Introduction
Systemic lupus erythematosus (SLE) is a chronic
autoimmune disease characterized by a mild rash, joint pain and
multi-organ damage. The global SLE incidence and prevalence are
estimated to be 5.14 (1.4 to 15.13) per 100,000 person-years and
43.7 (15.87 to 108.92) per 100,000 individuals (1). The hallmark of SLE is the defective
clearance of dead cells by the immune system and the loss of
tolerance to endogenous antigens such as double-stranded (ds)DNA,
resulting in the abnormal activation of the immune system. This
activation leads to immune-mediated attacks on organs and tissues,
including lung, gut and kidney involvement (2). In particular, the kidneys are
affected, leading to the development of lupus nephritis (LN) as the
most severe manifestation of SLE and the common cause of morbidity
and mortality in patients with SLE. In patients with SLE, >60%
suffer from LN, and 40% of them progress to end-stage renal disease
(ESRD) (3,4), which endangers the lives and health
of the individuals, with the exact incidence contingent upon
ethnicity and sex (5). The immune
system contributes to the development of SLE. The two main cell
types in the adaptive immune system, B cells and T cells, are both
indispensable for the development of LN. B cells are pathogenic in
SLE through the autoantibodies (such as anti-dsDNA antibodies and
anti-nucleosome antibodies) and cytokines they produce. T cells
drive both the systemic and intra-renal activation of B cells
(6). In addition, genes for SLE
also cause LN. Renal aggression during SLE is triggered by genes
that undermine immune tolerance and stimulate autoantibody
production. These genes may collaborate with other genetic factors
to amplify innate immune signaling and type α interferon (IFN-α)
production, consequently leading to the recruitment of leucocytes,
inflammatory mediators and autoantibodies into end organs, notably
the kidneys (7). Currently,
high-doses of hormones (glucocorticosteroid) combined with
immunosuppressants (methotrexate) are first line therapeutics for
LN treatment. Short-term complete renal response rates are only
10–40% at 12 months, long-term outcomes have not improved further,
and as many as 30% of LN patients will still progress to ESRD
(8,9).
Podocyte injury is involved in the occurrence and
development of LN, a disease that is associated with the
progressive loss of renal function and various renal pathological
features, including mesangial cell proliferation and subendothelial
immune complexes. Podocytes are terminal epithelial cells that
protrude into the glomerular urinary space and constitute an
important component of the glomerular filtration barrier. Immune
complex deposition in the renal tissues activates the complement
system and thereby contributes to the failure in the efficient
clearance of cellular fragments, triggering both the innate and
adaptive immune systems. The production of cytokines such as IFN-α
stimulates the antigen presentation and overactivation of T and B
cells (7). Ultimately, additional
inflammatory cytokines and chemokines promote the recruitment of
inflammatory cells to the kidney and cause podocyte injury in the
glomerulus (10). Numerous studies
have reported the involvement of podocyte injury in LN (11–13)
and have suggested that podocytes can serve as biomarkers of LN
activity (14). A new concept
called ‘lupus podocytopathy’ has been proposed (15), which refers to patients with LN
with normal glomeruli but a diffuse disappearance of foot processes
and podocyte damage. However, podocyte injury in patients with LN
with normal glomeruli is often overlooked in clinical practice,
which delays timely treatment and accelerates the deterioration of
renal function. Therefore, investigating the role of podocyte
injury in LN is warranted.
The immune microenvironment is considered as the
environment of the local immune response. It is composed of diverse
populations including infiltrating immune cells, immune molecules
and humoral components. Immune complex deposition, local complement
activation, along with immune cell recruitment and local intrarenal
cytokine signaling account for glomerular injury in the LN immune
microenvironment (10). Renal
immune cells accumulate in the kidneys of the patients with LN,
which involves the formation of tertiary lymphoid structures (TLS)
(16). The TLS consists of immune
cells, cytokines and resident renal cells in LN, and the activation
of immune cells triggers a transient aggravation of resident cell
injury and even the production of autoantibodies within the
kidneys, exacerbating disease progression (17).
LN develops from a loss of immune balance to
ubiquitous autoantigens, which is a result of inflammation and
immune responses. Although the immunomolecules and immune cells in
the renal immune microenvironment have important roles, the
underlying mechanisms of the immune responses in the podocyte
injury of LN remain unclear. The present review aimed to summarize
the current understanding of the immune microenvironment of
podocytes in LN, provide an update on their interaction mechanisms
and offer the rationale for the podocytes as novel therapeutic
targets in the treatment of LN.
Pathogenesis of LN
LN is the most common complication of SLE, and
proteinuria and hematuria are the primary clinical manifestations.
The salient features of LN are associated with the deposition of
immune complexes, which cause an inflammatory and immune response
in the kidney (18). In
vivo apoptosis or secondary necrosis (19) is responsible for the chromatin
fragment exposure. The exposed DNA or nucleosomes bind with
corresponding autoantibodies to form immune complexes and deposit
onto the glomerular basement membrane. Concurrently, after
associating with immune complexes in the glomeruli, nucleosomes
within necrotic intrinsic glomeruli cells form in situ
immune complexes containing both DNA and nucleosomes (20).
The nucleic acid components of the immune complexes
collectively stimulate intrarenal inflammation by binding to
toll-like receptors (TLRs) and Fc receptors (FcRs) or by activating
immune responses through the complement cascade. Additionally, the
ligation of TLRs induces the maturation of plasmacytoid dendritic
cells (pDCs) and facilitates the secretion of proinflammatory
cytokines and chemokines including interferon (IFN)-α (21,22).
Secreted IFN-α promotes the activation of antigen-presenting
dendritic cells (DCs), thereby promoting the differentiation of
self-reactive B cells to plasma cells and enhancing the production
of memory T cells, which form germinal center structures (23).
B-cell activating factor (BAFF) emerges as an
inducer of B cell proliferation, differentiation and maturation
through the CD40 ligand (CD40L) and CD28, on the surface of T cells
migrating into the glomeruli, binding with CD40 and B7 on B cells,
respectively (17). Subsequently,
the autoantibodies produced bind to autoantigens and deposit in
situ in the kidney. In addition to secreting antibodies,
activated B cells, as a type of antigen-presenting cell (24), promotes the activation of
pathogenic T cells to secrete proinflammatory cytokines such as
IL-6 and TNF-α, and facilitate the recruitment of macrophages and
DCs into the glomerulus and the tubulointerstitium (25). Immune cells also undergo intrarenal
proliferation and activation by binding to the FcRs of immune
complexes. Activated neutrophils and macrophages secrete reactive
oxygen species and proteases, which directly damage the kidneys
(26). In addition, immune
complexes also activate the complement pathway to form membrane
attack complexes, releasing anaphylatoxins to promote inflammatory
reactions and accelerate the progression of LN (27) (Fig.
1).
 | Figure 1.Pathogenesis of LN. Immune complexes,
formed by the combination of autoantigens and autoantibodies, are
deposited into the kidney. These immune complexes through FcR and
TLR stimulate pDCs to secrete inflammatory cytokines (such as
IFN-α). Inflammatory cytokines stimulate DC activation, inducing
DCs to present antigens to T cells, thereby activating T cells and
subsequently triggering B cell activation through CD40L and CD28
binding with CD40 and B7 respectively. B cells infiltrate the
kidney in LN to secrete autoantibodies that attack and damage the
renal intrinsic cells such as podocytes. FcR, Fc receptor; IFN,
interferon; TLR, toll-like receptors; DC, dendritic cell; pDC,
plasmacytoid DC; MHC, major histocompatibility complexes; TCR, T
cell receptor; GBM, glomerular basement membrane; CD40L, CD40
ligand; LN, lupus nephritis. |
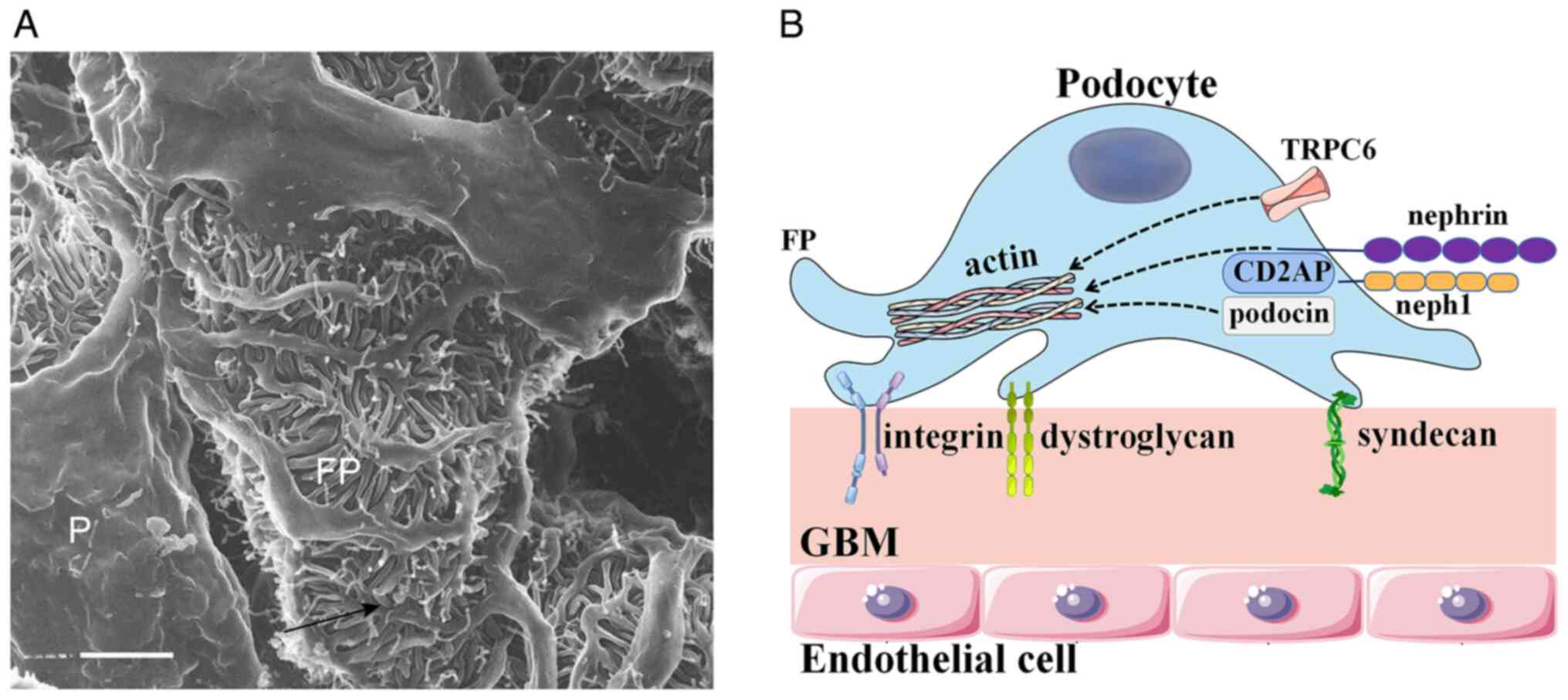
Basic structure and function of
podocytes
The podocyte, a terminally and highly differentiated
epithelial cell located in the urinary space, consists of a foot
process and an apical surface domain. Given that podocytes act as
essential components of the glomerular filtration barrier, the
apical surface domain carries a negative charge and restricts the
passage of negatively charged proteins (28). Foot processes attach to the
glomerular basement membrane through integrins, syndecans,
dystroglycan and other adhesion molecules (28). During podocyte development, a
number of large extensions are formed by the epithelial cells,
therefore, primary foot processes split into secondary and tertiary
processes, which are abundant in microtubule structures (29). The foot processes are primarily
composed of actin, which constitutes the cellular cytoskeleton of
the podocyte. The function of actin is to connect the apical and
basal membrane domains, as well as the slit diaphragm. The parallel
contractile actin bundles are controlled to regulate the
permeability of the filtration barrier (30). Destruction of podocyte actin leads
to podocyte injury, disappearance of foot process fusion, damage to
the filtration barrier and to proteinuria. Adjacent foot processes
interdigitate with each other, forming slit diaphragms that anchor
to the glomerular basement membrane. Nephrin, podocin,
CD2-associated protein (CD2AP) and other molecules compose the slit
diaphragm, participating in intracellular signaling and the
formation and maintenance of the filtration barrier (Fig. 2).
Nephrin, a member of the immunoglobulin superfamily,
interacts with the actin cytoskeleton through Nck adapter proteins
and maintains the integrity of the podocyte structure and the
filtration barrier (29). Nephrin
affects the assembly of actin polymers in cell membranes. Decreased
levels of nephrin leads to abnormal tertiary podocytes, loss of
normal polarity and abnormal intercellular junctions, as a result
of proteinuria. Neph1 is another transmembrane protein located near
to nephrin in the cell membrane (31). Neph1, a major regulator of actin
dynamics, is indispensable in maintaining normal slit diaphragm
function. The phosphorylated nephrin-Neph1 complexes can lead to
the reassembly of actin complexes, exerting irreversible effects on
podocyte filtration function (32). Podocin is a membrane-associated
protein that is crucial for signal transduction of the
nephrin-Neph1 complex. The reduction in nephrin level caused by
podocin mutations will alter the signaling of the nephrin-Nephl
complex and result in an impaired podocyte slit diaphragm (33). Podocin also regulates cellular
cytoskeletal dynamics through the activity of transient receptor
potential cation channel subfamily C member 6 (TRPC6). TRPC6 is a
non-selective, calcium-permeable cation channel in the plasma
membrane of podocytes, which stabilizes the actin cytoskeleton of
podocytes to sense changes in pressure, fluid flow or filtration
rate (34). CD2AP is a cytoplasmic
protein that interacts with nephrin and podocin and also takes part
in the actin filament assembly in the slit diaphragm of podocytes
through cellular signal transduction (35). The downregulation of these slit
diaphragm proteins can lead to the structural and functional damage
of podocytes and the production of proteinuria.
Podocytes in the immune system
The occurrence and progression of LN involves
multiple pathways, including abnormal cell death, autoantibody
production, immune complex deposition, complement activation and
the increased activation of immune cells (for example, T and B
cells). Immune complex deposition predominates in the development
of LN, and the majority of LN classifications by the International
Society of Nephrology and the Renal Pathology Society involve
immune complex deposition (Table
I). Additionally, foot process fusion and podocyte injury have
been observed in different classifications of LN (36). The deposition of immune complexes
in the kidneys takes place by various mechanisms and activates
complement components, inducing damage to podocytes through both
the innate and adaptive immune responses. Furthermore, podocytes
express immunomolecules and participate in immune responses
(37) (Fig. 3).
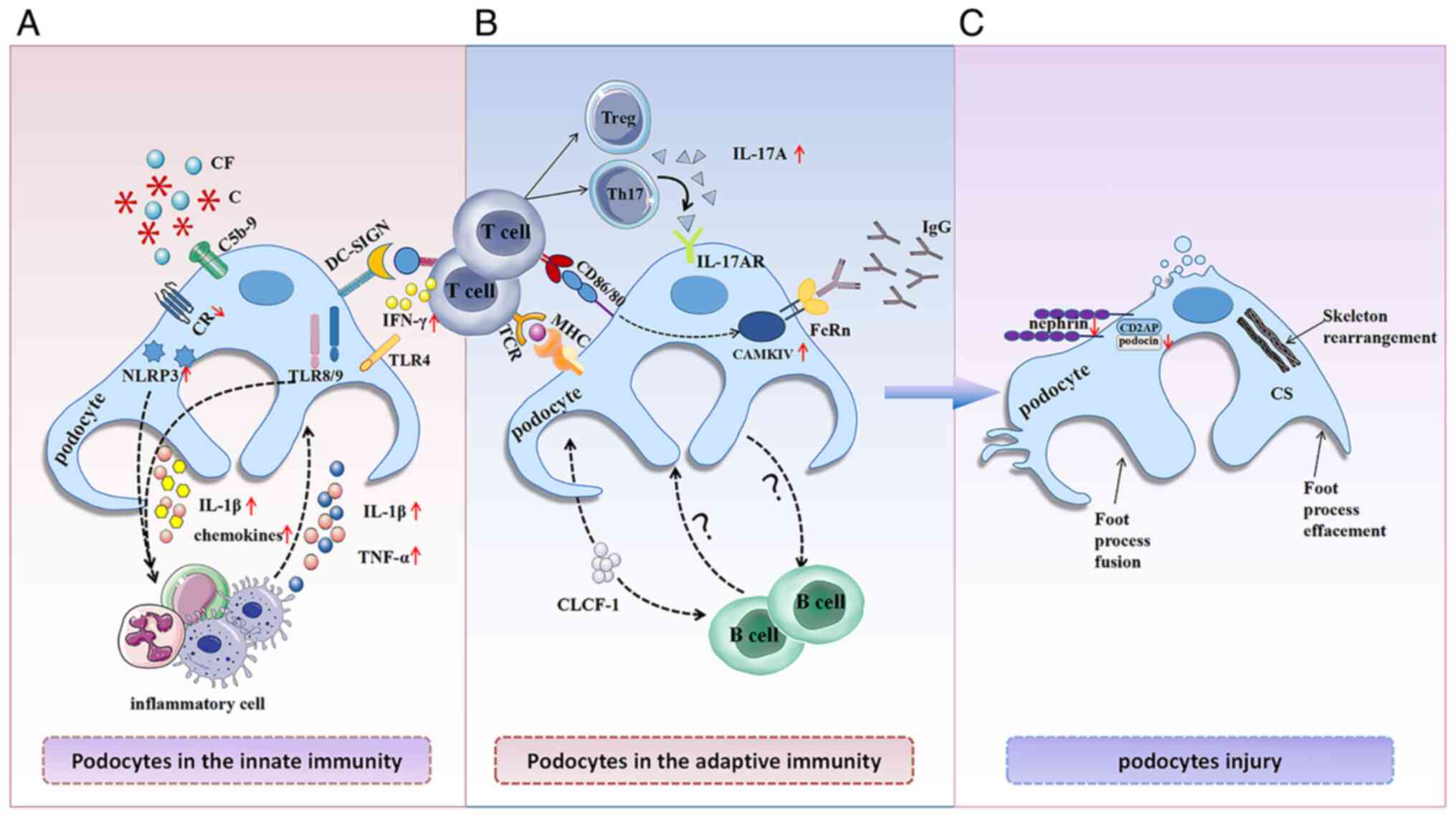 | Figure 3.Immune microenvironment of lupus
nephritis podocyte injury. In the immunity microenvironment, (A)
innate immune components and (B) adaptive immune components
stimulate (C) podocyte injury, and the damaged podocytes can
interact as immune cells with inflammatory cells (such as
macrophages, neutrophils and monocytes) and T cells; the specific
role of podocytes with B cells is unclear. C, complement; CF,
complement factor; CR, complement receptor; TLR, toll-like
receptor; DC-SIGN, dendritic cell-specific intercellular adhesion
molecule-3-grabbing non-integrin; NLRP3, NOD-like receptor thermal
protein domain associated protein 3; TNF-α, tumor necrosis
factor-α; Treg, regulatory T cell; Th17, CD4+ T
helper-17 cell; IFN, interferon; IL-17AR, IL-17A receptor; MHC,
major histocompatibility complexes; TCR, T cell receptor; FcRn,
neonatal Fc receptor; CAMKIV, Ca2+/calmodulin-dependent
protein kinase IV; CLCF-1, cardiotrophin-like cytokine factor 1;
CS, cytoskeletal; CD2AP, CD2-associated protein. |
 | Table I.Classification of lupus nephritis and
the condition of the podocytes after damage. |
Table I.
Classification of lupus nephritis and
the condition of the podocytes after damage.
| Type | Disease name | Pathological
manifestations | Podocyte condition
after damage |
|---|
| Class I | Minimal mesangial
lupus nephritis | Mesangial immune
complexes | Unknown |
| Class II | Mesangial
proliferative lupus nephritis | Mesangial immune
complexes and a small number of subepithelial or subendothelial
complexes | Extensive podocyte
foot process effacement |
| Class III | Focal lupus
nephritis | Subendothelial
immune complexes | Extensive podocyte
foot process effacement |
| Class IV | Diffuse lupus
nephritis | Subendothelial
immune complexes | Podocyte foot
process effacement |
| Class V | Membranous lupus
nephritis | Subepithelial
immune complexes | Marked
disappearance of podocytes |
| Class VI | Advanced sclerotic
lupus nephritis | Glomerulosclerosis
in ≥90% of the glomeruli | Disappearance of
podocytes |
Podocytes in the innate immunity
Podocytes interact with the complement
system
Multiple immune pathways engage in the pathogenesis
of LN. The complement system serves a positive role in maintaining
tolerance against LN for the efficient clearance of cellular
fragments. Increasing evidence suggests that complement can mediate
podocyte injury (38–40). In membranous nephropathy,
complement mediates podocyte injury by inducing cell scorch death
through mitochondrial dysfunction (40). When the formation of immune
complexes exceeds clearance pathways, complement components C1q,
C5a and C5b-9 are released and deposited in the kidney. Subiytic
C5b-9 stimulates the podocytes to release cytokines, proteases and
oxidants. It can also induce DNA damage, leading to restricted
podocyte proliferation (41). In
LN, activated intrinsic renal cells can also promote the release of
proinflammatory mediators (IL-1β), and express multiple complement
components.
Complement receptor 1 (CR1), which is exclusively
expressed on podocytes, is reduced in severe glomerular lesions
(42,43). Reduced expression levels of CR1 are
considered to result from decreased synthesis due to podocyte
injury rather than excessive depletion. The decrease in CR1
synthesis increases the sensitivity of podocytes to complement
attack, further exacerbating podocyte injury (44). In Murphy Roths
Large/lymphoproliferative (MRL/lpr) lupus mice, complement factor H
(CFH) deficiency leads to immune complex deposition in the
subendothelial and subepithelial regions of the kidney,
disappearance of podocyte foot processes, and accelerated
progression of LN (45). Podocytes
were revealed as the targets and sources of kidney injury due to
their production of complement components. Therefore, complement
pathway inhibition has been considered as a potential treatment
strategy for LN in clinical trials (46,47).
Podocytes interact with the TLRs
TLRs are expressed in innate immune cells (such as
monocytes, macrophages and dendritic cells), which recognize
pathogen-associated molecular patterns (PAMPs) and can also be
activated by endogenous ligands. Studies have revealed that TLRs
are expressed in mouse glomeruli and perform different
physiological functions (48,49).
TLR4 upregulated in podocytes in mice with membranoproliferative
glomerulonephritis (MPGN) can destroy the kidney by directly
releasing the chemokines, which may promote the recruitment of
inflammatory cells and exacerbate glomerular injury (37).
TLR8 and TLR9 are overexpressed in BXSB/Yaa (a
genetic mutation located on the Y chromosome, namely, Y-linked
autoimmune acceleration) SLE mice models (49). TLR8 is mainly located in podocytes
and its expression level is negatively correlated with nephrin
expression and positively correlated with proteinuria levels in
glomerulonephritis, suggesting that excessive levels of TLR8 are
associated with podocyte injury progression (50). Therefore, it is important to
monitor the changes of the TLR8 mRNA levels in the urine of
patients, which reflects the podocyte injury status. In human LN
pDCs recognize single-stranded RNA, 5′-C-phosphate-G-3′ DNA from
bacteria and viruses as well as altered eukaryotic nucleic acids
via TLR9 (51), which induces the
release of type I IFNs and promotes local and systemic immune
responses via increased expression levels of costimulatory
molecules (52). TLR9 coexists
with injury podocyte proteins, and its expression is associated
with podocyte injury and the development of MPGN. TLR9 is only
expressed in damaged podocytes during active LN while it is not
detected in healthy human kidneys. This expression may be
associated with increased levels of dsDNA antibodies and may be
involved in the process of podocyte injury in LN (53,54).
In summary, TLRs can combine podocytes with the
innate immunity, induce podocyte injury and mediate LN. Future
research could focus on the role of TLRs in LN podocyte injury in
order to inhibit the TLR-induced podocyte injury and prevent the
progression of LN.
Podocytes interact with the innate immune
cells
Macrophages are antigen-presenting cells that have
the capacity to process and present antigens to T cells. Cytokines,
such as TNF-α and IL-1β, produced by activated macrophages can
directly inhibit the expression of the podocyte-specific protein
nephrin, leading to podocyte injury and the induction of
glomerulonephritis and proteinuria (55). The polarization of macrophages from
a proinflammatory phenotype to an anti-inflammatory phenotype can
prevent podocyte injury (55,56).
Sung and Fu (57) revealed that
infiltrating macrophages in the glomerulus can activate T cells and
interact with podocytes through cytokines. Subsequently, the
injured podocytes and mesangial cells produce the inflammatory
cytokines IL-1β and IL-6, leading to an upregulation of adhesion
molecules and chemokines from podocytes and thereby promoting the
recruitment of macrophages to the glomerulus (58). After migrating to the glomerulus,
macrophages are stimulated by immune complexes, complement and
cytokines in the local glomerular environment to produce TNF-α and
induce podocyte injury. This process represents a cascade
amplification reaction that ultimately leads to severe glomerular
damage in LN. Thus, inhibiting the process of macrophage-induced
podocyte injury is necessary to prevent the progression of LN.
Podocytes interact with innate immune
molecules
Nucleotide oligomerization domain (NOD)-like
receptor thermal protein domain associated protein 3 (NLRP3)
inflammasome is a multimeric protein that is associated with the
secretion of inflammatory factors such as IL-1β (59). Previous studies have revealed that
patients and mice with LN demonstrate NLRP3 activation in
podocytes, which induces the secretion of IL-1β and inhibits the
expression of the podocyte-specific protein nephrin, thereby
disrupting the integrity of the podocyte filtration barrier,
leading to proteinuria (60,61).
Thus, inhibiting NLRP3 has been demonstrated to prevent the loss of
foot processes in podocytes and prevent renal tissue damage,
thereby reducing proteinuria (61).
DC-specific intercellular adhesion
molecule-3-grabbing non-integrin (DC-SIGN) is an innate immune
molecule with immune recognition functions that can be expressed on
podocytes in LN. When podocytes are exposed in vitro to
serum from mice with LN, the expression levels of DC-SIGN are
upregulated, which promotes T cell proliferation and increases
secretion of IFN-γ from CD4+ T cells. Therefore, it
seems that DC-SIGN-expressing podocytes may share similar functions
with DCs, stimulating T cell proliferation and exerting
corresponding effects (62). In
summary, the interaction between podocyte injury and the local
innate immune responses in the kidney are both involved in the
progression of LN.
Podocytes in the adaptive immunity
Podocytes interact with
autoantibodies
In LN, IgG-based autoantibodies bind to antigens to
form immune complexes that deposit in the kidneys, subsequently
causing glomerular injury and proteinuria. Simultaneously,
rearrangement of cytoskeletal components in podocytes induced by
IgG reduces the expression levels of podocyte proteins (nephrin and
synaptopodin) with structural podocyte damage in patients with LN.
As the Fc receptor (FcR) is overexpressed in LN podocytes, IgG
bound with FcR is endocytosed by the podocytes (63). Furthermore, endocytosed IgG
upregulates Ca2+/calmodulin-dependent kinase IV in
podocytes, which inhibits the expression of nephrin and results in
podocyte injury. Additionally, the expression levels of CD86 on
podocytes is increased by endocytosed IgG (64,65).
CD86 is one of the co-stimulatory molecules in T cell activation,
implying the potential engagement of podocytes in the initiation of
renal T cell activation.
Bruschi et al (66) reported that patients with LN have
antibodies that directly target podocytes, which are associated
with high proteinuria in these patients. dsDNA antibodies are
present at increased concentrations in renal tissue compared with
that of the systemic circulation. It forms immune complexes and
deposits them in the glomerulus, exhibiting podocyte injury and
local immune reactions. These antibodies can also cross-react with
-actinin-4 on podocytes, causing cytoskeletal rearrangement and
complement activation, which directly leads to podocyte injury
(67,68). Therefore, in addition to immune
complex formation, the direct binding of podocyte-targeting
autoantibodies present in the serum of patients can aggravate
LN-associated proteinuria, leading to podocyte injury.
Overall, podocytes may provide antigenic targets for
autoantibodies. Furthermore, podocyte injury is generated when
autoantibodies form immune complexes that deposit in situ
within the kidney in LN. In future studies, it will be important to
identify other relevant antibodies that directly target intrinsic
renal cells, such as podocytes, mesangial cells and the basement
membrane, to further elucidate new mechanisms of kidney injury in
LN.
Podocytes interact with adaptive
immune cells
In addition to reacting with autoantibodies in the
immune microenvironment, podocytes can also interact with immune
cells. In crescentic glomerulonephritis, rupture of Bowman's
capsule can release CD8+ T cells into the glomerulus,
leading to podocyte injury (69).
Compared with healthy mice and people, the podocytes of lupus mice
and patients with LN have high expression levels of CD80 and CD86,
which may promote T cell expansion and aggregation within the renal
parenchyma. Increases in CD80 levels are positively associated with
the severity of proteinuria (70).
In vitro experiments have revealed that CD80 activation
leads to the reorganization of slit diaphragm proteins, nephrin and
podocin, and the disruption of actin filaments, and also leads to
integrin inactivation to promote podocyte migration and damage
(71,72). CD80 and CD86 expressed in podocytes
are a potentially rich source of biomarkers that may capture
various aspects of the renal injury.
Coers et al (73) and Goldwich et al (74) demonstrated that podocytes can
express major histocompatibility complex (MHC) I and II, as well as
macrophage markers (CD68, F4/80, and CD206) and co-stimulatory
molecules (CD80). In the inflammatory environment, podocytes came
into close contact with glomerular infiltrating T cells (74), which can activate CD4+ T
cells and CD8+ T cells (74). Podocytes can cross-present
endocytosed IgG to local infiltrating T cells via MHC, activating T
cells to induce podocyte injury and apoptosis (75). Therefore, these findings suggest
that podocytes in LN participate in the local immune response,
which is identical to the role of antigen-presenting cells,
contributing to the pathogenesis of LN.
Local infiltration of CD4+ T cells in the
kidney promotes the progression of nephritis (76). Lipopolysaccharide (LPS)-induced
podocytes polarize naive CD4+ T cells into T helper-17
(Th17) and regulatory T (Treg) cells, affecting the Th17/Treg
balance and producing large amounts of proinflammatory cytokines.
Th17 cells can secrete IL-17A, which has the potential to promote
podocyte cytoskeletal rearrangement (77), and is probably the reason for the
positive correlation between IL-17A levels and proteinuria in
patients with LN (78,79). Following the stimulation of IL-17A
secretion by Th17 cells, podocytes can express IL-17A receptors and
produce the NLRP3 inflammasome, resulting in increased secretion of
IL-1β. Additionally, IL-17A can disrupt podocyte morphology and
induce podocyte injury (80).
Preventing CD4+ T cell activation in the renal immune
microenvironment and maintaining Th17/Treg balance may provide a
new potential therapeutic strategy for LN. However, further
research is required for in vivo validation and
investigation of the mechanisms of action (81).
In LN, intrarenal B cells can form germinal
center-like structures and locally produce pathogenic antibodies
(82,83). Kolovou et al (84) described that oligoclonal B cells
are associated with podocyte injury and glomerulosclerosis in LN,
but oligoclonal expansion of B cells failed to be detected in the
peripheral blood of patients with LN. This is possibly due to the
inflammatory stimulus promoting B cell proliferation in the local
renal environment. The interaction between B cells and podocytes in
the immune microenvironment leads to podocyte injury, but the
specific mechanism is unclear and further research is needed to
elucidate the role of intrarenal B cells in podocyte injury.
Cardiotrophin-like cytokine factor 1 (CLCF-1), also known as B
cell-stimulating factor, can regulate B cell differentiation and Ig
class switching when overexpressed (85). Moreover, CLCF-1 is currently
considered as a potential therapeutic target as it can affect
kidney development (86), and
activate the Janus kinase (JAK)/STAT pathway and change podocyte
morphology in focal segmental glomerulosclerosis, leading to renal
dysfunction and proteinuria (87).
Therefore, the progression of LN may be caused by
the interaction between substances in the immune microenvironment
and podocytes and it is necessary to explore this interaction.
Immune microenvironment of podocytes in
animal models
Previously, four lupus mouse models have been
established, including spontaneous lupus mouse models, induced
lupus mouse models, genetically modified lupus mouse models and
humanized lupus mouse models. Currently, the spontaneous lupus
mouse model is commonly used to study the interaction between
podocytes and the immune microenvironment. The spontaneous lupus
mouse models include New Zealand Black (NZB), NZB × New Zealand
White (NZB/W) first filial generation (F1), MRL/lpr and BXSB/Mp
(BXSB/Yaa). These models produce SLE-like autoimmunity, with the
production of autoantibodies including anti-nuclear, anti-dsDNA and
anti-histone. Clinical manifestations of SLE have also been
observed, such as immune complex-mediated glomerulonephritis and
polyclonal hypergammaglobulinemia and foot process effacement
(88) (Table II).
| Table II.Summary of LN mouse models. |
Table II.
Summary of LN mouse models.
| Mouse model | Autoantibodies | Main clinical
features | Age of mice at
mortality | Main research
area | (Refs.) |
|---|
| NZB | Anti-dsDNA,
anti-RBC and anti-gp70 | LN, IC-type GN,
autoimmune hemolytic anemia and hypocomplementemia | 15 and 18 months
because of autoimmune hemolytic anemia | Inflammatory
factors and immune complex deposition | (60,61,102,103) |
| NZB/WF1 | ANA, anti-dsDNA,
anti-Ro, anti-La, anti-Sm, anti-RBC and anti-RNA | LN,
lymphadenopathy, splenomegaly, mild vasculitis, lymphadenopathy and
hemolytic anemia | 10 and 12 months
because of renal failure | Inflammatory
factors, podocyte injury and immune complex deposition | (89) |
| MRL/lpr | ANA, anti-dsDNA,
anti-ssDNA, anti-Sm, anti-Ro, anti-La, rheumatoid factor, anti-RBC
and snRNP | LN,
hypergammaglobulinemia, high titers of autoantibodies, circulating
ICs, lymphadenopathy, polyarthritis, vasculitis and
splenomegaly | 5 and 12 months due
to renal failure | Immune complex
deposition, complement system, podocyte injury and specific
pathogenic mechanisms involved in kidney disease | (45,90,91) |
| BXSB/Yaa | ANA, anti-dsDNA and
anti-RBC | LN, IC-mediated GN,
secondary lymphoid node hyperplasia, hypergammaglobulinemia and
monocytosis | 5-8 months for
BXSB/Yaa male mice and ~15 months for BXSB female mice complex
because of | Expression levels
of the TLR, podocyte injury and immune deposition renal
failure | (46,50,54,93) |
NZB/W F1 mice are a model for studying renal lesions
in SLE. These mice have a susceptibility to autoimmunity and
exhibit podocyte damage, reduced nephrin and podocin expression
levels and proteinuria production. Immune complex deposition and
crescent formation can be observed in the glomeruli, with mild
mesangial hyperplasia in the early stages of murine development and
focal and diffuse proliferative histological forms in the late
stages of murine development, culminating in renal failure
(89).
MRL/lpr mice are a model of spontaneous recessive
mutant lymphocyte proliferation that can present with immune
complex deposition-mediated glomerulonephritis, with a
proliferative LN histological pattern characterized by endothelial
and mesangial cell proliferation and thickening of the basement
membrane (90). MRL/lpr mice are
commonly used to investigate the mechanisms of podocyte injury and
specific pathogenic mechanisms in diseases affecting the kidneys.
CFH deficiency in this mouse model leads to immune complex
deposition, loss of podocyte foot processes, accelerated renal
injury and LN progression (45).
When MRL/lpr mice are stimulated with LPS, levels of
proinflammatory cytokines increase in the serum, and there is
damage to the podocytes in the kidney, as well as increased urinary
albumin (91).
BXSB/Yaa mice are a male lupus-like autoimmune
disease model (92) that develops
severe immune complex-mediated glomerulonephritis with podocyte
injury and foot process effacement. This model also develops
membranoproliferative LN with IgG and C3 deposition in the
mesangium (46). The
overexpression of the TLR was correlated with urinary albumin
levels and mRNA levels of the nephrin in the BXSB-Yaa mice, which
are commonly used to investigate the interaction between TLRs and
podocytes. The BXSB/Yaa mouse model is the result of a gene
mutation that causes translocation of the terminal region of the X
chromosome to the Y chromosome, leading to TLR7 gene duplication,
which increases TLR7 expression levels (93). Additionally, BXSB/Yaa mice
demonstrate overexpression of the TLR8 in the glomerulus, which is
negatively correlated with podocyte markers (nephrin, podocin and
synaptopodin), inducing autoimmune responses. The mRNA expression
level of TLR8 is positively correlated with urinary albumin,
suggesting the involvement of TLR8 in the process of podocyte
injury (50).
New animal models are expected to be established to
explore the specific pathogenic mechanisms of renal intrinsic cell
autoantibodies in kidney disease. This will shed light on novel
pathways leading to renal damage in LN.
Application of drug-targeted podocytes
In the last decade, prominent advances have been
made in studying the structure and function of podocytes.
Anifrolumab is a human monoclonal antibody targeting the type I IFN
receptor subunit 1 and is the first type I IFN receptor antagonist
approved by the US Food and Drug Administration for the treatment
of SLE in adult patients. Meanwhile, anifrolumab has been
investigated as a promising strategy for the treatment of LN in
clinical trials (94–96). A large randomized
placebo-controlled trial suggested that neutralizing type I IFN
receptors expressed by podocytes can effectively reduce proteinuria
in patients with LN (97).
Tacrolimus, a calcineurin inhibitor, can reduce proteinuria and
improve kidney function in mice with lupus and patients with LN
(98), while stabilizing the
podocyte cytoskeleton and suppressing podocyte apoptosis, partially
protecting podocytes from injury in LN (99). Baricitinib, a selective inhibitor
of JAK1 and JAK2, is commonly used for rheumatoid arthritis
treatment. Recent a study has revealed that baricitinib
demonstrates benefits in inhibiting systemic autoimmunity in
MRL/lpr mice and improves the lupus-like phenotype. It has been
identified that baricitinib can inhibit the JAK/STAT pathway in
podocytes, restore the abnormal podocyte structure caused by
inflammation, and thus prevent renal damage (100). Greka et al (71) revealed that abatacept, a
co-stimulatory blocker of B7-1, suppresses T cell activation and
promotes integrin activation in podocytes. It also inhibits
podocyte migration and prevents podocyte damage, which improves
proteinuria in patients with LN.
Podocyte damage is reversible in LN, and actin is
able to restore foot processes and reorganize the podocyte
cytoskeleton. These studies illustrate a therapeutic target of
podocytes in the glomerulus and immune cells in the immune
microenvironment for precise treatment of LN, as they reduce
systemic side effects of drugs, relieve proteinuria symptoms and
improve disease progression.
Conclusion and future perspectives
Podocytes are glomerular epithelial cells, and the
majority of all kidney diseases lead to podocyte injury. There is
evidence that podocytes are important intrinsic cells of the
kidney, which confer immune cell functions to promote the
occurrence and development of LN. The manifestation and pathology
of SLE in murine models, especially NZB/W F1 and MRL/lpr mice, are
identical to that in patients with LN and have similar immune
mechanisms. These help to further investigate the interaction
mechanisms between podocytes and the renal immune microenvironment
in LN. Recently, a number of studies have revealed that current
therapies used to treat autoimmune diseases exhibit a direct
protective effect on podocytes, alleviating the occurrence of
proteinuria. These findings provide insights into targeted therapy
for kidney diseases. Numerous studies have confirmed that LN
podocytes participate in the innate and adaptive immune processes
and interact directly with cells and molecules in the immune
microenvironment. However, the mechanism of their interaction has
not been thoroughly investigated. Therefore, further studies are
needed to elucidate the interactions between LN podocytes and the
local immune cells of the kidney, especially T and B cells.
Targeting these pathogenic pathways might enable a more
personalized approach to the treatment of LN and lead to improved
outcomes for patients with LN.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China and Shanxi Province Applied Basic Research
Project (grant nos. 82202005 and 201901D211511), the National
Natural Science Foundation of China (grant no. 81871292), the Key
Research and Development (R&D) Projects of Shanxi Province
(grant no. 201803D31136), the Four ‘Batches’ Innovation Project of
Invigorating Medical Through Science and Technology of Shanxi
Province (grant no. 2023XM002), and the Shanxi Province
Postgraduate Practice Innovation Project (grant no. 2023SJ135).
Availability of data and materials
Not applicable.
Authors' contributions
RL and XP wrote the manuscript. MZ acquired and
interpreted the data. LM and JL conceptualized and designed the
manuscript. XW and KX reviewed and revised the manuscript. All
authors read and approved the final version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tian J, Zhang D, Yao X, Huang Y and Lu Q:
Global epidemiology of systemic lupus erythematosus: A
comprehensive systematic analysis and modelling study. Ann Rheum
Dis. 82:351–356. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kaul A, Gordon C, Crow MK, Touma Z,
Urowitz MB, van Vollenhoven R, Ruiz-Irastorza G and Hughes G:
Systemic lupus erythematosus. Nat Rev Dis Primers. 2:160392016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tektonidou MG, Dasgupta A and Ward MM:
Risk of end-stage renal disease in patients with lupus nephritis,
1971–2015: A systematic review and bayesian meta-analysis.
Arthritis Rheumatol. 68:1432–1441. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Seligman VA, Lum RF, Olson JL, Li H and
Criswell LA: Demographic differences in the development of lupus
nephritis: A retrospective analysis. Am J Med. 112:726–729. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aguirre A, Izadi Z, Trupin L, Barbour KE,
Greenlund KJ, Katz P, Lanata C, Criswell L, Dall'Era M and Yazdany
J: Race, ethnicity, and disparities in the risk of end-organ lupus
manifestations following a systemic lupus erythematosus diagnosis
in a multiethnic cohort. Arthritis Care Res (Hoboken). 75:34–43.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsokos GC, Lo MS, Costa RP and Sullivan
KE: New insights into the immunopathogenesis of systemic lupus
erythematosus. Nat Rev Rheumatol. 12:716–730. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mohan C and Putterman C: Genetics and
pathogenesis of systemic lupus erythematosus and lupus nephritis.
Nat Rev Nephrol. 11:329–341. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Costenbader KH, Desai A, Alarcón GS,
Hiraki LT, Shaykevich T, Brookhart MA, Massarotti E, Lu B, Solomon
DH and Winkelmayer WC: Trends in the incidence, demographics, and
outcomes of end-stage renal disease due to lupus nephritis in the
US from 1995 to 2006. Arthritis Rheum. 63:1681–1688. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Parikh SV and Rovin BH: Current and
emerging therapies for lupus nephritis. J Am Soc Nephrol.
27:2929–2939. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Davidson A: What is damaging the kidney in
lupus nephritis? Nat Rev Rheumatol. 12:143–153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kraft SW, Schwartz MM, Korbet SM and Lewis
EJ: Glomerular podocytopathy in patients with systemic lupus
erythematosus. J Am Soc Nephrol. 16:175–179. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bomback AS and Markowitz GS: Lupus
podocytopathy: A distinct entity. Clin J Am Soc Nephrol.
11:547–548. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wright RD and Beresford MW: Podocytes
contribute, and respond, to the inflammatory environment in lupus
nephritis. Am J Physiol Renal Physiol. 315:F1683–F1694. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moustafa FE, Soliman NA, Bakr AM and El
Shwaf IM: Assessment of detached podocytes in the Bowman's space as
a marker of disease activity in lupus nephritis. Lupus. 23:146–150.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu W, Chen Y, Wang S, Chen H and Liu Z,
Zeng C, Zhang H and Liu Z: Clinical-Morphological features and
outcomes of lupus podocytopathy. Clin J Am Soc Nephrol. 11:585–592.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jamaly S, Rakaee M, Abdi R, Tsokos GC and
Fenton KA: Interplay of immune and kidney resident cells in the
formation of tertiary lymphoid structures in lupus nephritis.
Autoimmun Rev. 20:1029802021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kang S, Fedoriw Y, Brenneman EK, Truong
YK, Kikly K and Vilen BJ: BAFF induces tertiary lymphoid structures
and positions T cells within the glomeruli during lupus nephritis.
J Immunol. 198:2602–2611. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schwartz N, Goilav B and Putterman C: The
pathogenesis, diagnosis and treatment of lupus nephritis. Curr Opin
Rheumatol. 26:502–509. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dieker J, Tel J, Pieterse E, Thielen A,
Rother N, Bakker M, Fransen J, Dijkman HB, Berden JH, de Vries JM,
et al: Circulating apoptotic microparticles in systemic lupus
erythematosus patients drive the activation of dendritic cell
subsets and prime neutrophils for NETosis. Arthritis Rheumatol.
68:462–472. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Elkon KB: Review: Cell death, nucleic
acids, and immunity: Inflammation beyond the grave. Arthritis
Rheumatol. 70:805–816. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Salvi V, Gianello V, Busatto S, Bergese P,
Andreoli L, D'Oro U, Zingoni A, Tincani A, Sozzani S and Bosisio D:
Exosome-delivered microRNAs promote IFN-α secretion by human
plasmacytoid DCs via TLR7. JCI Insight. 3:e982042018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Leonard D, Eloranta ML, Hagberg N,
Berggren O, Tandre K, Alm G and Rönnblom L: Activated T cells
enhance interferon-α production by plasmacytoid dendritic cells
stimulated with RNA-containing immune complexes. Ann Rheum Dis.
75:1728–1734. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wen L, Zhang B, Wu X, Liu R, Fan H, Han L,
Zhang Z, Ma X, Chu CQ and Shi X: Toll-like receptors 7 and 9
regulate the proliferation and differentiation of B cells in
systemic lupus erythematosus. Front Immunol. 14:10932082023.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schrezenmeier E, Jayne D and Dörner T:
Targeting B cells and plasma cells in glomerular diseases:
Translational perspectives. J Am Soc Nephrol. 29:741–758. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Flores-Mendoza G, Sansón SP,
Rodríguez-Castro S, Crispín JC and Rosetti F: Mechanisms of tissue
injury in lupus nephritis. Trends Mol Med. 24:364–378. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Parikh SV, Almaani S, Brodsky S and Rovin
BH: Update on lupus nephritis: Core curriculum 2020. Am J Kidney
Dis. 76:265–281. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sharma M, Vignesh P, Tiewsoh K and Rawat
A: Revisiting the complement system in systemic lupus
erythematosus. Expert Rev Clin Immunol. 16:397–408. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pavenstädt H, Kriz W and Kretzler M: Cell
biology of the glomerular podocyte. Physiol Rev. 83:253–307. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garg P: A review of podocyte biology. Am J
Nephrol. 47 (Suppl 1):S3–S13. 2018. View Article : Google Scholar
|
|
30
|
Humphries JD, Wang P, Streuli C, Geiger B,
Humphries MJ and Ballestrem C: Vinculin controls focal adhesion
formation by direct interactions with talin and actin. J Cell Biol.
179:1043–1057. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sellin L, Huber TB, Gerke P, Quack I,
Pavenstädt H and Walz G: NEPH1 defines a novel family of podocin
interacting proteins. FASEB J. 17:115–117. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garg P, Verma R, Nihalani D, Johnstone DB
and Holzman LB: Neph1 cooperates with nephrin to transduce a signal
that induces actin polymerization. Mol Cell Biol. 27:8698–8712.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huber TB, Simons M, Hartleben B, Sernetz
L, Schmidts M, Gundlach E, Saleem MA, Walz G and Benzing T:
Molecular basis of the functional podocin-nephrin complex:
Mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft
microdomains. Hum Mol Genet. 12:3397–3405. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dryer SE and Reiser J: TRPC6 channels and
their binding partners in podocytes: Role in glomerular filtration
and pathophysiology. Am J Physiol Renal Physiol. 299:F689–F701.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ha TS: Roles of adaptor proteins in
podocyte biology. World J Nephrol. 2:1–10. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang Y, Yu F, Song D, Wang SX and Zhao MH:
Podocyte involvement in lupus nephritis based on the 2003 ISN/RPS
system: A large cohort study from a single centre. Rheumatology
(Oxford). 53:1235–1244. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Banas MC, Banas B, Hudkins KL, Wietecha
TA, Iyoda M, Bock E, Hauser P, Pippin JW, Shankland SJ, Smith KD,
et al: TLR4 links podocytes with the innate immune system to
mediate glomerular injury. J Am Soc Nephrol. 19:704–713. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li X, Ding F, Zhang X, Li B and Ding J:
The expression profile of complement components in podocytes. Int J
Mol Sci. 17:4712016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gao S, Cui Z and Zhao MH: Complement C3a
and C3a receptor activation mediates podocyte injuries in the
mechanism of primary membranous nephropathy. J Am Soc Nephrol.
33:1742–1756. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang H, Lv D, Jiang S, Hou Q, Zhang L, Li
S, Zhu X, Xu X, Wen J, Zeng C, et al: Complement induces podocyte
pyroptosis in membranous nephropathy by mediating mitochondrial
dysfunction. Cell Death Dis. 13:2812022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pippin JW, Durvasula R, Petermann A,
Hiromura K, Couser WG and Shankland SJ: DNA damage is a novel
response to sublytic complement C5b-9-induced injury in podocytes.
J Clin Invest. 111:877–885. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Appay MD, Kazatchkine MD, Levi-Strauss M,
Hinglais N and Bariety J: Expression of CR1 (CD35) mRNA in
podocytes from adult and fetal human kidneys. Kidney Int.
38:289–293. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Teixeira JE, Costa RS, Lachmann PJ,
Würzner R and Barbosa JE: CR1 stump peptide and terminal complement
complexes are found in the glomeruli of lupus nephritis patients.
Clin Exp Immunol. 105:497–503. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Moll S, Miot S, Sadallah S, Gudat F,
Mihatsch MJ and Schifferli JA: No complement receptor 1 stumps on
podocytes in human glomerulopathies. Kidney Int. 59:160–168. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bao L, Haas M and Quigg RJ: Complement
factor H deficiency accelerates development of lupus nephritis. J
Am Soc Nephrol. 22:285–295. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pickering MC, Ismajli M, Condon MB,
McKenna N, Hall AE, Lightstone L, Terence Cook H and Cairns TD:
Eculizumab as rescue therapy in severe resistant lupus nephritis.
Rheumatology (Oxford). 54:2286–2288. 2015.PubMed/NCBI
|
|
47
|
Coppo R, Peruzzi L, Amore A, Martino S,
Vergano L, Lastauka I, Schieppati A, Noris M, Tovo PA and Remuzzi
G: Dramatic effects of eculizumab in a child with diffuse
proliferative lupus nephritis resistant to conventional therapy.
Pediatr Nephrol. 30:167–172. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Patole PS, Pawar RD, Lech M, Zecher D,
Schmidt H, Segerer S, Ellwart A, Henger A, Kretzler M and Anders
HJ: Expression and regulation of Toll-like receptors in lupus-like
immune complex glomerulonephritis of MRL-Fas(lpr) mice. Nephrol
Dial Transplant. 21:3062–3073. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Devarapu SK and Anders HJ: Toll-like
receptors in lupus nephritis. J Biomed Sci. 25:352018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kimura J, Ichii O, Miyazono K, Nakamura T,
Horino T, Otsuka-Kanazawa S and Kon Y: Overexpression of Toll-like
receptor 8 correlates with the progression of podocyte injury in
murine autoimmune glomerulonephritis. Sci Rep. 4:72902014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Marshak-Rothstein A and Rifkin IR:
Immunologically active autoantigens: The role of toll-like
receptors in the development of chronic inflammatory disease. Annu
Rev Immunol. 25:419–441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Anders HJ, Lichtnekert J and Allam R:
Interferon-alpha and -beta in kidney inflammation. Kidney Int.
77:848–854. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Machida H, Ito S, Hirose T, Takeshita F,
Oshiro H, Nakamura T, Mori M, Inayama Y, Yan K, Kobayashi N and
Yokota S: Expression of Toll-like receptor 9 in renal podocytes in
childhood-onset active and inactive lupus nephritis. Nephrol Dial
Transplant. 25:2530–2537. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Masum MA, Ichii O, Hosny Ali Elewa Y,
Nakamura T, Otani Y, Hosotani M and Kon Y: Overexpression of
toll-like receptor 9 correlates with podocyte injury in a murine
model of autoimmune membranoproliferative glomerulonephritis.
Autoimmunity. 51:386–398. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Takano Y, Yamauchi K, Hayakawa K,
Hiramatsu N, Kasai A, Okamura M, Yokouchi M, Shitamura A, Yao J and
Kitamura M: Transcriptional suppression of nephrin in podocytes by
macrophages: Roles of inflammatory cytokines and involvement of the
PI3K/Akt pathway. FEBS Lett. 581:421–426. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang Z, Niu L, Tang X, Feng R, Yao G,
Chen W, Li W, Feng X, Chen H and Sun L: Mesenchymal stem cells
prevent podocyte injury in lupus-prone B6.MRL-Faslpr mice via
polarizing macrophage into an anti-inflammatory phenotype. Nephrol
Dial Transplant. 34:597–605. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sung SJ and Fu SM: Interactions among
glomerulus infiltrating macrophages and intrinsic cells via
cytokines in chronic lupus glomerulonephritis. J Autoimmun.
106:1023312020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zoja C, Wang JM, Bettoni S, Sironi M,
Renzi D, Chiaffarino F, Abboud HE, Van Damme J, Mantovani A,
Remuzzi G, et al: Interleukin-1 beta and tumor necrosis
factor-alpha induce gene expression and production of leukocyte
chemotactic factors, colony-stimulating factors, and interleukin-6
in human mesangial cells. Am J Pathol. 138:991–1003.
1991.PubMed/NCBI
|
|
59
|
Latz E, Xiao TS and Stutz A: Activation
and regulation of the inflammasomes. Nat Rev Immunol. 13:397–411.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Fu R, Guo C, Wang S, Huang Y, Jin O, Hu H,
Chen J, Xu B, Zhou M, Zhao J, et al: Podocyte activation of NLRP3
inflammasomes contributes to the development of proteinuria in
lupus nephritis. Arthritis Rheumatol. 69:1636–1646. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Guo C, Fu R, Zhou M, Wang S, Huang Y, Hu
H, Zhao J, Gaskin F, Yang N and Fu SM: Pathogenesis of lupus
nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome
activation. J Autoimmun. 103:1022862019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Cai M, Zhou T, Wang X, Shang M, Zhang Y,
Luo M, Xu C and Yuan W: DC-SIGN expression on podocytes and its
role in inflammatory immune response of lupus nephritis. Clin Exp
Immunol. 183:317–325. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Haymann JP, Levraud JP, Bouet S, Kappes V,
Hagège J, Nguyen G, Xu Y, Rondeau E and Sraer JD: Characterization
and localization of the neonatal Fc receptor in adult human kidney.
J Am Soc Nephrol. 11:632–639. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ichinose K, Ushigusa T, Nishino A,
Nakashima Y, Suzuki T, Horai Y, Koga T, Kawashiri SY, Iwamoto N,
Tamai M, et al: Lupus Nephritis IgG induction of
calcium/calmodulin-dependent protein kinase IV expression in
podocytes and alteration of their function. Arthritis Rheumatol.
68:944–952. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Bhargava R, Lehoux S, Maeda K, Tsokos MG,
Krishfield S, Ellezian L, Pollak M, Stillman IE, Cummings RD and
Tsokos GC: Aberrantly glycosylated IgG elicits pathogenic signaling
in podocytes and signifies lupus nephritis. JCI Insight.
6:e1477892021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Bruschi M, Moroni G, Sinico RA,
Franceschini F, Fredi M, Vaglio A, Cavagna L, Petretto A, Pratesi
F, Migliorini P, et al: Serum IgG2 antibody multi-composition in
systemic lupus erythematosus and in lupus nephritis (Part 2):
Prospective study. Rheumatology (Oxford). 60:3388–3397. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Mason LJ, Ravirajan CT, Rahman A,
Putterman C and Isenberg DA: Is alpha-actinin a target for
pathogenic anti-DNA antibodies in lupus nephritis? Arthritis Rheum.
50:866–870. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Renaudineau Y, Deocharan B, Jousse S,
Renaudineau E, Putterman C and Youinou P: Anti-alpha-actinin
antibodies: A new marker of lupus nephritis. Autoimmun Rev.
6:464–468. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chen A, Lee K, D'Agati VD, Wei C, Fu J,
Guan TJ, He JC, Schlondorff D and Agudo J: Bowman's capsule
provides a protective niche for podocytes from cytotoxic CD8+ T
cells. J Clin Invest. 128:3413–3424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Reiser J, von Gersdorff G, Loos M, Oh J,
Asanuma K, Giardino L, Rastaldi MP, Calvaresi N, Watanabe H,
Schwarz K, et al: Induction of B7-1 in podocytes is associated with
nephrotic syndrome. J Clin Invest. 113:1390–1397. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Greka A, Weins A and Mundel P: Abatacept
in B7-1-positive proteinuric kidney disease. N Engl J Med.
370:1263–1266. 2014.PubMed/NCBI
|
|
72
|
Khullar B, Balyan R, Oswal N, Jain N,
Sharma A, Abdin MZ, Bagga A, Bhatnagar S, Wadhwa N, Natchu UCM, et
al: Interaction of CD80 with Neph1: A potential mechanism of
podocyte injury. Clin Exp Nephrol. 22:508–516. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Coers W, Brouwer E, Vos JT, Chand A,
Huitema S, Heeringa P, Kallenberg CG and Weening JJ: Podocyte
expression of MHC class I and II and intercellular adhesion
molecule-1 (ICAM-1) in experimental pauci-immune crescentic
glomerulonephritis. Clin Exp Immunol. 98:279–286. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Goldwich A, Burkard M, Olke M, Daniel C,
Amann K, Hugo C, Kurts C, Steinkasserer A and Gessner A: Podocytes
are nonhematopoietic professional antigen-presenting cells. J Am
Soc Nephrol. 24:906–916. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Li S, Liu Y, He Y, Rong W, Zhang M, Li L,
Liu Z and Zen K: Podocytes present antigen to activate specific T
cell immune responses in inflammatory renal disease. J Pathol.
252:165–177. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Okamoto A, Fujio K, Tsuno NH, Takahashi K
and Yamamoto K: Kidney-infiltrating CD4+ T-cell clones promote
nephritis in lupus-prone mice. Kidney Int. 82:969–979. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
May CJ, Welsh GI, Chesor M, Lait PJ,
Schewitz-Bowers LP, Lee RWJ and Saleem MA: Human Th17 cells produce
a soluble mediator that increases podocyte motility via signaling
pathways that mimic PAR-1 activation. Am J Physiol Renal Physiol.
317:F913–F921. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Cheng Y, Yang X, Zhang X and An Z:
Analysis of expression levels of IL-17 and IL-34 and influencing
factors for prognosis in patients with lupus nephritis. Exp Ther
Med. 17:2279–2283. 2019.PubMed/NCBI
|
|
79
|
Wang N, Gao C, Cui S, Qin Y, Zhang C, Yi
P, Di X, Liu S, Li T, Gao G and Zheng Z: Induction therapy
downregulates the expression of Th17/Tfh cytokines in patients with
active lupus nephritis. Am J Clin Exp Immunol. 7:67–75.
2018.PubMed/NCBI
|
|
80
|
Yan J, Li Y, Yang H, Zhang L, Yang B, Wang
M and Li Q: Interleukin-17A participates in podocyte injury by
inducing IL-1β secretion through ROS-NLRP3 inflammasome-caspase-1
pathway. Scand J Immunol. 87:e126452018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yuan DH, Jia Y, Hassan OM, Xu LY and Wu
XC: LPS-Treated podocytes polarize naive CD4(+) T Cells into Th17
and treg cells. Biomed Res Int. 2020:85879232020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Chang A, Henderson SG, Brandt D, Liu N,
Guttikonda R, Hsieh C, Kaverina N, Utset TO, Meehan SM, Quigg RJ,
et al: In situ B cell-mediated immune responses and
tubulointerstitial inflammation in human lupus nephritis. J
Immunol. 186:1849–1860. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Tsokos GC: Autoimmunity and organ damage
in systemic lupus erythematosus. Nat Immunol. 21:605–614. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Kolovou K, Laskari K, Roumelioti M,
Tektonidou MG, Panayiotidis P, Boletis JN, Marinaki S and Sfikakis
PP: B-cell oligoclonal expansions in renal tissue of patients with
immune-mediated glomerular disease. Clin Immunol. 217:1084882020.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Senaldi G, Stolina M, Guo J, Faggioni R,
McCabe S, Kaufman SA, Van G, Xu W, Fletcher FA, Boone T, et al:
Regulatory effects of novel neurotrophin-1/b cell-stimulating
factor-3 (cardiotrophin-like cytokine) on B cell function. J
Immunol. 168:5690–5698. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Schmidt-Ott KM, Yang J, Chen X, Wang H,
Paragas N, Mori K, Li JY, Lu B, Costantini F, Schiffer M, et al:
Novel regulators of kidney development from the tips of the
ureteric bud. J Am Soc Nephrol. 16:1993–2002. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Savin VJ, Sharma M, Zhou J, Gennochi D,
Fields T, Sharma R, McCarthy ET, Srivastava T, Domen J, Tormo A and
Gauchat JF: Renal and Hematological Effects of CLCF-1, a
B-Cell-Stimulating Cytokine of the IL-6 Family. J Immunol Res.
2015:7149642015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Dos Santos M, Poletti PT, Milhoransa P,
Monticielo OA and Veronese FV: Unraveling the podocyte injury in
lupus nephritis: Clinical and experimental approaches. Semin
Arthritis Rheum. 46:632–641. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Andrews BS, Eisenberg RA, Theofilopoulos
AN, Izui S, Wilson CB, McConahey PJ, Murphy ED, Roths JB and Dixon
FJ: Spontaneous murine lupus-like syndromes. Clinical and
immunopathological manifestations in several strains. J Exp Med.
148:1198–1215. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
McGaha TL and Madaio MP: Lupus Nephritis:
Animal modeling of a complex disease syndrome pathology. Drug
Discov Today Dis Models. 11:13–18. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Pawar RD, Castrezana-Lopez L, Allam R,
Kulkarni OP, Segerer S, Radomska E, Meyer TN, Schwesinger CM, Akis
N, Gröne HJ and Anders HJ: Bacterial lipopeptide triggers massive
albuminuria in murine lupus nephritis by activating Toll-like
receptor 2 at the glomerular filtration barrier. Immunology. 128 (1
Suppl):e206–e221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Maibaum MA, Haywood ME, Walport MJ and
Morley BJ: Lupus susceptibility loci map within regions of BXSB
derived from the SB/Le parental strain. Immunogenetics. 51:370–372.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Pisitkun P, Deane JA, Difilippantonio MJ,
Tarasenko T, Satterthwaite AB and Bolland S: Autoreactive B cell
responses to RNA-related antigens due to TLR7 gene duplication.
Science. 312:1669–1672. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Jayne D, Rovin B, Mysler EF, Furie RA,
Houssiau FA, Trasieva T, Knagenhjelm J, Schwetje E, Chia YL,
Tummala R and Lindholm C: Phase II randomised trial of type I
interferon inhibitor anifrolumab in patients with active lupus
nephritis. Ann Rheum Dis. 81:496–506. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Parodis I and Houssiau FA: From sequential
to combination and personalised therapy in lupus nephritis: Moving
towards a paradigm shift? Ann Rheum Dis. 81:15–19. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Steiger S, Ehreiser L, Anders J and Anders
HJ: Biological drugs for systemic lupus erythematosus or active
lupus nephritis and rates of infectious complications. Evidence
from large clinical trials. Front Immunol. 13:9997042022.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Markowitz GS, Nasr SH, Stokes MB and
D'Agati VD: Treatment with IFN-{alpha}, -{beta}, or -{gamma} is
associated with collapsing focal segmental glomerulosclerosis. Clin
J Am Soc Nephrol. 5:607–615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Liao R, Liu Q, Zheng Z, Fan J, Peng W,
Kong Q, He H, Yang S, Chen W, Tang X and Yu X: Tacrolimus protects
podocytes from injury in lupus nephritis partly by stabilizing the
cytoskeleton and inhibiting podocyte apoptosis. PLoS One.
10:e1327242015. View Article : Google Scholar
|
|
99
|
Yasuda H, Fukusumi Y, Ivanov V, Zhang Y
and Kawachi H: Tacrolimus ameliorates podocyte injury by restoring
FK506 binding protein 12 (FKBP12) at actin cytoskeleton. FASEB J.
35:e219832021. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Lee J, Park Y, Jang SG, Hong SM, Song YS,
Kim MJ, Baek S, Park SH and Kwok SK: Baricitinib attenuates
autoimmune phenotype and podocyte injury in a murine model of
systemic lupus erythematosus. Front Immunol. 12:7045262021.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Rice WL, Van Hoek AN, Păunescu TG, Huynh
C, Goetze B, Singh B, Scipioni L, Stern LA and Brown D: High
resolution helium ion scanning microscopy of the rat kidney. PLoS
One. 8:e570512013. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Howie JB and Helyer BJ: The immunology and
pathology of NZB mice. Adv Immunol. 9:215–266. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Hall AM, Ward FJ, Shen CR, Rowe C, Bowie
L, Devine A, Urbaniak SJ, Elson CJ and Barker RN: Deletion of the
dominant autoantigen in NZB mice with autoimmune hemolytic anemia:
Effects on autoantibody and T-helper responses. Blood.
110:4511–4517. 2007. View Article : Google Scholar : PubMed/NCBI
|