Introduction
Alzheimer's disease (AD) is a progressive
neurodegenerative disorder characterized by cognitive and memory
impairments (1). There are >35
million individuals with AD worldwide, thus resulting in marked
emotional and financial burdens on patients and society (2). The accumulation of β-amyloid (Aβ)
peptide in the brain is strongly implicated as a main hallmark of
sporadic and familial forms of AD (3,4).
Amyloid precursor protein (APP) -derived toxic peptides are found
at autopsy in the brains of individuals with AD (5). Aβ is cleaved from APP by β-secretase
1 (BACE1) and subsequently by γ-secretase. BACE1 is considered the
rate-limiting enzyme in the production of Aβ (6,7). In
addition, accumulation of BACE1 is observed in normal and
dystrophic presynaptic terminals surrounding the amyloid plaques in
brains of AD mouse models and patients (8,9).
The effects of medications that are currently
approved by the USA Food and Drug Administration for the treatment
of AD manifestations are modest, transient and provide only
symptomatic treatment (10).
Recently, researchers have been paying more attention to herbal
extracts for their potential therapeutic effects on AD. Resveratrol
(Res) is a natural polyphenol with strong effects, including
anti-oxidative, anti-inflammatory, cardiovascular protective,
neuroprotective and cancer chemopreventive activities (11–13).
It has been suggested that Res can act as a potent antioxidant in
neurodegenerative disorders. Previous studies have reported that
Res has the ability to regulate Aβ toxicity or significantly
increase the Aβ clearance rate in a mouse model of AD (14,15).
Res can also increase resistance against nerve inflammation and
exerts anti-oxidant effects that contribute to its neuroprotective
effects on the nervous system (16,17).
Tg6799 mice are APP/presenelin 1 (PS1) double
transgenic mice that coexpress five familial AD (FAD) mutations,
which are also known as 5XFAD mice. This mouse strain is a useful
model of AD that recapitulates the relevant pathogenic features of
AD amyloid pathology. Intraneuronal Aβ42 accumulates in
the brain starting at 1.5 months, and the levels of brain
Aβ42 and Aβ40 increase with age, whereas, the
Aβ42/Aβ40 ratio decrease. Amyloid deposition
starts at 2 months. Tg6799 mice develop memory deficits by 4 months
of age, as assessed using a Y-maze test compared with nontransgenic
control mice (18). The present
study aimed to determine the protective effects and underlying
mechanisms of Res in AD treatment.
Materials and methods
Animals and Res treatment
This study was approved by The Animal Ethics
Committee of Nanfang Hospital, Southern Medical University
(application no. NFYY-2015-43; Guangzhou, China). The experiments
were performed using male Tg6799 mice (Jackson Laboratory, Bar
Harbor, ME, USA), which express the human APP and PS1 genes and
five FAD mutations on a C57/B6XSJL background (18). These mice were group-housed under
standard environmental conditions (12-h light/dark cycle, 23±1°C
and 55±5% relative humidity), with free access to food and water.
The procedures were performed in strict accordance with the
National Institutes of Health Guidelines for the Care and Use of
Laboratory Animals (19). At 2.5
months of age, 40 male Tg6799 mice (weight, 18–22 g) were randomly
divided into the control and Res groups (n=20/group). According to
a previous study (20), the Res
group was treated daily with 0.5% Res (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany; 60 mg/kg/day) by oral gavage for 60 consecutive
days, whereas the control group was treated with volume-matched
normal saline by gavage.
Behavioral testing
Open field test
The open field apparatus consisted of a rectangular
chamber (40×40×30 cm) made of gray polyvinyl chloride. A video
camera, a loudspeaker that provided masking noise and a 25 W red
light bulb (illumination density at the center of the maze was 0.3
lx) were placed 180 cm above the center of the apparatus. The mice
were gently placed at the center and were allowed to explore the
area for 5 min. The digitized image of the path taken by each mouse
was recorded and locomotor activity was subsequently analyzed using
EthoVision 7.0 software (Noldus Information Technology, Wageningen,
The Netherlands).
Y-maze test
A black Y-maze was used with an arm length of 40 cm,
width of 3 cm and wall height of 12.5 cm. Each mouse was placed in
the center of the symmetrical Y-maze and allowed to explore freely
through the maze until they entered the arms 22 times; the sequence
of 22 entries was recorded. Each mouse was placed at the end of one
arm facing the center and its movements were recorded for 8 min.
Data were analyzed to determine the number of arm entries without
repetition. Successful alternations were defined as sequential
entries into a new arm. A mouse was excluded from the analysis if
no entries into new arms were recorded for 2 consecutive min. Mean
alternation percentage (%) was calculated as the number of
successful alternations /20 ×100.
Morris water maze
The basic protocol and apparatus for the hidden
platform version of the water maze have previously been described
(21). Each mouse underwent four
trials a day for 6 consecutive days. All mice underwent a probe
test for 90 sec, where the platform was removed from the pool on
day 7. The number of crosses over the platform location (crosses),
the time spent in the target quadrant and the speed were recorded
using a video tracking system.
Thioflavin S staining
Four mice from each group were sacrificed with 0.75%
pentobarbitone sodium (50 mg/kg) by intraperitoneal injection. The
deeply anesthetized mice exhibiting loss of reflexes and muscle
relaxation were perfused with 4% paraformaldehyde in PBS.
Subsequently, the hippocampus was extracted and frozen sections,
measuring 35 µm in thickness, were prepared at −25°C. For
thioflavin S immunostaining, brain sections were allowed to defrost
at room temperature and were then washed with PBS for 5 min. The
sections were then stained with freshly prepared 0.4% thioflavin S
solution (Sigma-Aldrich; Merck KGaA) for 5 min in the dark at room
temperature, washed twice for 3 min with 70% alcohol, washed twice
for 5 min with PBS and mounted. Image acquisition was performed
under a fluorescence microscope. Plaque counts were quantified by
counting thioflavin S-positive plaques in the hippocampus. The
region of interest was manually selected under ×4 magnification and
all parameters were kept constant during the analysis.
Aβ40 and Aβ42
ELISA
Another four mice from each group were used to
conduct Aβ ELISA analyses. Following Res treatment for 60 days,
animals were anesthetized by pentobarbitone sodium (50 mg/kg) and
were decapitated. The required amount of homogenization buffer was
prepared prior to use. The hemisphere tissues (50 mg) were isolated
and homogenized using a Potter-Elvehjem homogenizer (DWK Life
Sciences, Millville, NJ, USA). Brain homogenates were treated with
PBS/protease inhibitor (Thermo Fisher Scientific, Inc., Waltham,
MA, USA) and were supplemented with guanidine HCl to a final
concentration of 5 mol/l. The brain tissue samples were then added
to an Eppendorf tube containing the brain extraction buffer
provided in the ELISA kits (cat. nos. KHB3481, KHB3441 and KHB3442;
Invitrogen; Thermo Fisher Scientific, Inc.). The sample was ground
thoroughly with a tissue homogenizer, and then centrifuged at
16,000 × g for 20 min at 4°C. The supernatant was carefully
transferred to a new tube and diluted with the buffer provided in
the BioRad protein assay kit (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). Final guanidine HCl concentrations were <0.1 mol/l.
The protein content was measured using the Bio-Rad protein assay
kit. Duplicate samples were subsequently analyzed using
Aβ40- and Aβ42-specific sandwich colorimetric
ELISA kits (cat. nos. KHB3481, KHB3441 and KHB3442; Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. The absorbance was measured at 450 nm, and curve-fitting
software (TableCurve 2D; version 6; Jandel Scientific Software, San
Rafael, CA) was used to generate the standard curve.
Western blotting
For western blot analysis, hippocampal tissues were
dissected from four mice in each group and disrupted by sonication
(three pulses at 1 Hz for 20 sec with 30 sec intervals between
pulses) for 2 min in ice-cold radioimmunoprecipitation assay buffer
with a complete protease inhibitor cocktail (Roche Diagnostics,
Indianapolis, IN, USA). The lysates were centrifuged at 16,900 × g
for 40 min at 4°C, and the supernatant was boiled in loading buffer
for 10 min. The protein content was measured using a Bio-Rad
protein assay kit (Bio-Rad Laboratories, Inc.). A total of 50 µg
protein was loaded into each lane and separated by SDS-PAGE with
10% polyacrylamide gels. Following transfer onto Immobilon
polyvinylidene difluoride membranes, nonspecific binding was
blocked with 5% nonfat milk in TBS containing 0.05% Tween 20 for
1.5 h at room temperature. After the membranes were washed, they
were incubated with the following primary antibodies: Anti-APP
(1:1,000; cat. no. SIG-39300; BioLegend, Inc., San Diego, CA, USA),
anti-BACE1 (1:1,000; cat. no. ab108394; Abcam, Cambridge, UK),
anti-secreted (s)APPα (1:500; cat. no. 2B3; IBL, Japan), anti-sAPPβ
(1:500, cat. no. 6A1; Immuno-Biological Laboratories Co., Ltd.,
Fujioka, Japan) and anti-sirtuin 1 (SIRT1; 1:1,000; cat. no. 8469s;
Cell Signaling Technology, Inc., Danvers, MA, USA) at 4°C
overnight. The membranes were then incubated with horseradish
peroxidase-conjugated goat anti-mouse or anti-rabbit secondary
antibodies (1:2,000; cat. nos. TA130003 and TA130023; OriGene
Technologies, Inc., Beijing, China) for 1 h at room temperature.
Bands were visualized using enhanced chemiluminescence western blot
detection reagents (Bio-Rad Laboratories, Inc.). ImageJ software
(version 1.50; National Institutes of Health, Bethesda, MD, USA)
was used to semi-quantify the relative expression levels of target
proteins, which were normalized to the internal control GAPDH
(1:1,000; cat. no. 5174; Cell Signaling Technology, Inc.).
Statistical analysis
Data are presented as the means ± standard error of
the mean, and all experiments were repeated at least three times.
Data analysis was conducted using SPSS 18.0 statistical software
(SPSS, Inc., Chicago, IL, USA). Comparisons between two groups were
performed using two independent samples Student's t-test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Res treatment rescues cognitive
impairment but does not affect the motor function of Tg6799
mice
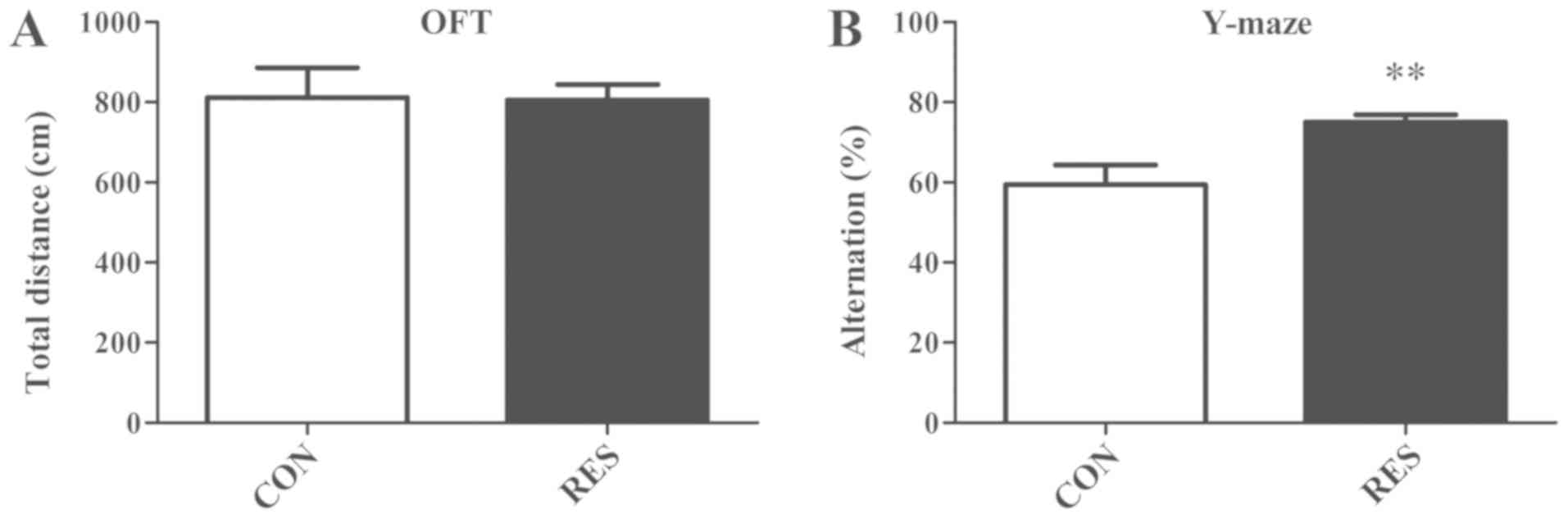
Open field tests were used to measure locomotor
activity, which served as a control to reflect motor function. In
the open field test, the total distances of the two groups over 5
min were not significantly different (Fig. 1A). In addition, the spontaneous
alternation performance of mice was determined using the Y-maze
test, which is used to assess their spatial working memory. The
results revealed that the Res group had an elevated mean
alternation percentage compared with the control group (Fig. 1B). These findings indicated that
the administration of Res improved spatial working memory.
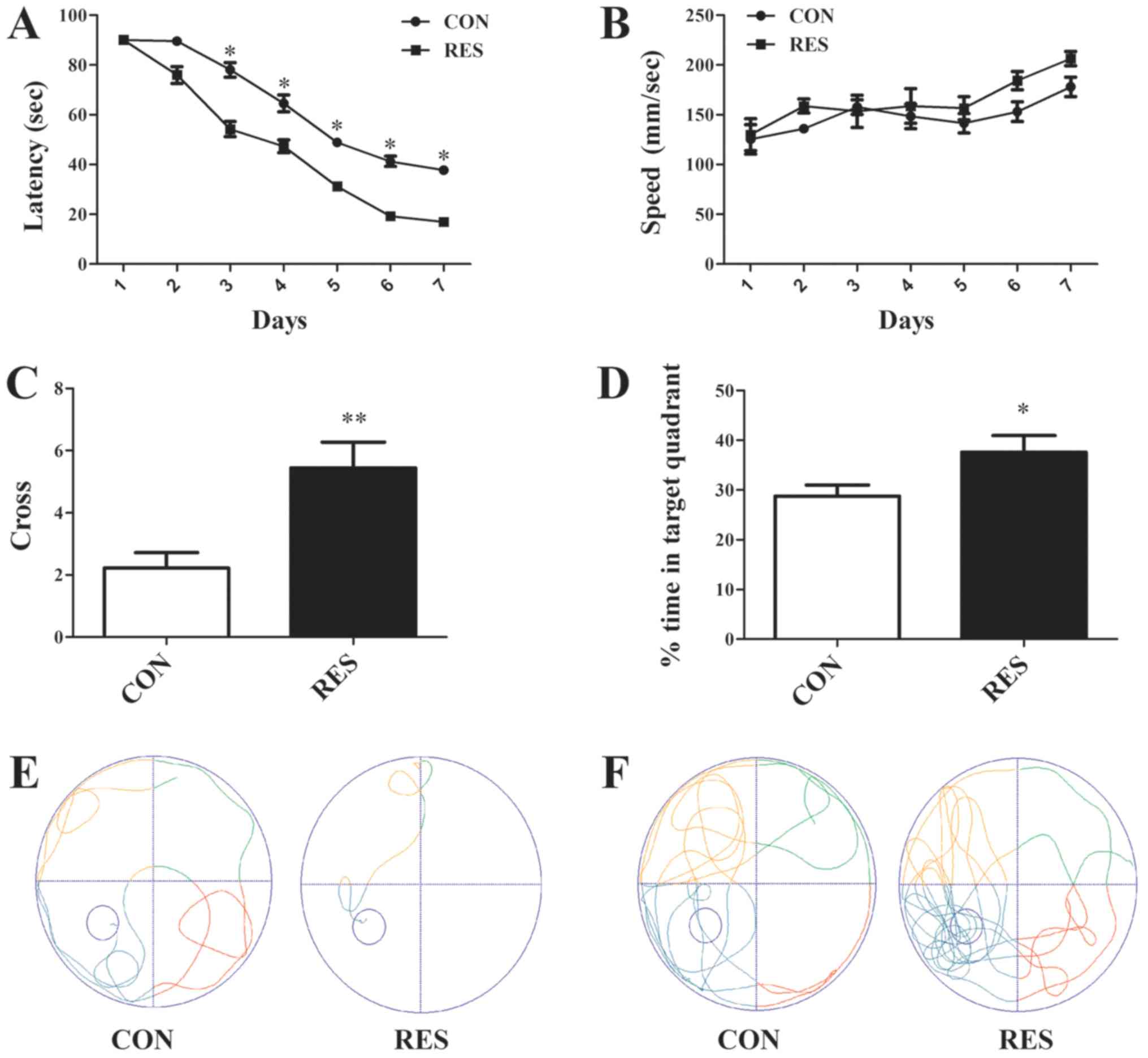
The effects of Res on learning and memory were also
determined in AD mice using the Morris water maze. The Morris water
maze test is most widely used to test spatial memory in AD mouse
models. Over the 6-day training phase, the escape latencies of all
mice gradually decreased; however, the Res group mice exhibited
significantly lower escape latencies compared with the controls
(Fig. 2A). The average swimming
speed during training and the probe test did not differ between the
two groups (Fig. 2B). In addition,
the administration of Res significantly increased the number of
crosses and time spent in the target quadrant (Fig. 2C and D). The swimming path during
the training phase and the probe test reflected the improved
learning and memory of the mice in the Res group (Fig. 2E and F).
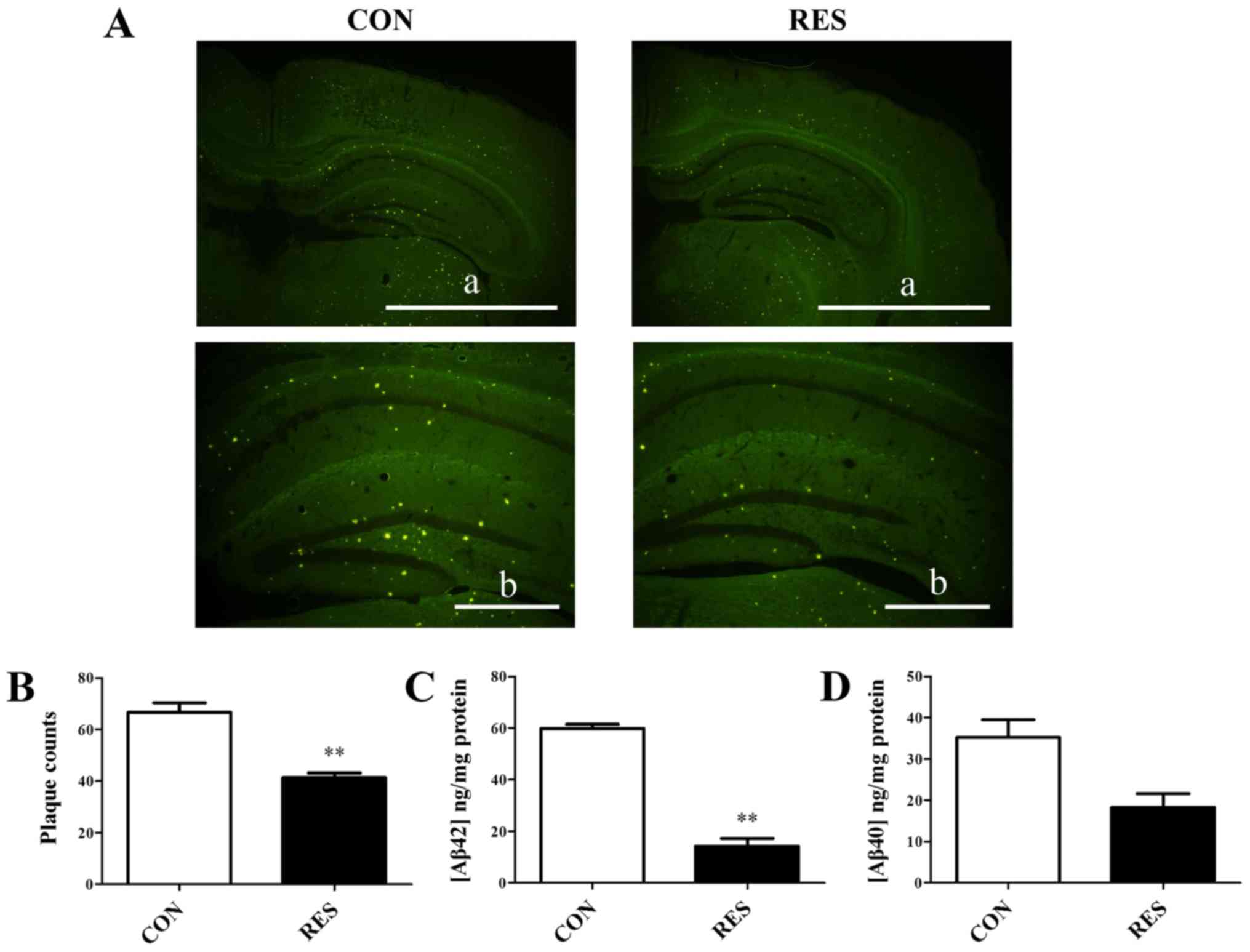
Administration of Res reduces amyloid
plaques and Aβ42 levels in Tg6799 mice
Thioflavin S staining was performed to examine
amyloid plaque pathology. Amyloid deposition was positive after
staining with thioflavin S. The results revealed that the plaque
loads in the Res group were quantitatively lower compared with in
the control group. Amyloid plaques were significantly reduced in
the hippocampus of the Res group compared with in the control group
(Fig. 3A and B). Cerebral levels
of Aβ42 and Aβ40 were also detected in
hippocampal homogenates using ELISA kits. The Res group had lower
levels of Aβ42 and Aβ40 compared with the
control group. For Aβ42 levels, this alteration was
statistically significant (Fig. 3C and
D), which was consistent with the thioflavin S staining
results.
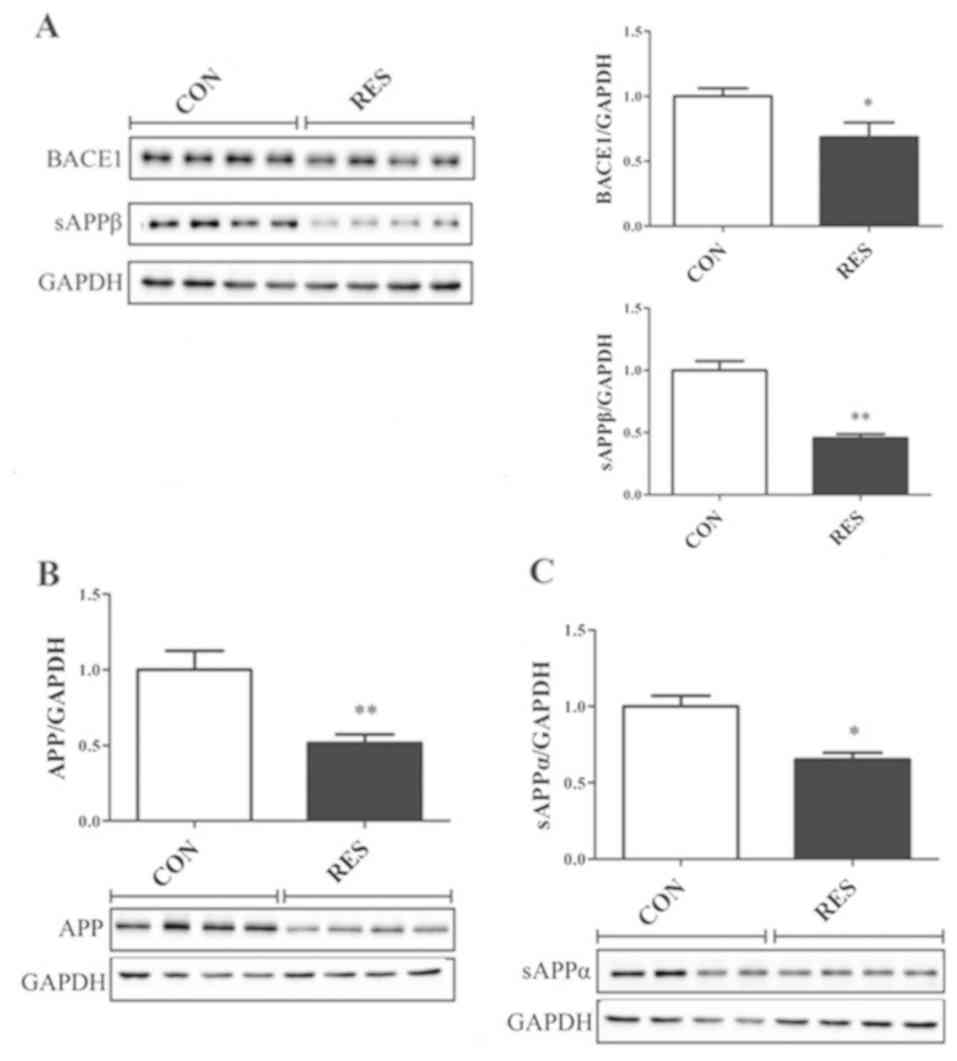
Administration of Res alters the
levels of BACE1, APP, sAPPα and sAPPβ in Tg6799 mice
Western blotting was used to determine APP, cleaved
APP products and BACE1 expression, in order to investigate the
mechanism underlying the reduction of amyloid plaques in the Res
group. The results of western blot analysis revealed that the
expression levels of APP, sAPPα, sAPPβ and BACE1 were significantly
reduced in the Res group compared with in the control group
(Fig. 4).
Res treatment did not affect SIRT1
levels in Tg6799 mice
A previous study (22) suggested that activation of the
sirtuin family member SIRT1 is an important pharmacological effect
of Res. Therefore, SIRT1 levels were determined by western
blotting; however, the administration of Res did not affect the
protein expression levels of SIRT1 (Fig. 5).
Discussion
The pathogenesis of AD continues to be debated, and
there are numerous hypotheses, including the amyloid cascade
hypothesis, neurotrophic factor hypothesis and oxidative stress
hypothesis (23). The pathological
hallmarks of AD include intraneuronal neurofibrillary tangles,
extracellular amyloid plaques, neuronal loss and gliosis. The
aggregation of Aβ forms amyloid plaques. Numerous clinical,
cytobiological and experimental animal studies (24,25)
support the causative role of Aβ in the pathogenesis of AD.
Notably, Aβ can induce loss of synaptic terminals and calcium
imbalance, and can drive tau aggregation, thus inducing
neurofibrillary tangles (26,27).
Aβ is cleaved from APP, genetic mutations of which cause a familial
form of AD. BACE1 cuts APP at the N-terminus of the Aβ domain to
produce C99 and sAPPβ, after which, C99 is further cleaved by
γ-secretase to generate Aβ (28).
Notably, previous studies have demonstrated that Res is able to
cross the blood-brain barrier, thus making oral gavage an efficient
route of administration (29,30).
The present study demonstrated that the
administration of Res significantly diminished the number and
intensity of amyloid plaques, as measured by thioflavin S staining.
Senile plaques are formed by the accumulation of APP-derived toxic
peptides (predominantly Aβ42). Previous data support the
hypothesis that AD neurodegeneration is initiated by an imbalance
between Aβ42 production and clearance in selected brain
regions (4). Tg6799 mice
accumulate large amounts of cerebral Aβ42 at a young
age, and generate much more Aβ42 than Aβ40.
The ELISA results in this study demonstrated that the cerebral
levels of Aβ42 and Aβ40 were decreased in
hippocampal homogenates in response to Res, particularly
Aβ42 levels, which were significantly reduced.
In vitro, Res promotes Aβ clearance
potentially by increasing intracellular proteasomal activity
(31). In a previous study, the
Tg2576 mouse transgenic model of AD was fed red wine (containing
Res), after which cognitive improvement or attenuation of amyloid
brain pathology was assessed based on nonamyloidogenic processing
of βAPP or the oligomerization of Aβ molecules (15,32).
To investigate the mechanism underlying the reduction of plaque
pathology by Res, the present study detected the expression levels
of APP-associated enzymes by western blotting. A significant
decrease in BACE1 levels may alter the APP processing pathway,
leading to a reduction in amyloid burden. BACE1 is considered the
rate-limiting enzyme in the production of Aβ. High-molecular weight
APP and sAPPα levels were also decreased in response to Res. A
number of previous studies (33,34)
have suggested that Res can promote Aβ clearance without affecting
Aβ-producing enzyme activities (γ-secretase and BACE1). This
mechanism could be synergistic with a reduction in BACE1.
A previous study (35) suggested that Res is able to induce
activation of the sirtuin family member SIRT1, which is beneficial
for preventing neurodegeneration and improving memory. In addition,
Res can counteract Aβ toxicity in cellular models due to its
natural antioxidant properties and SIRT1 activation (36–38).
Other findings have suggested that Res is able to reduce the
harmful process that occurs in the APP/PS1 mouse hippocampus, which
is mainly mediated by increased activation of SIRT1 and 5′
AMP-activated protein kinase pathways (39). However, in the present study, Res
did not induce activation of SIRT1.
The present study assessed hippocampus-dependent
spatial working memory in mice using the Y-maze test. This learning
task does not involve any training, reward or punishment, and
allows for the assessment of hippocampus-dependent spatial working
memory. The hippocampus is the hub for learning and memory. The
results demonstrated that treatment with Res resulted in a
significant increase in the mean alternation percentage. It may be
hypothesized that reduced amyloid plaques are responsible for the
improved working memory in the Res group. Furthermore, the effects
of Res treatment on learning and memory in Tg6799 mice were
evaluated using the Morris water maze test. In the hidden platform
version of the water maze test, animals learn to locate a submerged
platform in a pool filled with opaque water. This spatial
navigation performance is also known to be hippocampus dependent.
This study confirmed that learning and memory were significantly
improved by the administration of Res.
Numerous previous studies (40,41)
have described the neuroprotective properties of Res; however, Res
treatment as a therapy for AD still has many limitations, including
poor bioavailability. The complex interaction and mechanisms of Res
remain to be explored. In future studies, we aim to investigate the
expression of α and γ secretases (ADAM metallopeptidase domain 9,
10 and 17, and PS1), and to investigate whether a modified
treatment with Res may induce alterations in SIRT1 activity. In the
present study, mice were treated with 60 mg/kg/day Res by oral
gavage; this dosage per day is much higher than that ingested by
humans via diet and wine, this is also an important issue to be
considered in future work.
In conclusion, the present study demonstrated that
the administration of Res for 60 consecutive days protected against
Aβ plaque formation and cognitive loss in Tg6799 mice. The exact
underlying mechanism has yet to be elucidated; however, the animal
behavioral data suggested that Res may serve a neuroprotective role
in a mouse model of AD.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81471388 and
81771484), the Natural Science Foundation of Guangdong Province,
China (grant nos. 2015A030313235 and 2014A030313351), the Guangdong
Science & Technology Planning Project, China (grant no.
2014A020211016), the Undergraduate Innovation and Entrepreneurship
Training Program of Southern Medical University (grant no.
C1051222), and the Program for Changjiang Scholars and Innovative
Research Team in University (grant no. IRT_16R37).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
XMZ, FW and YC conceived and designed the
experiments, and revised the final version of the paper. YC, ZML
and GWS performed the experiments. YC and YSS analyzed the data and
wrote the paper. LP, SYS and YPW prepared reagents and materials,
and performed part of the experiments. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the animal ethics
committee of Nanfang Hospital, Southern Medical University
(application no. NFYY-2015-43).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
McKhann G, Drachman D, Folstein M, Katzman
R, Price D and Stadlan EM: Clinical diagnosis of Alzheimer's
disease: Report of the NINCDS-ADRDA work group under the auspices
of department of health and human services task force on
Alzheimer's disease. Neurology. 34:939–944. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alzheimer's Association: 2011 Alzheimer's
disease facts and figures. Alzheimers Dement. 7:208–244. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Selkoe DJ and Schenk D: Schenk,
Alzheimer's disease: Molecular understanding predicts amyloid-based
therapeutics. Annu Rev Pharmacol Toxicol. 43:545–584. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hardy J and Selkoe DJ: The amyloid
hypothesis of Alzheimer's disease: Progress and problems on the
road to therapeutics. Science. 297:353–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan
M, Wisniewski HM and Binder LI: Abnormal phosphorylation of the
microtubule-associated protein tau (tau) in Alzheimer cytoskeletal
pathology. Proc Natl Acad Sci USA. 83:4913–4917. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cole SL and Vassar R: BACE1 structure and
function in health and Alzheimer's disease. Curr Alzheimer Res.
5:100–120. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin X, Koelsch G, Wu S, Downs D, Dashti A
and Tang J: Human aspartic protease memapsin 2 cleaves the
beta-secretase site of beta-amyloid precursor protein. Proc Natl
Acad Sci USA. 97:1456–1460. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B,
O'Connor T, Logan S, Maus E, Citron M, Berry R, et al: Beta-site
amyloid precursor protein cleaving enzyme 1 levels become elevated
in neurons around amyloid plaques: Implications for Alzheimer's
disease pathogenesis. J Neurosci. 27:3639–3649. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kandalepas PC, Sadleir KR, Eimer WA, Zhao
J, Nicholson DA and Vassar R: The Alzheimer's β-secretase BACE1
localizes to normal presynaptic terminals and to dystrophic
presynaptic terminals surrounding amyloid plaques. Acta
Neuropathol. 126:329–352. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
De Strooper B: Proteases and proteolysis
in Alzheimer disease: A multifactorial view on the disease process.
Physiol Rev. 90:465–494. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Robb EL, Page MM, Wiens BE and Stuart JA:
Molecular mechanisms of oxidative stress resistance induced by
resveratrol: Specific and progressive induction of MnSOD. Biochem
Biophys Res Commun. 367:406–412. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baltaci AK, Arslangil D, Mogulkoc R and
Patlar S: Effect of resveratrol administration on the element
metabolism in the blood and brain tissues of rats subjected to
acute swimming exercise. Biol Trace Elem Res. 175:421–427. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baltaci SB, Mogulkoc R and Baltaci AK:
Resveratrol and Exercise. Biomed Rep. 5:525–530. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ladiwala AR, Lin JC, Bale SS,
Marcelino-Cruz AM, Bhattacharya M, Dordick JS and Tessier PM:
Resveratrol selectively remodels soluble oligomers and fibrils of
amyloid Abeta into off-pathway conformers. J Biol Chem.
285:24228–24237. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang J, Ho L, Zhao Z, Seror I, Humala N,
Dickstein DL, Thiyagarajan M, Percival SS, Talcott ST and Pasinetti
GM: Moderate consumption of Cabernet Sauvignon attenuates Abeta
neuropathology in a mouse model of Alzheimer's disease. FASEB J.
20:2313–2320. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Capiralla H, Vingtdeux V, Zhao H,
Sankowski R, Al-Abed Y, Davies P and Marambaud P: Resveratrol
mitigates lipopolysaccharide- and Aβ-mediated microglial
inflammation by inhibiting the TLR4/NF-κB/STAT signaling cascade. J
Neurochem. 120:461–472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Crandall JP and Barzilai N: Exploring the
promise of resveratrol: Where do we go from here? Diabetes.
62:1022–1023. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oakley H, Cole SL, Logan S, Maus E, Shao
P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik
L, et al: Intraneuronal beta-amyloid aggregates, neurodegeneration,
and neuron loss in transgenic mice with five familial Alzheimer's
disease mutations: Potential factors in amyloid plaque formation. J
Neurosci. 26:10129–10140. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
The NIH guidelines for the care and use of
laboratory animals, . NIH Publication. 85–23, 1985. 1985.
|
|
20
|
Menet MC, Baron S, Taghi M, Diestra R,
Dargère D, Laprévote O, Nivet-Antoine V, Beaudeux JL, Bédarida T
and Cottart CH: Distribution of trans-resveratrol and its
metabolites after acute or sustained administration in mouse heart,
brain, and liver. Mol Nutr Food Res. 61:2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ohno M, Tseng W, Silva AJ and Disterhoft
JF: Trace eyeblink conditioning requires the hippocampus but not
autophosphorylation of alphaCaMKII in mice. Learn Mem. 12:211–215.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hong YA, Bae SY, Ahn SY, Kim J, Kwon YJ,
Jung WY and Ko GJ: Resveratrol ameliorates contrast induced
nephropathy through the activation of SIRT1-PGC-1α-Foxo1 signaling
in mice. Kidney Blood Press Res. 42:641–653. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Area-Gomez E and Schon EA: Alzheimer
disease. Adv Exp Med Biol. 997:149–156. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Barage SH and Sonawane KD: Amyloid cascade
hypothesis: Pathogenesis andtherapeutic strategies in Alzheimer's
disease. Neuropeptides. 52:1–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Andreeva TV, Lukiw WJ and Rogaev EI:
Biological basis for amyloidogenesis in Alzheimer's disease.
Biochemistry (Mosc). 82:122–139. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Billings LM, Oddo S, Green KN, McGaugh JL
and LaFerla FM: Intraneuronal Abeta causes the onset of early
Alzheimer's disease-related cognitive deficits in transgenic mice.
Neuron. 45:675–688. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Selkoe DJ and Schenk D: Alzheimer's
disease: Molecular understanding predicts amyloid-based
therapeutics. Annu Rev Pharmacol Toxicol. 43:545–584. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fukumoto H, Takahashi H, Tarui N, Matsui
J, Tomita T, Hirode M, Sagayama M, Maeda R, Kawamoto M, Hirai K, et
al: A noncompetitive BACE1 inhibitor TAK-070 ameliorates Abeta
pathology and behavioral deficits in a mouse model of Alzheimer's
disease. J Neurosci. 30:11157–11166. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Han YS, Bastianetto S, Dumont Y and
Quirion R: Specific plasma membrane binding sites for polyphenols,
including resveratrol, in the rat brain. J Pharmacol Exp Ther.
318:238–245. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gilgun-Sherki Y, Melamed E and Offen D:
Oxidative stress induced-neurodegenerative diseases: The need for
antioxidants that penetrate the blood brain barrier.
Neuropharmcology. 40:959–975. 2001. View Article : Google Scholar
|
|
31
|
Marambaud P, Zhao H and Davies P:
Resveratrol promotes clearance of Alzheimer's disease amyloid-beta
peptides. J Biol Chem. 280:37377–37382. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ho L, Chen LH, Wang J, Zhao W, Talcott ST,
Ono K, Teplow D, Humala N, Cheng A, Percival SS, et al:
Heterogeneity in red wine polyphenolic contents differentially
influences Alzheimer's disease-type neuropathology and cognitive
deterioration. J Alzheimers Dis. 16:59–72. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jia Y, Wang N and Liu X: Resveratrol and
amyloid-beta: Mechanistic insights. Nutrients. 9(pii): E11222017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Deng H and Mi MT: Resveratrol attenuates
Aβ25–35 caused neurotoxicity by inducing autophagy through the
TyrRS-PARP1-SIRT1 signaling pathway. Neurochem Res. 41:2367–2379.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen CJ, Yu W, Fu YC, Wang X, Li JL and
Wang W: Resveratrol protects cardiomyocytes from hypoxia-induced
apoptosis through the SIRT1-FoxO1 pathway. Biochem Biophys Res
Commun. 378:389–393. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jang JH and Surh YJ: Protective effect of
resveratrol on beta-amyloid-induced oxidative PC12 cell death. Free
RadicBiol Med. 34:1100–1110. 2003. View Article : Google Scholar
|
|
37
|
Kim D, Nguyen MD, Dobbin MM, Fischer A,
Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, et
al: SIRT1 deacetylase protects against neurodegeneration in models
for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J.
26:3169–3179. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lagouge M, Argmann C, Gerhart-Hines Z,
Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P,
Elliott P, et al: Resveratrol improves mitochondrial function and
protects against metabolic disease by activating SIRT1 and
PGC-1alpha. Cell. 127:1109–1122. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Porquet D, Griñán-Ferré C, Ferrer I,
Camins A, Sanfeliu C, Del Valle J and Pallàs M: Neuroprotective
role of trans-resveratrol in a murine model of familial Alzheimer's
disease. J Alzheimers Dis. 42:1209–1220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bastianetto S, Ménard C and Quirion R:
Neuroprotective action of resveratrol. Biochim Biophys Acta.
1852:1195–1201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hou Y, Wang K, Wan W, Cheng Y, Pu X and Ye
X: Resveratrol provides neuroprotection by regulating the
JAK2/STAT3/PI3K/AKT/mTOR pathway after stroke in rats. Genes Dis.
5:245–255. 2018. View Article : Google Scholar : PubMed/NCBI
|