of p53+/+ cells
express detectable levels of c-Myc protein (65). Apoptotic p53−/− cells
characteristically exhibit atypical chromosome morphology,
abnormally amplified centrosomes, aneuploidy, c-myc gene
amplification and elevated c-Myc protein levels, indicating that
c-Myc-overexpressing cells undergo apoptosis and numerous
genetically aberrant cells are eliminated by p53-independent
apoptosis in vivo (65).
The apoptotic control mechanism proposed in the
present study for MYCN-amplified neuroblastoma cells via
c-myb and E2F1-associated MYCN overexpression
can explain why the MYCN gene copy number increases despite
reductions in mRNA expression levels of MYCN following
c-myb RNAi treatment. In cisplatin-resistant UKF-NB-4
neuroblastoma cells with MYCN amplification, MYCN
expression levels increased although no significant change was
observed in the MYCN copy number following cisplatin
treatment, suggesting that alterations in MYCN expression
may not always be associated with variations in MYCN copy
number (66). MYCN
expression may be controlled by mechanisms independent from
MYCN copy number under discrete conditions in neuroblastoma
cell lines. Additional MYCN gene copies may also repress
their own transcription. By analyzing genome-wide data of the
regions that vary in copy number in humans and certain model
organisms, it has been reported that genes with varied copy number
in copy number variation (CNV) regions are expressed at lower and
more variable levels than genes mapped elsewhere; CNVs also exert a
global influence on the transcriptome (67). Alterations in copy number of CNV
regions can influence gene expression by several mechanisms,
including physical dissociation of the transcription unit from its
cis-acting regulators, modification of transcriptional
control by altering chromatin structure and position, and
perturbation of transcript structure (67). Additional copies of MYCN may
also impair MYCN transcription due to steric hindrance for
access to specific transcription factories, as previously described
in a general hypothesis regarding extra copies of any gene
(67,68).
In the present study, FISH analyses of the
metaphase spreads from neuroblastoma cells revealed near-diploid
(2n±), hyperhaploid (n+) and near-polyploid (3n-, 3n+ and 4n-)
aneuploidies. In addition, qPCR experiments revealed the expression
of CDK2 and MYCN genes in neuroblastoma cells.
Centrosomes with multiple copies, abnormal structure and function
have frequently been observed in the most common types of human
malignant tumors and tumor-derived cell lines (69). Hyperactive CDK2 and
MYCN overexpression induce centrosome amplification and
chromosomal instability, resulting in aneuploidy and formation of
micronuclei (a precursor to aneuploidy) in p53−/−
mouse embryonic fibroblasts and neuroblastoma cells following DNA
damage, respectively (70,71). However, enhanced expression of
MYCN without DNA damage in a neuroblastoma cell line did not
cause centrosome hyperamplification and micronuclei formation
(71).
Sugihara et al (71) predicted an increase in a fraction
of the cell population with DNA contents of 8N, resulting from
centrosome hyperamplification with DNA replication caused by the
failure of cell division following DNA damage. However, Sugihara
et al (71) also identified
that the fraction of MYCN-EGFP cells with 8N did not increase
compared with that of vector-EGFP cells, suggesting that
MYCN overexpression may contribute to the apoptotic
elimination of neuroblastoma cells with genomic DNA amplification
and formation of micronuclei. Similarly, the apoptotic cells of
p53−/− mice contain abnormally amplified centrosomes,
aneuploidy, high levels of c-Myc expression and gene amplification
(65).
Statistical analyses revealed that B-myb and
p21 expression are negatively correlated with 1p36 deletion
and imbalance, respectively (rs=−0.82, P=0.089;
rs=−1.00, P<0.001). Conversely, a significant
positive correlation was identified between c-myb expression
and 1p36-3/3 alteration in neuroblastoma cells (r=0.96,
P<0.02), suggesting that c-myb, B-myb and
p21 may serve a role against genomic instability in
chromosome 1p. p21 deficiency leads to abnormal centriole
replication (72), whereas p21
overexpression rescues p53-deficient human tumor cells from
endoreduplication and aneuploidy/polyploidy (73,74).
Following c-myb RNAi treatment,
L3MBTL1 expression was silenced in MYCN-amplified
Kelly, SIMA, IMR32 and MHH-NB-11 neuroblastoma cell lines in the
current study, indicating that L3MBTL1 may be a potential
c-Myb target gene. The c-myb gene may be involved in the
induction of L3MBTL1 expression, possibly via a putative
c-Myb-binding site in the L3MBTL1 promoter. In certain
cancer cell lines, including SW480, the L3MBTL1 transcript
is markedly reduced (24). In
addition, L3MBTL1 has been considered to be a candidate
tumor suppressor gene in myeloid malignancies associated with 20q12
deletions (25). Furthermore,
depletion of L3MBTL1 causes replicative stress, DNA breaks,
activation of the DNA damage response and genomic instability in
human cells, and leads to G2/M arrest in a variety of
cancerous and normal human cells (25). Considering the published
literature, the results of the present study suggested that
L3MBTL1 may act as a tumor suppressor gene in neuroblastoma
cells. Together, the results indicated that MYCN gene copy
number increased prior to G2/M arrest, potentially
following the loss of L3MBTL1 mRNA expression on treatment
with c-myb RNAi.
GMNN expression was also increased along
with MYCN gene copy number following c-myb RNAi
treatment in MYCN-amplified neuroblastoma cell lines in the
current study. In addition, a positive correlation was identified
between GMNN expression and MYCN gene copy number.
These results suggest that GMNN upregulation may lead to a
switch from genomic DNA replication to MYCN amplification by
suppressing re-replication. Similarly, in the follicle cells of
Dm geminin mutant ovaries, it was reported that a switch
from general genomic endoreplication to the amplification cycles
occurs during stage 10B (75). The
present study also reported a partially identical region of ~44%
between DNA fragments encompassing the sequence upstream of human
MYCN gene and ACE3 controlling chorion gene
amplification in D. melanogaster. This finding suggested
that the expression and/or amplification mechanisms of
developmentally-regulated genes may be conserved among different
organisms during the evolutionary process. As with c-myb
RNAi, E2F1 RNAi caused the upregulation of GMNN
expression in MYCN-amplified neuroblastoma cell lines (data
not shown). GMNN expression is transcriptionally repressed
by the activation of Rb throughout G1 (55). In view of the literature and the
findings of the present study, it can be hypothesized that the
E2F1 gene may be indirectly involved in controlling
GMNN expression via transcriptional activation of the
Rb promoter.
In addition, the results of the present study
suggested that E2F1 contributed to the induction of
hCdt1 expression in MYCN-amplified neuroblastoma
cells. Additionally, it was identified that an E2F1 RNAi
experiment in SIMA, as with c-myb RNAi, caused the
downregulation of hCdt1 expression (data not shown). The
promoter of hCdt1 is activated by E2F1 in HCT116 cells
(52). Taken together, the results
suggested that E2F1 may be implicated in controlling the
expression of GMNN and hCdt1 genes throughout the
cell cycle (Fig. 7B).
The mRNA expression levels of GMNN are
upregulated in S and G2/M phases (28,52),
followed by the degradation of geminin protein via
APC/CCdc20 and mitotic spindle checkpoint during
metaphase and early anaphase (26,76)
in human cells. In addition, loss of L3MBTL1 transcript
leads to G2/M arrest (25). B-Myb and c-Myb contribute to the
G2/M transition in normal human cells, embryonic stem
cells and cancer cells (47–49).
In conclusion, the results of the present study
revealed that c-myb and E2F1 genes may be involved in
controlling MYCN expression and amplification in
MYCN-amplified neuroblastoma cells. As described in the
model mechanism proposed in Fig.
7A, c-myb and E2F1 may contribute to the
apoptotic elimination of MYCN-amplified cells via the
induction of MYCN overexpression that indirectly activates
E2F1-induced apoptosis. However, it remains to be determined
whether c-myb and E2F1 are involved in the induction of apoptosis
in MYCN-amplified neuroblastoma cell lines. In addition,
c-myb may be associated with the upregulation of
L3MBTL1 (Fig. 7B).
Furthermore, L3MBTL1 may be considered as candidate tumor
suppressor gene in neuroblastoma cells. The results of the present
study also indicated that the cell cycle is stopped or delayed in
the G2/M transition of the MYCN-amplified
neuroblastoma cells following c-myb RNAi treatment (Fig. 7B). Therefore, it is concluded that
MYCN gene is amplified during S phase, possibly via a
replication-based mechanism (Fig.
7B).
In drug-resistant Chinese hamster sublines and two
neuroblastoma cell lines, SK-N-BE(2) and IMR-32, the HSRs
containing DHFR and MYCN, respectively, were
previously demonstrated to undergo relatively rapid and synchronous
replication prior to the midpoint of the S phase as observed by
tritiated thymidine radioautography (77). However, there is no clear evidence
on whether MYCN is amplified during S phase. Neuroblastoma
cell lines harbor a head-to-tail tandem array in the direct
orientation of DNA segments containing MYCN gene in
amplified regions (78),
suggesting that MYCN amplification may arise from a
mechanism other than those involving breakage-fusion-bridge cycles
that produce inverted duplications (79). Numerous replication-based
mechanisms for the formation of MYCN or general gene
amplification have been proposed (12,41,78,80–84).
Aygun (84)
recently reported that long inverted repeats (LIRs) are
significantly associated with 5′ and 3′ boundary regions of the
amplicon units containing MYCN gene in 14 neuroblastoma cell
lines and 42 other primary solid tumors, whose genomic data were
obtained from the catalogue of somatic mutations in cancer (COSMIC
database; cancer.sanger.ac.uk/cosmic/) (85). In addition, Aygun (86) previously reported a significant
association between LIRs and the breakpoint regions of gross
deletions in human cancers and inherited diseases. Numerous LIRs
inside and outside the MYCN amplicon units were identified,
suggesting that LIRs may extrude hairpin and cruciform structures
in single- and double-stranded DNA during replication, respectively
(84). The hairpin structure can
form at an interrupted LIR on leading and lagging strand templates
simultaneously during replication (87). The hairpin and cruciform
conformations can cause replication fork stalling (87,88).
In addition, a mean microhomology of 5.18 bp (range, 2–14 bp)
between DNA sequences of 150 bp encompassing the 5′ and 3′
boundaries of the MYCN amplicon units has been reported
(12,84). Microhomology between 0 and 15 bp
indicates nonhomologous end joining, microhomology-mediated end
joining, microhomology-mediated break-induced replication or fork
stalling and template switching mechanisms (83,89,90).
Consequently, the findings of the present study indicated that a
replication-based mechanism involving LIRs in a
microhomology-dependent manner may generate MYCN
amplification during S phase. Therapeutic targeting of MYCN
amplification during S phase may improve the prognosis of
neuroblastoma patients.
Acknowledgements
The authors would like to thank Professor Nur Olgun
(Division of Pediatric Oncology, Oncology Institute, Dokuz Eylul
University, Izmir, Turkey) for purchasing the Kelly and IMR32 cell
lines.
Funding
The present study was supported by the Research
Fund of Dokuz Eylul University (grant no. 2005.KB.SAG.077, to
OA).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
NA conceived the study, performed the experiments,
analyzed the data, wrote the manuscript and prepared all figures
and tables. OA supervised the project and conducted the
research.
Ethics approval and consent to
participate
All the experiments were performed in established
human neuroblastoma cell lines. The present study was approved by
the local Ethics Committee of Clinical and Laboratory Research of
the Faculty of Medicine, Dokuz Eylul University (04.10.2005/223,
protocol no. 188).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Seeger RC, Brodeur GM, Sather H, Dalton A,
Siegel SE, Wong KY and Hammond D: Association of multiple copies of
the N-myc oncogene with rapid progression of neuroblastomas. N Engl
J Med. 313:1111–1116. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maris JM and Matthay KK: Molecular biology
of neuroblastoma. J Clin Oncol. 17:2264–2279. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Altungoz O, Aygun N, Tumer S, Ozer E,
Olgun N and Sakizli M: Correlation of modified Shimada
classification with MYCN and 1p36 status detected by fluorescence
in situ hybridization in neuroblastoma. Cancer Genet Cytogenet.
172:113–119. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gurney JG, Ross JA, Wall DA, Bleyer WA,
Severson RK and Robison LL: Infant cancer in the U.S.:
Histology-specific incidence and trends, 1973 to 1992. J Pediatr
Hematol Oncol. 19:428–432. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brodeur GM, Seeger RC, Schwab M, Varmus HE
and Bishop JM: Amplification of N-myc in untreated human
neuroblastomas correlates with advanced disease stage. Science.
224:1121–1124. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bartram CR and Berthold F: Amplification
and expression of the N-myc gene in neuroblastoma. Eur J Pediatr.
146:162–165. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Valent A, Guillaud-Bataille M, Farra C,
Lozach F, Spengler B, Terrier-Lacombe MJ, Valteau-Couanet D,
Danglot G, Lenoir GM, Brison O and Bernheim A: Alternative pathways
of MYCN gene copy number increase in primary neuroblastoma tumors.
Cancer Genet Cytogenet. 153:10–15. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schwab M, Alitalo K, Klempnauer KH, Varmus
HE, Bishop JM, Gilbert F, Brodeur G, Goldstein M and Trent J:
Amplified DNA with limited homology to myc cellular oncogene is
shared by human neuroblastoma cell lines and a neuroblastoma
tumour. Nature. 305:245–248. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Edsjö A, Nilsson H, Vandesompele J,
Karlsson J, Pattyn F, Culp LA, Speleman F and Påhlman S:
Neuroblastoma cells with overexpressed MYCN retain their capacity
to undergo neuronal differentiation. Lab Invest. 84:406–417. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
White PS, Thompson PM, Gotoh T, Okawa ER,
Igarashi J, Kok M, Winter C, Gregory SG, Hogarty MD, Maris JM and
Brodeur GM: Definition and characterization of a region of 1p36.3
consistently deleted in neuroblastoma. Oncogene. 24:2684–2694.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Henrich KO, Schwab M and Westermann F:
1p36 tumor suppression-a matter of dosage? Cancer Res.
72:6079–6088. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aygun N: Biological and genetic features
of neuroblastoma and their clinical importance. Curr Pediatr Rev.
14:73–90. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Raschellà G, Negroni A, Skorski T, Pucci
S, Nieborowska-Skorska M, Romeo A and Calabretta B: Inhibition of
proliferation by c-myb antisense RNA and oligodeoxynucleotides in
transformed neuroectodermal cell lines. Cancer Res. 52:4221–4226.
1992.PubMed/NCBI
|
|
14
|
Thiele CJ, Reynolds CP and Israel MA:
Decreased expression of N-myc precedes retinoic acid-induced
morphological differentiation of human neuroblastoma. Nature.
313:404–406. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thiele CJ, Cohen PS and Israel MA:
Regulation of c-myb expression in human neuroblastoma cells during
retinoic acid-induced differentiation. Mol Cell Biol. 8:1677–1683.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abemayor E and Sidell N: Human
neuroblastoma cell lines as models for the in vitro study of
neoplastic and neuronal cell differentiation. Environ Health
Perspect. 80:3–15. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aktas S, Altun Z, Erbayraktar Z, Aygun N
and Olgun N: Effect of cytotoxic agents and retinoic acid on Myc-N
protein expression in neuroblastoma. Appl Immunohistochem Mol
Morphol. 18:86–89. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guglielmi L, Cinnella C, Nardella M,
Maresca G, Valentini A, Mercanti D, Felsani A and D'Agnano I: MYCN
gene expression is required for the onset of the differentiation
programme in neuroblastoma cells. Cell Death Dis. 5:e10812014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nomura N, Takahashi M, Matsui M, Ishii S,
Date T, Sasamoto S and Ishizaki R: Isolation of human cDNA clones
of myb-related genes, A-myb and B-myb. Nucleic Acids Res.
16:11075–11089. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Davidson CJ, Tirouvanziam R, Herzenberg LA
and Lipsick JS: Functional evolution of the vertebrate Myb gene
family: B-Myb, but neither A-Myb nor c-Myb, complements
drosophila Myb in hemocytes. Genetics. 169:215–229. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Beall EL, Manak JR, Zhou S, Bell M,
Lipsick JS and Botchan MR: Role for a drosophila
Myb-containing protein complex in site-specific DNA replication.
Nature. 420:833–837. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu L, Zhang H and Tower J: Functionally
distinct, sequence-specific replicator and origin elements are
required for Drosophila chorion gene amplification. Genes
Dev. 15:134–146. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lewis PW, Beall EL, Fleischer TC,
Georlette D, Link AJ and Botchan MR: Identification of a
Drosophila Myb-E2F2/RBF transcriptional repressor complex.
Genes Dev. 18:2929–2940. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Koga H, Matsui S, Hirota T, Takebayashi S,
Okumura K and Saya H: A human homolog of Drosophila
lethal(3)malignant brain tumor (l(3)mbt) protein associates with
condensed mitotic chromosomes. Oncogene. 18:3799–3809. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gurvich N, Perna F, Farina A, Voza F,
Menendez S, Hurwitz J and Nimer SD: L3MBTL1 polycomb protein, a
candidate tumor suppressor in del(20q12) myeloid disorders, is
essential for genome stability. Proc Natl Acad Sci USA.
107:22552–22557. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fujita M: Cdt1 revisited: Complex and
tight regulation during the cell cycle and consequences of
deregulation in mammalian cells. Cell Div. 1:222006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Karakaidos P, Taraviras S, Vassiliou LV,
Zacharatos P, Kastrinakis NG, Kougiou D, Kouloukoussa M, Nishitani
H, Papavassiliou AG, Lygerou Z and Gorgoulis VG: Overexpression of
the replication licensing regulators hCdt1 and hCdc6 characterizes
a subset of non-small-cell lung carcinomas: Synergistic effect with
mutant p53 on tumor growth and chromosomal instability-evidence of
E2F-1 transcriptional control over hCdt1. Am J Pathol.
165:1351–1365. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xouri G, Lygerou Z, Nishitani H, Pachnis
V, Nurse P and Taraviras S: Cdt1 and geminin are down-regulated
upon cell cycle exit and are over-expressed in cancer-derived cell
lines. Eur J Biochem. 271:3368–3378. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wohlschlegel JA, Kutok JL, Weng AP and
Dutta A: Expression of geminin as a marker of cell proliferation in
normal tissues and malignancies. Am J Pathol. 161:267–273. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen L, Iraci N, Gherardi S, Gamble LD,
Wood KM, Perini G, Lunec J and Tweddle DA: p53 is a direct
transcriptional target of MYCN in neuroblastoma. Cancer Res.
70:1377–1388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eckerle I, Muth D, Batzler J, Henrich KO,
Lutz W, Fischer M, Witt O, Schwab M and Westermann F: Regulation of
BIRC5 and its isoform BIRC5-2B in neuroblastoma. Cancer Lett.
285:99–107. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Petroni M, Veschi V, Prodosmo A, Rinaldo
C, Massimi I, Carbonari M, Dominici C, McDowell HP, Rinaldi C,
Screpanti I, et al: MYCN sensitizes human neuroblastoma to
apoptosis by HIPK2 activation through a DNA damage response. Mol
Cancer Res. 9:67–77. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu DX, Biswas SC and Greene LA: B-Myb and
C-Myb play required roles in neuronal apoptosis evoked by nerve
growth factor deprivation and DNA damage. J Neurosci. 24:8720–8725.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aygun N and Altungoz O: Novel structural
abnormalities involving chromosomes 1, 17 and 2 identified by
fluorescence in situ hybridization (FISH) and/or cytogenetic
karyotyping in Kelly and SH-SY5Y human neuroblastoma cell lines,
respectively. J Can Res Updates. 6:46–55. 2017.
|
|
35
|
Protocol for DNA extraction. DNA
preparation from adherent cells. Section of Cancer Genomics,
Genetics Branch, NCI National Institutes of Health 2 pages. 2006,
https://ccr.cancer.gov/sites/default/files/dna_prep_from_adherent_cells.pdf
|
|
36
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Babicki S, Arndt D, Marcu A, Liang Y,
Grant JR, Maciejewski A and Wishart DS: Heatmapper: Web-enabled
heat mapping for all. Nucleic Acids Res. 44:W147–W153. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rasmussen R: Quantification on the
LightCycler. Meuer S, Wittwer C and Nakagawara K: Rapid cycle
real-time PCR, methods and applications Heidelberg: Springer Press;
pp. 21–34. 2001, View Article : Google Scholar
|
|
39
|
Aksakoglu G: Korelasyon ve regresyon
(Correlation and regression): Sağlıkta araştırma teknikleri ve
analiz yöntemleri (Research techniques and analysis methods in
health). 1st. Dokuz Eylul University Press; Izmir: pp. 305–346.
2001, (In Turkish).
|
|
40
|
Hiller S, Breit S, Wang ZQ, Wagner EF and
Schwab M: Localization of regulatory elements controlling human
MYCN expression. Oncogene. 6:969–977. 1991.PubMed/NCBI
|
|
41
|
Tower J: Developmental gene amplification
and origin regulation. Annu Rev Genet. 38:273–304. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wright JA, Smith HS, Watt FM, Hancock MC,
Hudson DL and Stark GR: DNA amplification is rare in normal human
cells. Proc Natl Acad Sci USA. 87:1791–1795. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Himoudi N, Yan M, Papanastasiou A and
Anderson J: MYCN as a target for cancer immunotherapy. Cancer
Immunol Immunother. 57:693–700. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Orr-Weaver TL and Spradling AC:
Drosophila chorion gene amplification requires an upstream
region regulating s18 transcription. Mol Cell Biol. 6:4624–4633.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Imamura Y, Iguchi-Ariga SM and Ariga H:
The upstream region of the mouse N-myc gene: Identification of an
enhancer element that functions preferentially in neuroblastoma
IMR32 cells. Biochim Biophys Acta. 1132:177–187. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen H, Li H, Liu F, Zheng X, Wang S, Bo X
and Shu W: An integrative analysis of TFBS-clustered regions
reveals new transcriptional regulation models on the accessible
chromatin landscape. Sci Rep. 5:84652015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhu W, Giangrande PH and Nevins JR: E2Fs
link the control of G1/S and G2/M transcription. EMBO J.
23:4615–4626. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nakata Y, Shetzline S, Sakashita C, Kalota
A, Rallapalli R, Rudnick SI, Zhang Y, Emerson SG and Gewirtz AM:
c-Myb contributes to G2/M cell cycle transition in human
hematopoietic cells by direct regulation of cyclin B1 expression.
Mol Cell Biol. 27:2048–2058. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tarasov KV, Tarasova YS, Tam WL, Riordon
DR, Elliott ST, Kania G, Li J, Yamanaka S, Crider DG, Testa G, et
al: B-MYB is essential for normal cell cycle progression and
chromosomal stability of embryonic stem cells. PLoS One.
3:e24782008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kreis NN, Sanhaji M, Rieger MA, Louwen F
and Yuan J: p21Waf1/Cip1 deficiency causes multiple mitotic defects
in tumor cells. Oncogene. 33:5716–5728. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Strieder V and Lutz W: E2F proteins
regulate MYCN expression in neuroblastomas. J Biol Chem.
278:2983–2989. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yoshida K and Inoue I: Regulation of
Geminin and Cdt1 expression by E2F transcription factors. Oncogene.
23:3802–3812. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Radhakrishnan SK, Feliciano CS, Najmabadi
F, Haegebarth A, Kandel ES, Tyner AL and Gartel AL: Constitutive
expression of E2F-1 leads to p21-dependent cell cycle arrest in S
phase of the cell cycle. Oncogene. 23:4173–4176. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sala A, Casella I, Bellon T, Calabretta B,
Watson RJ and Peschle C: B-myb promotes S phase and is a downstream
target of the negative regulator p107 in human cells. J Biol Chem.
271:9363–9367. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Markey M, Siddiqui H and Knudsen ES:
Geminin is targeted for repression by the retinoblastoma tumor
suppressor pathway through intragenic E2F sites. J Biol Chem.
279:29255–29262. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sala A, Nicolaides NC, Engelhard A, Bellon
T, Lawe DC, Arnold A, Graña X, Graña X, Giordano A and Calabretta
B: Correlation between E2F-1 requirement in the S phase and E2F-1
transactivation of cell cycle-related genes in human cells. Cancer
Res. 54:1402–1406. 1994.PubMed/NCBI
|
|
57
|
Brodeur GM: Neuroblastoma: Biological
insights into a clinical enigma. Nat Rev Cancer. 3:203–216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Stiewe T and Pützer BM: Role of the
p53-homologue p73 in E2F1-induced apoptosis. Nat Genet. 26:464–469.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Goldschneider D, Blanc E, Raguénez G,
Barrois M, Legrand A, Le Roux G, Haddada H, Bénard J and Douc-Rasy
S: Differential response of p53 target genes to p73 overexpression
in SH-SY5Y neuroblastoma cell line. J Cell Sci. 117:293–301. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Fontana L, Fiori ME, Albini S, Cifaldi L,
Giovinazzi S, Forloni M, Boldrini R, Donfrancesco A, Federici V,
Giacomini P, et al: Antagomir-17-5p abolishes the growth of
therapy-resistant neuroblastoma through p21 and BIM. PLoS One.
3:e22362008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tang XX, Zhao H, Kung B, Kim DY, Hicks SL,
Cohn SL, Cheung NK, Seeger RC, Evans AE and Ikegaki N: The MYCN
enigma: Significance of MYCN expression in neuroblastoma. Cancer
Res. 66:2826–2833. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jost CA, Marin MC and Kaelin WG Jr: p73 is
a human p53-related protein that can induce apoptosis. Nature.
389:191–194. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kaghad M, Bonnet H, Yang A, Creancier L,
Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, et al:
Monoallelically expressed gene related to p53 at 1p36, a region
frequently deleted in neuroblastoma and other human cancers. Cell.
90:809–819. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lau LM, Wolter JK, Lau JT, Cheng LS, Smith
KM, Hansford LM, Zhang L, Baruchel S, Robinson F and Irwin MS:
Cyclooxygenase inhibitors differentially modulate p73 isoforms in
neuroblastoma. Oncogene. 28:2024–2033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Fukasawa K, Wiener F, Vande Woude GF and
Mai S: Genomic instability and apoptosis are frequent in p53
deficient young mice. Oncogene. 15:1295–1302. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Prochazka P, Hrabeta J, Vicha A, Cipro S,
Stejskalova E, Musil Z, Vodicka P and Eckschlager T: Changes in
MYCN expression in human neuroblastoma cell lines following
cisplatin treatment may not be related to MYCN copy numbers. Oncol
Rep. 29:2415–2421. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Henrichsen CN, Chaignat E and Reymond A:
Copy number variants, diseases and gene expression. Hum Mol Genet.
18:R1–R8. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sexton T, Umlauf D, Kurukuti S and Fraser
P: The role of transcription factories in large-scale structure and
dynamics of interphase chromatin. Semin Cell Dev Biol. 18:691–697.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Pihan GA, Purohit A, Wallace J, Knecht H,
Woda B, Quesenberry P and Doxsey SJ: Centrosome defects and genetic
instability in malignant tumors. Cancer Res. 58:3974–3985.
1998.PubMed/NCBI
|
|
70
|
Adon AM, Zeng X, Harrison MK, Sannem S,
Kiyokawa H, Kaldis P and Saavedra HI: Cdk2 and Cdk4 regulate the
centrosome cycle and are critical mediators of centrosome
amplification in p53-null cells. Mol Cell Biol. 30:694–710. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Sugihara E, Kanai M, Matsui A, Onodera M,
Schwab M and Miwa M: Enhanced expression of MYCN leads to
centrosome hyperamplification after DNA damage in neuroblastoma
cells. Oncogene. 23:1005–1009. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Mantel C, Braun SE, Reid S, Henegariu O,
Liu L, Hangoc G and Broxmeyer HE: p21(cip-1/waf-1) deficiency
causes deformed nuclear architecture, centriole overduplication,
polyploidy, and relaxed microtubule damage checkpoints in human
hematopoietic cells. Blood. 93:1390–1398. 1999.PubMed/NCBI
|
|
73
|
Stewart ZA, Leach SD and Pietenpol JA:
p21(Waf1/Cip1) inhibition of cyclin E/Cdk2 activity prevents
endoreduplication after mitotic spindle disruption. Mol Cell Biol.
19:205–215. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chen J, Willingham T, Shuford M, Bruce D,
Rushing E, Smith Y and Nisen PD: Effects of ectopic overexpression
of p21(WAF1/CIP1) on aneuploidy and the malignant phenotype of
human brain tumor cells. Oncogene. 13:1395–1403. 1996.PubMed/NCBI
|
|
75
|
Quinn LM, Herr A, McGarry TJ and
Richardson H: The Drosophila geminin homolog: Roles for
Geminin in limiting DNA replication, in anaphase and in
neurogenesis. Genes Dev. 15:2741–2754. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Clijsters L, Ogink J and Wolthuis R: The
spindle checkpoint, APC/C(Cdc20), and APC/C(Cdh1) play distinct
roles in connecting mitosis to S phase. J Cell Biol. 201:1013–1026.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Biedler JL and Spengler BA: A novel
chromosome abnormality in human neuroblastoma and
antifolate-resistant Chinese hamster cell lines in culture. J Natl
Cancer Inst. 57:683–695. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Amler LC and Schwab M: Amplified N-myc in
human neuroblastoma cells is often arranged as clustered tandem
repeats of differently recombined DNA. Mol Cell Biol. 9:4903–4913.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Lo AW, Sabatier L, Fouladi B, Pottier G,
Ricoul M and Murnane JP: DNA amplification by
breakage/fusion/bridge cycles initiated by spontaneous telomere
loss in a human cancer cell line. Neoplasia. 4:531–538. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Schwab M: Human neuroblastoma: From basic
science to clinical debut of cellular oncogenes.
Naturwissenschaften. 86:71–78. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Watanabe T, Tanabe H and Horiuchi T: Gene
amplification system based on double rolling-circle replication as
a model for oncogene-type amplification. Nucleic Acids Res.
39:e1062011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Blumrich A, Zapatka M, Brueckner LM,
Zheglo D, Schwab M and Savelyeva L: The FRA2C common fragile site
maps to the borders of MYCN amplicons in neuroblastoma and is
associated with gross chromosomal rearrangements in different
cancers. Hum Mol Genet. 20:1488–1501. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Slack A, Thornton PC, Magner DB, Rosenberg
SM and Hastings PJ: On the mechanism of gene amplification induced
under stress in Escherichia coli. PLoS Genet. 2:e482006. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Aygun N: Acquired chromosomal
abnormalities and their potential formation mechanisms in solid
tumours. In: Chromosomal abnormalities-a hallmark manifestation of
genomic instability. Larramendy M: InTech. pp. 27–70. 2017,
https://www.intechopen.com/books/chromosomal-abnormalities-a-hallmark-manifestation-of-genomic-instability/acquired-chromosomal-abnormalities-and-their-potential-formation-mechanisms-in-solid-tumours
|
|
85
|
Forbes SA, Beare D, Boutselakis H, Bamford
S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al:
COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids
Res. 45:D777–D783. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Aygun N: Correlations between long
inverted repeat (LIR) features, deletion size and distance from
breakpoint in human gross gene deletions. Sci Rep. 5:83002015.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Lai PJ, Lim CT, Le HP, Katayama T, Leach
DR, Furukohri A and Maki H: Long inverted repeat transiently stalls
DNA replication by forming hairpin structures on both leading and
lagging strands. Genes Cells. 21:136–145. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Voineagu I, Narayanan V, Lobachev KS and
Mirkin SM: Replication stalling at unstable inverted repeats:
Interplay between DNA hairpins and fork stabilizing proteins. Proc
Natl Acad Sci USA. 105:9936–9941. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Conrad DF, Bird C, Blackburne B, Lindsay
S, Mamanova L, Lee C, Turner DJ and Hurles ME: Mutation spectrum
revealed by breakpoint sequencing of human germline CNVs. Nat
Genet. 42:385–391. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
90
|
Hastings PJ, Ira G and Lupski JR: A
microhomology-mediated break-induced replication model for the
origin of human copy number variation. PLoS Genet. 5:e10003272009.
View Article : Google Scholar : PubMed/NCBI
|















