Introduction
Cutaneous squamous cell carcinoma (CSCC) is the
second most common cancer with an annual incidence of over one
million worldwide (1). CSCC
develops mainly in skin with chronic sun exposure. Ultraviolet (UV)
radiation is considered to be the major cause of CSCC due to
UV-induced DNA damage and epigenetic changes (2). In response to UV radiation, skin
cells activate several pathways including mitogen-activated protein
kinase (MAPK), phosphoinositide 3-kinase (PI3K), extracellular
signal-regulated kinase (ERK), p38 kinase, and c-Jun N-terminal
kinase (JNK) signaling cascades (3,4).
These pathways can promote tumorigenesis at an early stage, and
facilitate the development of CSCC to a poorly differentiated
cancer. Furthermore, metastases can eventually develop in the lymph
system or other organs (5).
For invasive CSCC, surgical excision and Mohs
micrographic surgery are typically the primary treatment options
(6). Chemotherapy is considered to
be adjuvant therapy to block the aforementioned pro-carcinogenic
pathways in select high risk cases of CSCC to provide improved
locoregional control (5). The
selection of appropriate targets is critical for the success of
chemotherapy.
The epidermal growth factor receptor (EGFR), an
activator of MAPK and PI3K pathways, is stimulated upon exposure to
UV radiation (2). Blockade of the
EGFR inhibits the subsequent activation of EGFR downstream
signaling cascades and makes EGFR the optimal target for cancer
therapy, including CSCC (5,7).
Among the various EGFR inhibitors, gefitinib (ZD1839) is a
selective EGFR tyrosine kinase inhibitor with a significant
beneficial effect in various cancers (8,9).
Cell death induced by gefitinib is one of the major mechanisms by
which it inhibits cancer development. Apoptosis, type I programmed
cell death, was considered to be one of the most promising
antitumor mechanisms in cancer therapy. However, inherent and
acquired resistance to apoptosis play crucial roles in tumor
development and treatment failure. Therefore, novel strategies are
essential to improve treatment efficiency.
Autophagy, an evolutionarily conserved catabolic
process, sequesters proteins and damaged organelles into
autophagosomes, then fuses them with lysosomes to form
autolysosomes for bulk degradation of embedded materials (7). The role of autophagy is debatable and
has special importance in cancer research. Primarily, autophagy is
defined as type II programmed cell death (10). It is considered to be an
alternative tumor-suppressing mechanism induced by genomic
instability, suppression of cell growth, and degradation of vital
components (11). However,
recycled proteins and other components, along with the energy
saved, contribute to the maintenance of cellular homeostasis and
support tumor cell survival under stressed circumstances (9,11).
Therefore, the role of autophagy is complicated and may lead to
diametrically opposed consequences: tumor survival or death.
In this study, the tumor-suppressive role of
gefitinib was evaluated in A431 CSCC cells that are recognized for
their high expression of EGFR. Gefitinib treatment strongly
suppressed the proliferation and invasion of CSCC cells by
targeting EGFR to induce apoptosis and autophagy. Co-treatment of
these cells with gefitinib and chloroquine inhibited protective
autophagy and enhanced apoptosis, thereby improving the therapeutic
response of CSCC cells.
Materials and methods
Ethics statement
The present study was approved by the Institutional
Review Board of Nanfang Hospital, affiliated to Southern Medical
University. All patients provided written informed consent for the
use of surgical samples. This study was conducted in accordance
with the World Medical Association Declaration of Helsinki.
Cell culture and tumor samples
The CSCC cell line, A431 (China Center for Type
Culture Collection and Cell Bank of the Chinese Academy of
Sciences, Shanghai, China), and the human benign epidermal
keratinocyte cell line, HaCaT (China Center for Type Culture
Collection, Wuhan, China), were cultured in Dulbecco's modified
Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 100
U/ml penicillin, and streptomycin (Invitrogen; Thermo Fisher
Scientific, Inc.), and maintained at 37°C with 5% CO2 in
a humidified atmosphere. Twenty normal (age range from 25 to 67)
and 20 CSCC (age range from 29 to 65) samples were obtained from
patients diagnosed with CSCC from January 2009 to August 2011 at
the departments of Dermatology, Pathology and Oncology at Nanfang
Hospital and Zhujiang Hospital, affiliated to Southern Medical
University, and The Third Affiliated Hospital to Sun Yat-sen
University.
UV irradiation
HaCaT cells were irradiated when grown to ~80–90%
confluence. Prior to UVB exposure (30 mJ/cm2), cells
were washed twice with phosphate-buffered saline (PBS) and covered
with a thin layer of PBS. In parallel, non-irradiated cells were
treated similarly and maintained in the dark in an incubator. The
UVB light source was produced by Shanghai Gucun Electro-Optical
Instrument Factory. The dose rate was assessed by Shanghai
Institute of Measurement and Testing Technology. The wavelength
peak of the UVB lamp used was 305 nm, and the power density was
16.5 µW/cm2.
Drug treatment
Gefitinib (cat. no. S1025) was purchased from
Selleck Chemicals. Chloroquine (cat. no. A506569) was obtained from
Sangon Biotech Co., Ltd. Various concentrations (0.1, 1, 10, 50 and
100 µM) of gefitinib, alone or combined with chloroquine, were
added to the cells at the indicated time-points at 37°C with 5%
CO2 in a humidified incubator. Then, cellular proteins
were collected for further analysis. 3-Methyladenine (3-MA), an
autophagy (PI3K) inhibitor, was used at 5 mM to evaluate the
accumulation of LC3-II induced by gefitinib.
Immunoblotting and
immunohistochemistry (IHC) assays
Total cell extracts were prepared and assayed by
western blotting as previously described (12), with minor modifications. Briefly,
total cellular proteins (20 µg) were electrophoresed through a 10%
denaturing polyacrylamide gel and transferred to a nitrocellulose
membrane (Schleicher & Schuell BioScience, Inc.). The blots
were probed with the following primary antibodies and dilutions:
LC3-II (Cell Signaling Technology, Inc., 3868, 1:2,000), and p62
(Santa Cruz Biotechnology, Inc., sc-25575, 1:5,000), GAPDH (Santa
Cruz Biotechnology, Inc., sc-32233, 1:5,000), caspase-3 (Santa Cruz
Biotechnology, Inc., sc-7148, 1:5,000), phospho (p)-EGFR (Tyr 1173)
(Santa Cruz Biotechnology, Inc., sc-101668, 1:2,000), and EGFR
(Cell Signaling Technology, Inc., 4267, 1:2,000). Anti-mouse
IgG-horseradish peroxidase (HRP) (Thermo Scientific, Inc., 31430,
1:5,000) and anti-rabbit IgG-HRP (Thermo Scientific, Inc., 31460,
1:5,000) were used as secondary antibodies. Bound antibodies were
visualized with the Luminata Forte Western HRP substrate (EMD
Millipore) according to the manufacturer's protocol. Densimetric
quantification of the western bands was performed using Quantity
One software (Bio-Rad Laboratories, Inc.) and presented as
histograms.
IHC staining on formalin-fixed paraffin-embedded
CSCC sections, were performed using EGFR (1:100) and p-EGFR
(1:100). At least 10 representative images of all IHC stained
sections were captured using an Olympus IX51 microscope (Olympus
Corporation) for statistical analysis.
RNA isolation and the quantitative
real-time polymerase chain reaction
Total RNA from cells was extracted using TRIzol
reagent (Life Technologies; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. cDNAs were prepared
using Moloney murine leukemia virus reverse transcriptase (Life
Technologies; Thermo Fisher Scientific, Inc.) and an oligo(dT)20
primer. mRNA expression analysis was performed using SYBR Green
Master Mix (Life Technologies; Thermo Fisher Scientific, Inc.) on a
LightCycler 96 Detection System (Roche Diagnostics) using GAPDH for
normalization. The cycling parameters were: 95°C for 10 min,
followed by 40 cycles at 95°C for 15 sec and an annealing/extension
step at 60°C for 40 sec. The primer pairs used in this study were:
N-cadherin (F: 5′-ATCCTCCAGAGTTTACTGCCATGA-3′ and R:
5′-TGCAGCAACAGTAAGGACAAACA-3′); vimentin (F:
5′-AAGAGAACTTTGCCGTTGAAGCT-3′ and R: 5′-CCTCAGGTTCAGGGAGGAAAA-3′);
glyceraldehyde 3-phosphatedehydrogenase (GAPDH) (F:
5′-TTGCCATCAATGACCCCTTCA-3′ and R: 5′-CGCCCCACTTGATTTTGGA-3′). The
fold change was calculated by the 2−ΔΔCq method
(13). All experiments were
performed in triplicate.
Cell viability assay
Crystal violet staining was used to measure cell
viability in cultures as previously described (14). In this procedure, the attached
cells are stained with crystal violet that binds to proteins and
DNA. Dead cells lose their adherence and are subsequently washed
away from the population of cells. This reduces the amount of
crystal violet staining in the culture. At least 10 representative
bright field images of randomly selected fields were captured using
an Olympus IX51 microscope for statistical analysis.
Wound healing assay
Cells were seeded into six-well plates and allowed
to form confluent monolayers. Then, the cells were exposed to
various concentrations (0, 50 and 100 µM) of agents. Next, the
monolayers were scratched horizontally using a sterile 200-µl
pipette tip. Cells were washed with PBS and cultured in DMEM.
Progression of migration was observed and images were captured at
0, 3, 6, and 9 h after wounding.
Cell invasion and migration
assays
Cells were plated on the top side of an uncoated or
Matrigel-coated polycarbonate Transwell filter in the upper chamber
and incubated at 37°C for ~12–16 h, followed by the removal of
cells inside the upper chamber with a cotton swab. Migrated and
invaded cells on the lower membrane surface were fixed in 4%
paraformaldehyde for 10 min at room temperature and stained with
0.1% crystal violet for 5 min at room temperature. The number of
migrated or invading cells was counted in five fields under an ×200
magnification.
Colony formation assay
Exponentially growing cells were collected by
trypsinization and plated at a low density (500 cells/plate).
Various concentrations (0, 10 and 20 µM) of gefitinib were then
added in complete DMEM for 6 days. Colonies were fixed in 1%
crystal violet (w/v) in 100% methanol for 20 min and then counted
visually.
Cell Counting Kit (CCK)-8 cell
proliferation assay
Cells were plated in triplicate in 96-well plates at
a density of 8×103 cells/well. After 24 h, the cells
were placed in complete DMEM containing the indicated drug
concentrations, or vehicle control. Then, the plates were incubated
at 37°C. After 12 h, each well was incubated with 100 µl DMEM
containing 10 µl CCK-8 reagent. After 1 h, the absorbance was
measured at 450 nm using a microplate reader (Bio-Rad Laboratories,
Inc.).
Apoptosis analysis
A431 cells were treated with the indicated
concentrations (0.1, 1, 10, 50 and 100 µM) of gefitinib.
Trypsinized cells were collected and washed with cold PBS. The
cells were then collected and resuspended in 50 µl of binding
buffer containing 2.5 µl of Annexin V-fluorescein isothiocyanate
and 2.5 µl of propidium iodide. After incubation for 15 min in the
dark at room temperature, ~200 µl buffer were added to the
solution. Fluorescence was measured on a flow cytometer and
analyzed using ModFit software (version 3.1, Verity Software House,
Inc.).
Morphologic analysis by electron
microscopy
Transmission electron microscopic analysis is
considered the gold standard in cell death research. Cells were
harvested by trypsinization, washed with PBS, then fixed with a
solution containing 2.5% glutaraldehyde in PBS for at least 1 h at
4°C. The samples were rinsed three times for 15 min each in 0.1 M
cacodylate buffer containing 7.5% sucrose, and fixed in 1% osmium
in cacodylate buffer for 1 h. After being washed three times in
0.11 M veronal acetate buffer for 15 min each, the samples were
incubated with 0.5% uranyl acetate in veronal acetate buffer for 1
h at room temperature. Specimens were then dehydrated in an
ascending series of ethanol (35, 70, and 95%, then twice with 100%)
for 10 min each, followed by two changes of propylene oxide for 5
min each. The samples were incubated with a 1:1 mixture of 100%
resin and propylene oxide for 1 h, followed by two changes of 100%
resin, each for 30 min. Finally, the samples were embedded in resin
and polymerized at 60°C overnight. Areas containing cells were
block mounted and cut into 70-nm sections. The sections were
stained with uranyl acetate and lead citrate. Finally, the
ultrathin sections were examined under a transmission electron
microscope.
Autophagy analysis by acridine orange
(AO) staining
AO is a cell-permeable green fluorophore that can be
trapped and fluoresce red in acidic vesicular organelles (AVOs);
i.e., autolysosomes. This feature makes AO staining a quick and
reliable method to evaluate the volume of AVOs, which significantly
increase during the induction of autophagy. AO staining was
performed as previously described (15). Specifically, the cells were stained
with 1 µg/ml AO for 15 min, and then examined under a fluorescence
microscope.
Statistical analysis
Data are presented as the mean ± standard deviation.
Groups were compared by a one-way analysis of variance using the
SNK-q test, or by t-test with a two-tailed P-value. The expression
levels of proteins were analyzed by ANOVA based on the value of
densimetric quantification of the western bands. All statistical
tests were considered to reflect significant differences if
P<0.05.
Results
EGFR is induced by UV irradiation and
is highly expressed in CSCC cells and tumors
UV radiation is one of the major causal stimulators
leading to CSCC (6). To gain
insights into the molecular events within skin cells after exposure
to UV radiation, HaCaT keratinocytes were treated with 30
mJ/cm2 UVB. Western blotting revealed that the
expression of EGFR was induced by UVB irradiation (Fig. 1A).
The A431 cell line features an unusual EGFR gene
amplification (8) that contributes
to the markedly higher expression of EGFR in these cells than HaCaT
keratinocytes (Fig. 1B). To
determine whether EGFR expression was also dysregulated in CSCC
tumors, EGFR was detected in 20 normal and 20 CSCC tumor sections
by IHC. The results confirmed the presence of high levels of EGFR
protein in CSCC tumors (Fig. 1C).
Collectively, these results indicated that the induction of EGFR by
UV exposure may be involved in causing CSCC.
Gefitinib inhibits the activation of
EGFR
Since EGFR is highly upregulated in CSCC cells,
treatments designed to inhibit its activity are considered to be
useful. Gefitinib inhibits the EGFR tyrosine kinase domain by
binding to the ATP binding site of the enzyme thereby inhibiting
the anti-apoptotic signal transduction cascade (16). To assess whether gefitinib
functioned in CSCC cells, A431 cells were treated with 100 µM
gefitinib and the level of p-EGFR was probed by western blotting.
By analyzing the ratio of p-EGFR/total EGFR, the activation
(phosphorylation) of EGFR was significantly suppressed by gefitinib
treatment (Fig. 1D).
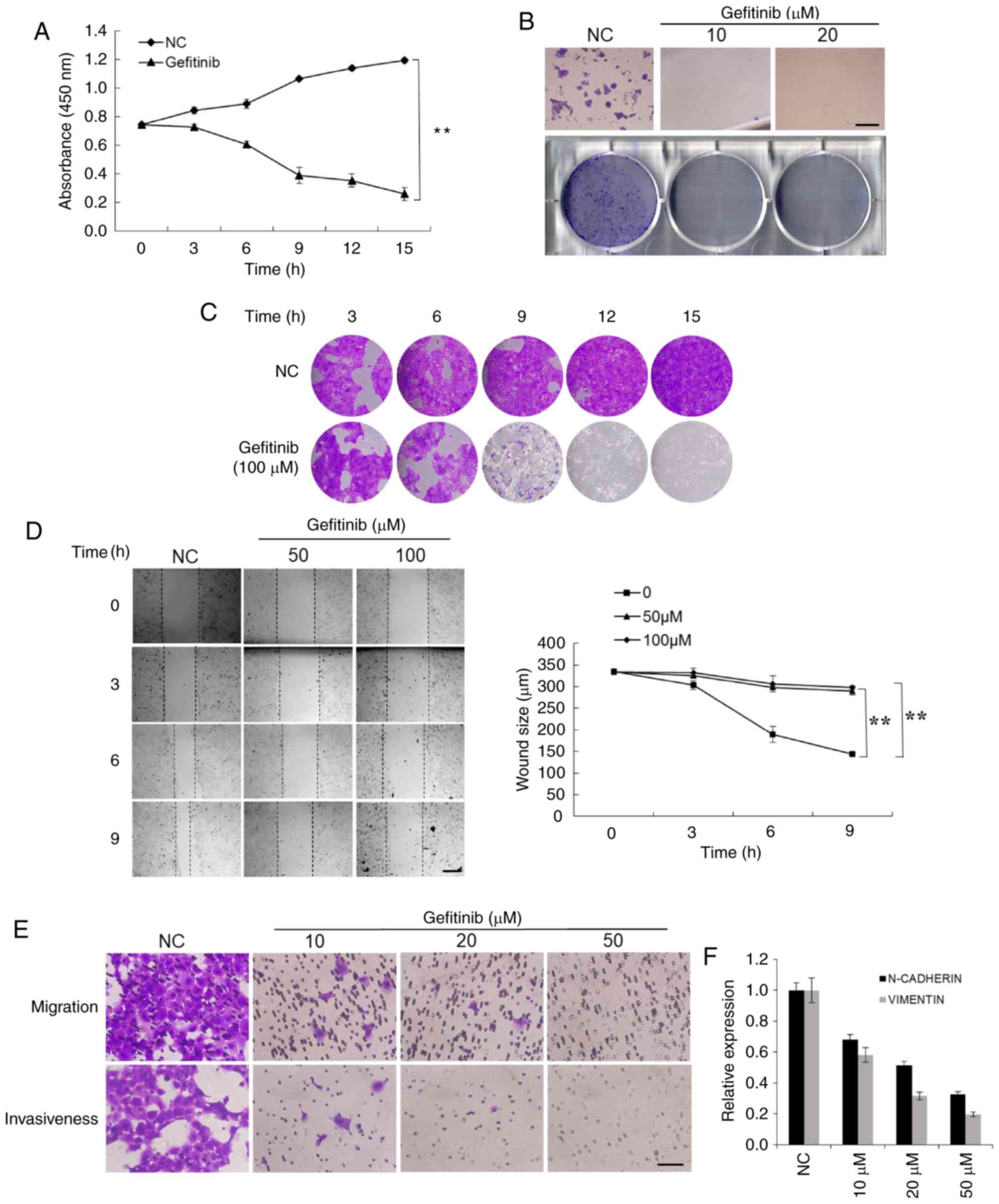
Gefitinib treatment strongly inhibits
the proliferation, mobility, and invasiveness of CSCC cells
To investigate the cellular effects of gefitinib,
the capacities of proliferation, mobility, and invasiveness of CSCC
cells were explored. Cell proliferation was assessed by both the
CCK-8 assay and crystal violet staining. The results revealed that
gefitinib significantly suppressed the proliferation of A431 cells
(Fig. 2A and B). The colony
formation assay revealed that there were significantly fewer
colonies formed in the gefitinib-treated group compared to the
control cells that formed relatively large and numerous colonies
(Fig. 2C).
The mobility of CSCC cells was evaluated by wound
healing and Transwell migration assays. The mobility of A431 cells
was significantly inhibited by gefitinib treatment (Fig. 2D). The Transwell assay revealed
that the invasiveness of CSCC cells was also significantly
suppressed by gefitinib (Fig. 2E).
To further address the effects of gefitinib on cellular migration
and invasiveness, the expression of the cellular adhesion and
migration markers N-cadherin and vimentin, were assessed,. Their
expression was decreased with increasing concentrations of
gefitinib (Fig. 2F). These
findings demonstrated that gefitinib inhibits various malignant
capacities of CSCC cells.
Gefitinib treatment induces apoptosis
as well as autophagy in CSCC cells
To explore the mechanism underlying the inhibition
of malignant features in CSCC cells treated with gefitinib, its
apoptotic effects were examined. Annexin/propidium iodide double
staining followed by flow cytometry revealed enhanced apoptosis
induced by gefitinib treatment (Fig.
3A and B). To confirm the apoptosis-inducing role of gefitinib,
the apoptotic phenotype wασ observed directly by transmission
electron microscopy. Clear condensation and apoptotic body
formation was observed in >50% of A431 cells treated with
gefitinib (Fig. 3C). Notably,
autophagosomes, direct evidence of ongoing autophagy, were widely
distributed in the cells (Fig.
3D). The time-dependent enhancement of autophagy by gefitinib
in A431 cells was supported by AO staining (Fig. 3E). Western blot results revealed
not only time-dependent enhanced cleavage of caspase-3 and
caspase-9 (which marked the development of apoptosis upon gefitinib
treatment), but also gradually increasing autophagy indicated by an
increased level of LC3-II (Fig.
3F). Furthermore, treatment of these cells with 3-MA, an
autophagy (PI3K) inhibitor, suppressed the accumulation of LC3-II
induced by gefitinib (Fig. 3G).
These results indicate both apoptosis and autophagy are induced by
gefitinib treatment.
Inhibition of the autophagy by
chloroquine promotes gefitinib-induced apoptosis
Autophagy is recognized as a survival mechanism in
cancer cells (9,12). In contrast, autophagy is also
considered to be type II programmed cell death (11). To assess the precise role of
gefitinib-induced autophagy (i.e., survival or death), chloroquine,
which can block the fusion of autophagosomes with lysosomes and
thus disrupt autophagic flux, was used to selectively inhibit
autophagy induced by gefitinib. Treatment of A431 cells with
chloroquine alone suppressed their proliferation, migration, and
invasiveness to some extent (Fig. 4A
and B) while promoting apoptosis (Fig. 4C). Notably, co-treatment of A431
cells with both gefitinib and chloroquine suppressed their
proliferation, mobility, and invasiveness to a greater extent than
either agent alone, and in a concentration-dependent manner
(Fig. 4A and B). Chloroquine also
concentration-dependently promoted gefitinib-induced apoptosis
(Fig. 4D). The
concentration-dependent enhancement of apoptosis and inhibition of
autophagic flux by chloroquine was also supported by the enhanced
cleavage of caspase-3 and the accumulation of LC3-II and SQSTM1
(p62) due to the blockade of autophagic flux (Fig. 4E).
Discussion
In the present study, the pro-apoptotic role of
gefitinib in CSCC cells was validated and autophagy as a survival
mechanism induced by gefitinib treatment was identified. The
pro-survival autophagic flux can be blocked by using chloroquine to
interfere with the fusion of autophagosomes with lysosomes. Based
on our results, the combined usage of gefitinib with chloroquine
was proposed to establish an efficient therapy for CSCC.
EGFR, also known as human epidermal receptor 1 or
ERBB1, is a transmembrane tyrosine kinase receptor that belongs to
the human epidermal receptor family (17). Binding with its ligands, EGF or
transforming growth factor-α, can lead to conformational changes
and homo- or hetero-dimerization. Subsequent autophosphorylation in
the cytoplasmic tyrosine kinase domain triggers downstream
signaling events. Due to its important roles in cell proliferation,
differentiation, and inflammation, EGFR is regarded as a crucial
focus of cell signaling pathways (18). In cancer, EGFR serves as a stimulus
supporting cancer proliferation and tumor growth. Overexpression of
the EGFR protein or mutations within its gene can activate
downstream pro-carcinogenic pathways, especially in lung cancer
(8).
Tyrosine kinase inhibitors (TKIs), including
gefitinib, can inhibit the tyrosine kinase domain of EGFR
reversibly by competitive binding to ATP, and are considered to
cause tumor cell death through BIM-mediated apoptosis (19). Thus, TKIs have been widely used to
treat cancers harboring aberrant expression of EGFR or EGFR
mutations.
In a prospective phase II clinical trial, gefitinib
was used as a neoadjuvant administered prior to standard treatment
of surgery and/or radiotherapy to treat aggressive CSCC (5). Gefitinib was active and
well-tolerated in these patients, and did not interfere with
definitive treatments. An 18% complete response rate was observed.
Despite the success reported in that study, we continue to wait for
new therapeutic advances using gefitinib as a neoadjuvant.
Non-small cell lung cancer (NSCLC) is closely
associated with activating mutations of EGFR that activate
pro-survival and anti-apoptotic signaling pathways, including
Ras/Raf/MEK/MAPK, PI3K/AKT/mammalian target of rapamycin, and
JAK/STAT (20). To induce the
intrinsic mitochondrial apoptotic pathway, the specific EGFR TKI,
gefitinib, has been approved by the United States Food and Drug
Administration to treat advanced NSCLC with classical EGFR
mutations. Nevertheless, most cases of NSCLC ultimately relapse
within one year due to acquired resistance to EGFR TKIs (21). To enhance the pro-apoptotic effects
of TKIs, methods of blocking the pro-survival pathway are under
intensive study.
As a catabolic process, autophagy assures that
cytosolic materials, including organelles, are sequestered into
double-membrane autophagosomes and fuse with lysosomes for bulk
degradation. The role of autophagy in the survival of cancer cells
is unclear. As type II programmed cell death, autophagy may act as
an alternative tumor-suppressing mechanism through the degradation
of vital components, limiting cell growth and genomic instability
(7). However, the recycling of
proteins, organelles, and energy contributes to the maintenance of
cellular homeostasis and can promote the survival of cells
(22). Thus, the role of autophagy
in regulating cancer cell death or survival under different
circumstances has not been fully explored (23).
In the present study, gefitinib was an effective TKI
to induce apoptosis in CSCC cells. However, the accompanying
induction of autophagy complicates the application of gefitinib.
Currently, the activation of pro-survival autophagy as an early
response to gefitinib treatment has been well-documented and
functions as a protective mechanism to mediate resistance during
anticancer therapy. This suggests using autophagy as a potential
therapeutic target in cancer treatment (23,24).
To test this possibility, chloroquine was used to inhibit
protective autophagy. This agent strongly suppressed autophagic
flux and enhanced gefitinib-induced apoptosis in CSCC cells.
The anti-proliferation function of a specific drug
for chemotherapy is determined by the combinatory effects of
pathophysiological cellular processes promoted or suppressed by
that drug, such as pro-apoptosis, anti-glycolysis or
anti-autophagy. Since induction of apoptosis is a natural effect of
gefinitib, gefinitib treatment must decrease the number of cells
and influence the proliferation, migration, invasion, wound healing
of tumor cells which is revealed by the decrease of the
proliferation rate, especially in the case of the combined
application of chloroquine to suppress pro-survival autophagy.
Another question is whether this combinatorial treatment of drugs
is harmful to normal cells? Owing to the natural effect of
gefinitib designed for anti-EGFR treatment, the lower expression of
EGFR in normal cells should markedly decrease the harmful effects
induced by gefinitib.
There is a continuing need to focus on the clinical
application of the concept of autophagy inhibition due to the
ongoing question of whether inhibition or induction of autophagy
improves the efficacy of anticancer therapy. One group revealed
that chloroquine treatment induced apoptotic cell death in
vitro and in vivo in nude mice. Conversely, chloroquine
treatment did not elicit tumor suppression or prolong survival in
immune-competent mice. This suggests that chloroquine-enhanced cell
death in immune cells may compromise anticancer immune responses
(25). Thus, in clinical
applications of this type of therapy, it is important to consider
the complex in vivo microenvironment and the impact on
immune responses to avoid negative influences.
In summary, the present findings demonstrated that
autophagy inhibition is an effective strategy for enhancing the
sensitivity of tumor cells to anticancer treatment. Manipulating
pro-survival autophagy by the combined application of chloroquine
promotes gefitinib-induced apoptosis. These results support the
combined use of EGFR and autophagy inhibitors for the treatment of
UV-induced CSCC. This is a promising approach for improving the
efficacy of EGFR inhibitors in cancer treatment. The results of the
present study may have practical implications for EGFR-targeted
therapeutic strategies in the near future.
Acknowledgements
Not applicable.
Funding
The present study was substantially supported by
grants from the National Natural Science Foundation of China (grant
nos. 81573076, 81172634 and 81772914; http://www.nsfc.gov.cn/), a grant from the Guangdong
Provincial Department of Science and Technology, China (grant no.
2016A030313738; http://www.gdstc.gov.cn/), a grant from the Science
and Technology Program of Guangzhou, China (grant no. 201904010063;
http://sop.gzsi.gov.cn/) and a grant from the
School of Public Health of Southern Medical University, China
(grant no. GW201612; http://web2.fimmu.com/phatm/).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LZ and ZD conceived and designed the experiments.
LZ, CO and HL performed the experiments. LZ, CO and HL analyzed the
data. LZ and ZD wrote the manuscript. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of Nanfang Hospital, affiliated to Southern Medical
University. All patients provided written informed consent for the
use of surgical samples.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lomas A, Leonardi-Bee J and Bath-Hextall
F: A systematic review of worldwide incidence of nonmelanoma skin
cancer. Br J Dermatol. 166:1069–1080. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Abaseri TB, Fuhrman J, Trempus C,
Shendrik I, Tennant RW and Hansen LA: Chemoprevention of UV
light-induced skin tumorigenesis by inhibition of the epidermal
growth factor receptor. Cancer Res. 65:3958–3965. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kyriakis JM and Avruch J: Protein kinase
cascades activated by stress and inflammatory cytokines. Bioessays.
18:567–577. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rosette C and Karin M: Ultraviolet light
and osmotic stress: Activation of the JNK cascade through multiple
growth factor and cytokine receptors. Science. 274:1194–1197. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lewis CM, Glisson BS, Feng L, Wan F, Tang
X, Wistuba II, El-Naggar AK, Rosenthal DI, Chambers MS, Lustig RA
and Weber RS: A phase II study of gefitinib for aggressive
cutaneous squamous cell carcinoma of the head and neck. Clin Cancer
Res. 18:1435–1446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alam M and Ratner D: Cutaneous
squamous-cell carcinoma. N Engl J Med. 344:975–983. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang CY, Kuan YH, Ou YC, Li JR, Wu CC,
Pan PH, Chen WY, Huang HY and Chen CJ: Autophagy contributes to
gefitinib-induced glioma cell growth inhibition. Exp Cell Res.
327:102–112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Merlino GT, Xu YH, Ishii S, Clark AJ,
Semba K, Toyoshima K, Yamamoto T and Pastan I: Amplification and
enhanced expression of the epidermal growth factor receptor gene in
A431 human carcinoma cells. Science. 224:417–419. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: therapeutic implications. Mol
Cancer Ther. 10:1533–1541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou L, Wang Y, Zhou M, Zhang Y, Wang P,
Li X, Yang J, Wang H and Ding Z: HOXA9 inhibits HIF-1α-mediated
glycolysis through interacting with CRIP2 to repress cutaneous
squamous cell carcinoma development. Nat Commun. 9:14802018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Feoktistova M, Geserick P and Leverkus M:
Crystal violet assay for determining viability of cultured cells.
Cold Spring Harbor Protocols. 2016:pdb.prot087379. 2016. View Article : Google Scholar
|
|
15
|
Thomé MP, Filippi-Chiela EC, Villodre ES,
Migliavaca CB, Onzi GR, Felipe KB and Lenz G: Ratiometric analysis
of acridine orange staining in the study of acidic organelles and
autophagy. J Cell Sci. 129:4622–4632. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ciardiello F and Tortora G: EGFR
antagonists in cancer treatment. N Engl J Med. 358:1160–1174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Johnston JB, Navaratnam S, Pitz MW,
Maniate JM, Wiechec E, Baust H, Gingerich J, Skliris GP, Murphy LC
and Los M: Targeting the EGFR pathway for cancer therapy. Curr Med
Chem. 13:3483–3492. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bethune G, Bethune D, Ridgway N and Xu Z:
Epidermal growth factor receptor (EGFR) in lung cancer: An overview
and update. J Thorac Dis. 2:48–51. 2010.PubMed/NCBI
|
|
20
|
Henson ES and Gibson SB: Surviving cell
death through epidermal growth factor (EGF) signal transduction
pathways: Implications for cancer therapy. Cell Signal.
18:2089–2097. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Galluzzi L, Aaronson SA, Abrams J, Alnemri
ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren
K, Borner C, et al: Guidelines for the use and interpretation of
assays for monitoring cell death in higher eukaryotes. Cell Death
Differ. 16:1093–1107. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Galluzzi L, Pietrocola F, Amaravadi RK,
Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno
P, Debnath J, Gewirtz DA, et al: Autophagy in malignant
transformation and cancer progression. EMBO J. 34:856–880. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dragowska WH, Weppler SA, Wang JC, Wong
LY, Kapanen AI, Rawji JS, Warburton C, Qadir MA, Donohue E, Roberge
M, et al: Induction of autophagy is an early response to gefitinib
and a potential therapeutic target in breast cancer. PLoS One.
8:e765032013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Masuelli L, Granato M, Benvenuto M,
Mattera R, Bernardini R, Mattei M, d'Amati G, D'Orazi G, Faggioni
A, Bei R and Cirone M: Chloroquine supplementation increases the
cytotoxic effect of curcumin against Her2/neu overexpressing breast
cancer cells in vitro and in vivo in nude mice while counteracts it
in immune competent mice. Oncoimmunology. 6:e13561512017.
View Article : Google Scholar : PubMed/NCBI
|