Introduction
Neuroglobin (Ngb) is an oxygen-carrying globin and
its high oxygen-binding capacity and specific expression in the
nervous system make it an important candidate in the study of novel
mechanisms underlying neural repair following cerebral ischemia and
hypoxia (1–3). It has been reported that hypoxia
induces Ngb expression (4) and
that hypoxia-inducible factor-1α (HIF-1α) positively regulates Ngb
expression (5). In addition,
increased expression of Ngb has been observed in the brain tissue
of patients with ischemic stroke (6). Ngb not only enhances cellular
survival by regulating cell signaling via G proteins (7), but also regulates apoptotic
mechanisms associated with hypoxia (8–10). A
previous study demonstrated that overexpression of Ngb reduces
brain injury induced by intracerebral hemorrhage, providing further
evidence that Ngb plays a vital role in neuroprotection (11). In addition, a recent in vivo
animal study revealed that exogenous Ngb protein (fused to the
11-amino-acid human immunodeficiency virus transactivator of
transcription protein transduction domain) might be efficiently
transduced into neurons in the mouse and protect the brain from
mild or moderate ischemic injury (12). The authors reported that Ngb
functions as an endogenous neuroprotective factor in brain ischemia
(13).
However, the mechanisms underlying the
neuroprotective properties of Ngb and the relevant signaling
pathways have not been fully elucidated. Ngb has been identified to
specifically bind to the Gα subunit of G proteins, consequently
releasing the Gβγ subunit, functioning as a signal transduction
factor important for nerve growth factor (NGF)-induced
neuroprotection via activation of the PI3K/Akt signaling cascade
(14). Of note, transducin, a
specific inhibitor of Gβγ, prevents NGF-induced neuroprotection,
consistent with a role for Gβγ in a PI3K/Akt-dependant
neuroprotection mechanism (15),
indicating that Ngb may be involved in this process. HIF-1α is
induced by ischemia and exerts a neuroprotective effect, at least
in part, via induction of insulin growth factor, which activates
the PI3K/Akt pathway to exert neuroprotection (16). As ischemia and specifically,
HIF-1α, directly induce Ngb expression (5), we hypothesize that the
neuroprotective effects of Ngb are mediated by PI3K/Akt signaling.
In the present study, the expression of Ngb and activation of
PI3K/Akt was examined in injured brains following focal cerebral
ischemia in adult rats and PI3K/Akt signaling was found to be
required for the neuroprotective effects of hemin-induced Ngb
expression.
Materials and methods
Experimental animals and grouping
Healthy adult male Sprague-Dawley rats, weighing
280±20 g (n=60), were provided by the Experimental Animal Center of
Xi’an Jiaotong University (Xi’an, China). This study was approved
by the Animal Ethics Committee of Xi’an Medical University (Xi’an,
China). The rats were randomly divided into five groups
(n=12/group): i) Sham control; ii) ischemia, with middle cerebral
artery occlusion (MCAO); iii) Hemin, which received intraperitoneal
injection of 50 mg/kg Hemin, a specific Ngb inducer, 1 h following
MCAO; iv) LY, in which tail vein injection of LY294002 (a PI3K/Akt
inhibitor, at a concentration of 0.3 mg/kg) was performed 15 min
prior to MCAO; v) Hemin + LY, which received LY294002 15 min prior
to MCAO and Hemin 1 hr following MCAO.
Reagents
Rabbit anti-rat polyclonal anti-Ngb antibody (Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA); rabbit anti-pAkt
antibody (Cell Signaling Technology, Inc., Danvers, MA, USA);
horseradish peroxidase (HRP)-labeled goat anti-rabbit IgG antibody
(Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing,
China); rabbit anti-rat actin monoclonal antibody (Abcam,
Cambridge, UK); RIPA lysis buffer (Bioon, Shanghai, China);
enhanced chemiluminescence kit (Santa Cruz Biotechnology, Inc.);
BCA protein quantitation kit (Bioon); TRIzol reagent kit
(Gibco-BRL, Carlsbad, CA, USA); RT-PCR and PCR amplification kits
and DNA ladder (both Takara Bio, Inc., Shiga, Japan); RNA
extraction kit (Tiangen Biotech Co., Ltd., Beijing, China); Hemin
(Lizhu Biotech Co. Ltd., Shanghai, China); and LY294002 (Santa Cruz
Biotechnology, Inc.).
Animal model of MCAO
Rats were anesthetized in a supine position by
intraperitoneal injection of 10% hydrochloride (0.3 mg/kg). The
right middle cerebral artery was permanently occluded as described
previously, with minor modifications (17). Briefly, the neck skin of the rat
was shaved and sterilized, and an incision was made in the midline
of the cervical skin. Next, the right common carotid artery (CCA),
external carotid artery (ECA) and internal carotid artery (ICA)
were exposed following muscle dissection, and the ECA and CCA were
ligated proximally. A small incision was made at the bifurcation of
the CCA and a 4–0 monofilament with heated blunt round end (0.3 mm,
sterilized by alcohol and soaked with heparin) was introduced
~18–20 cm into the origin of the middle cerebral artery via the
right ICA, followed by tight ligation of the monofilament around
the CCA. Sham surgery was performed by exposing the carotid artery
without occlusion of blood flow.
Neurological assessment
Evaluation of neurological function was performed 24
h prior to and following the induction of ischemia and scored on a
4-point scale as described previously (17). Scale: 0, normal; 1, incomplete
stretching of left forepaw; 2, circle to the left; 3, left limbs
limping while walking; 4, cannot walk, impaired. Rats exhibiting
any of these markers prior to surgery were excluded from the study
and rats were then randomly redistributed between groups. Scores
from 1–4 were regarded as successful establishment of the MCAO
model. Unconscious rats or animals with a score of 0 or mortality
within 24 h were discarded and additional surgeries were performed
to reach appropriate numbers of MCAO-positive animals for
experiments.
Measurement of volume of cerebral
infarction
Brains were dissected 24 h following surgery
(n=6/group) and a total of 6 sections were cut with an interval of
2 mm from Bregma +4.0 to −8.0 mm, excluding the olfactory bulb,
cerebrum and low brain stem. The sections were then stained with a
1% 2,3,5-triphenyltetrazolium chloride (TTC) solution (pH 7.4) and
incubated for 30 min at 37ºC. The infarct area appeared pale on a
background of red ‘normal’ brain. The sections were fixed with a 4%
paraformaldehyde solution. The total volume of each hemisphere and
infarction was determined by integration of the distance of the 6
sections. The infarct volume in cubic millimeters was calculated
with the following formula: V (infarct volume) = t × (A1 + A2 +
...An), where t represents the thickness of brain sections and A
represents infarction area.
RT-PCR analysis of mRNA expression of Ngb
and Akt
Brains were dissected 24 h following surgery
(n=6/group) and were immediately stored in liquid nitrogen. mRNA
expression of Ngb and Akt was tested in the sham surgery, ischemia
and Hemin groups. Total RNA was extracted according to the TRIzol
manufacturer’s instructions and RNA was reverse transcribed
according to the manufacturer’s instructions (Takara, Otsu, Japan).
The primers used for RT-PCR were designed using Primer 5.0 software
and synthesized by Sangon Biotech Co. Ltd., Shanghai, China: Ngb
forward, 5′-AGCCG CAGCCCTCTGGAACA-3′ and reverse, 5′-GCAG
CATCAATCACAAGCA-3′ (176 bp); Akt forward, 5′-CTGG
CCAGGCCCAAGCACCG-3′ and reverse, 5′-CGT TCACTGTCCACACACTC-3′ (109
bp); and GAPDH (internal control) forward,
5′-ACCACCATGGAGAAGGCTGG-3′ and reverse, 5′-CTCAGTGTAGCCCAGGATGC-3′
(528 bp). PCR conditions were as follows: 94ºC for 2 min, 94ºC for
1 min, 55ºC for 1 min and 72ºC for 1.5 min, total cycles were 28
and extension was at 72ºC for 7 min. PCR amplification yield
materials were visualized by 1.5% agarose gel electrophoresis. A UV
gel imaging system (Alphalmager2200; Alpha Innotech Corporation,
Santa Clara, CA, USA) was used for recording results of
electrophoresis. Image J software (NIH, Bethesda, MD, USA) was used
for quantitative analysis.
Western blot analysis of Ngb expression
and activation of PI3K/AKT
Total protein was extracted from dissected brain
tissue and concentration was determined with a BCA protein assay
kit. Proteins were separated by 20% SDS-PAGE gel electrophoresis
and transferred onto a PVDF membrane, then blocked at 4ºC
overnight. The membrane was incubated with primary antibodies
(rabbit anti-rat polyclonal anti-Ngb antibody (1:500) and rabbit
anti-pAkt antibody (1:200) at 4ºC overnight. Following washing, the
membrane was incubated with HRP-conjugated goat anti-rabbit IgG
antibody (Ngb, 1:10,000; pAkt, 1:2,000) and chemiluminescence
detection was used to quantify protein expression.
Statistical analysis
Data are expressed as the mean ± SD and analyzed by
t-test analysis with SPSS 17.0 software (SPSS, Inc., Chicago, IL,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Mortality
No animals died prior to tissue harvest in the sham
control group, one rat died in the ischemia group, two in the LY
group, one in the Hemin group (due to acute pulmonary edema
following damage to the vagus nerve during surgery) and one in the
Hemin + LY group. No significant difference in mortality was found
among groups. Animals that died prior to tissue harvest were
excluded from further analyses.
Neurological assessment
No neurological deficit was found in the sham
control group (score=0). Other groups presented with neurological
impairment to various extents 24 h following surgery. The score in
the Hemin group was significantly lower than that in the ischemia
group (Table I; P<0.05). By
contrast, the score in the LY group was higher than that in the
ischemia group (P<0.05), indicating that inhibition of the
PI3K/Akt pathway increases the severity of neurological impairment.
However, no significant difference was found between the Hemin + LY
and the ischemia group (Table I),
indicating that PI3K/Akt signaling is required for Hemin-mediated
neuroprotection.
 | Table INeurological deficit scores. |
Table I
Neurological deficit scores.
| Group | Score |
|---|
| Sham control | 0.00±0.00 |
| Ischemia | 2.98±0.76 |
| Hemin | 2.13±0.54a |
| LY | 3.42±1.01a |
| Hemin + LY | 2.87±0.62bc |
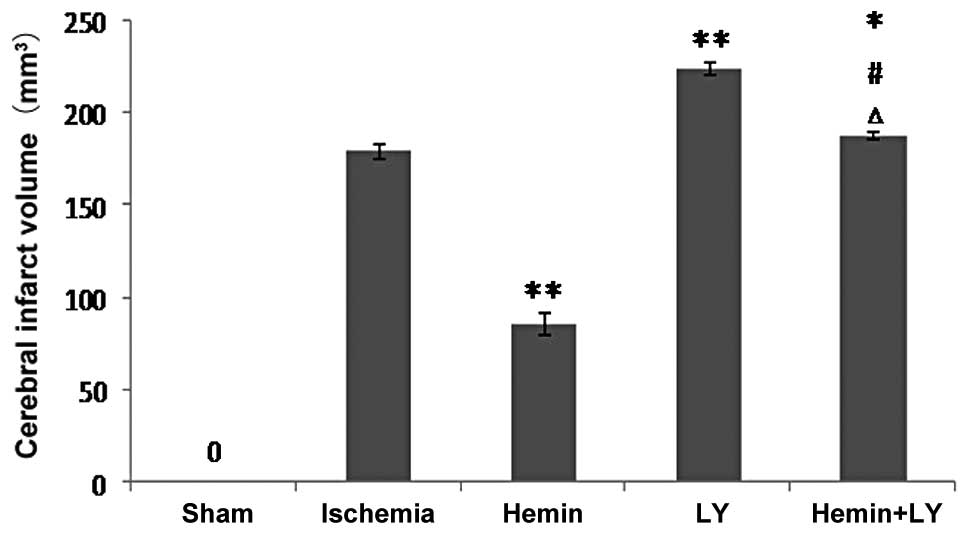
Treatment with Hemin but not LY294002
inhibits increased infarct volume induced by ischemia
Since measurement of infarct volume more accurately
reflects damage caused by cerebral brain ischemia compared with
assessment of neurological functions, infarct volume in the
ischemic brains of the rats was measured with or without Hemin or
LY treatment. Following TCC staining, no pale area was observed in
the brains from the sham surgery group. However, ischemia induced a
clear infarct volume (Fig. 1). Of
note, Hemin treatment significantly reduced ischemia-induced
infarct volume by ~2 fold (Fig. 1;
P<0.01), indicating that Hemin had a protective effect against
brain damage. By contrast, when compared with rats with ischemia
only, pre-treatment with LY294002 prior to ischemia was found to
significantly increase ischemia-induced infarct volume (Fig. 1; P<0.01). Consistent with these
observations, when compared with the Hemin group, pre-treatment
with LY in the ischemic rats treated with Hemin significantly
increased infarct volume by ~2 fold (Fig. 1; P<0.01). These results indicate
that inhibition of PI3K/Akt signaling exacerbates ischemia-induced
neurological damage. By contrast, when compared with LY alone, rats
treated with Hemin and LY revealed a small but significant decrease
in infarct volume, indicating that inhibition of PI3K/Akt signaling
was incomplete or that Hemin may mediate neuroprotection via
additional unknown mechanisms, but not solely through the PI3K/Akt
signaling pathway.
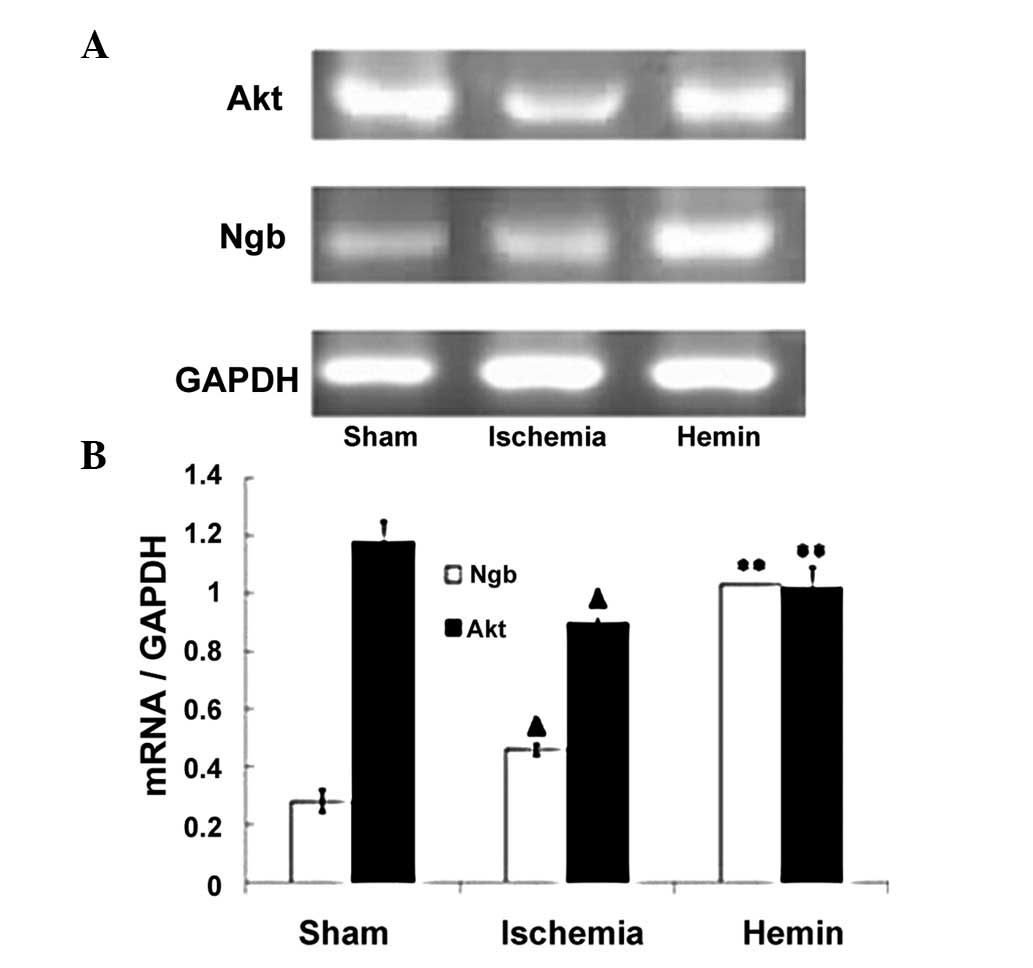
Hemin treatment enhances the expression
of Ngb and Akt mRNA
Ngb mRNA expression was low in the sham control
group; however, following ischemia, the expression of Ngb mRNA was
increased by 64% compared with sham controls (Fig. 2; P<0.01). By contrast, the
expression of Akt mRNA was decreased by 24% following ischemia
(Fig. 2; P<0.01). Of note,
Hemin treatment significantly increased the expression of Ngb and
Akt mRNA by 124 and 13%, respectively (Fig. 2; P<0.01).
Expression of Ngb protein and activation
of PI3K/Akt
To examine the expression of Ngb protein and
activation of PI3K/Akt, western blot analysis was performed. The
expression of Ngb protein 24 h following ischemia was increased by
102% and the levels of pAkt were decreased by 55% when compared
with sham controls. Hemin treatment increased Ngb and pAkt by 47
and 91%, respectively (Fig. 3;
P<0.01), whereas LY treatment inhibited pAkt levels but not Ngb
expression (Fig. 3, P<0.01).
When compared with Hemin treatment, the combination of Hemin and LY
treatment inhibited the increased pAkt levels induced by Hemin
treatment alone by 50% (Fig. 3;
P<0.01). When compared with LY alone, the combination of Hemin
and LY treatment induced an increase in pAkt and Ngb expression by
162 and 31%, respectively (Fig. 3;
P<0.01). These results indicate that PI3K/Akt signaling has no
effect on Ngb expression, but that Hemin treatment, presumably
through Ngb, is able to overcome the ischemia-induced reduction in
PI3K/Akt signaling and partially reverses the effects of LY
treatment. These results are consistent with our model indicating
that PI3K/Akt signaling is a downstream mediator of Ngb
neuroprotection.
Discussion
Ngb is a globin mainly found in neurons, which is
capable of reversibly binding oxygen. It is widely expressed in the
vertebrate cerebral cortex, hippocampus, thalamus, hypothalamus and
cerebellum (18). Ngb expression
in different regions is negatively associated with sensitivity to
oxygen. Upregulation of Ngb expression has been observed to varied
extents in response to various pathological conditions, including
cerebral ischemia, hypoxia, oxidation and toxicity. Ngb serves as
an endogenous neuroprotection factor to enhance tolerance of brain
tissues in response to ischemia and hypoxia. It has been revealed
that Hemin, a porphyrin containing ferric iron with a chlorine
ligand, specifically induces upregulation of Ngb in nerve cells
(19). In the present study,
intraperitoneal injection of Hemin prior to ischemia enhanced Ngb
mRNA expression in rat brains and reversed ischemia-induced
neurological impairment and infarction volume. These observations
further demonstrated that Ngb had neuroprotective effects on focal
cerebral ischemia and that PI3K/Akt signaling functioned downstream
of Ngb in this pathway.
However, the mechanisms underlying improvement of
neurological function and prognosis via endogenous Ngb remain
unclear. Currently, three hypotheses exist: i) Ngb plays an
important physiological role in oxygen absorption, usage and
transportation of oxygen to the mitochondria (13); ii) Ngb protects cells via clearance
of reactive oxygen species produced by oxidative stress (20); and iii) Ngb serves as an oxygen
sensor to regulate intracellular signal transduction according to
changes in oxygen concentration (7,21).
It is important to determine which, if any, of these mechanisms are
important for Ngb-mediated neuroprotection in future studies.
Signal transduction pathways associated with
apoptosis following cerebral ischemia mainly include
mitogen-activated protein kinase (MAPK) and PI3K/Akt. It has been
hypothesized that MAPK plays a role in regulating apoptosis
(22), whereas PI3K/Akt is mainly
involved in cellular survival (23). It is known that multiple
neurotrophic factors exert neuroprotective effects by inhibiting
apoptosis via activation of PI3K/Akt (24). Since it has been previously
demonstrated that PI3K/Akt is downregulated in ischemic penumbra
within 24 h of infarction (25),
it is likely that PI3K/Akt is important for the processing of
neurological damage. Thus, it appears that Ngb activation of
PI3K/Akt signaling is a mechanism underlying Ngb-mediated
neuroprotection.
The results of the present study indicate that Hemin
not only upregulates the expression of Ngb mRNA and protein, but
also increases levels of pAkt. In addition, Hemin was found to
reduce infarct volume and improve neurological functions. By
contrast, administration of the PI3K/Akt-specific inhibitor,
LY294002, prior to ischemia increased infarct volume and
exacerbated neurological damage, demonstrating the congruence of
Ngb and PI3K/Akt function in this system. RT-PCR and western blot
analysis revealed that inhibition of PI3K/Akt had no effect on
expression of Ngb mRNA and protein, respectively, but it is
required for the effects of upregulation of Ngb expression,
indicating that PI3K/Akt functions downstream of Ngb in the
signaling pathway associated with neuroprotection. In conclusion,
this study delineates a novel mechanism underlying Ngb-mediated
neuroprotection, which provides a theoretical basis for the
clinical application of Ngb and also demonstrates that PI3K/Akt
signaling may represent a novel target for therapeutic intervention
in hypoxic brain injury.
Acknowledgements
This study was supported by grants from Shaanxi
Natural Science Foundation of China (no. 2010JM4054) and research
Fund from Shaanxi Provience Education Department (no. 09JK713). The
authors would like to thank Medjaden Bioscience Limited for
assisting in the preparation of this manuscript.
References
|
1
|
Kelsen J, Hundahl CA and Hay-Schmidt A:
Neuroglobin: endogenous neuroprotectant or maintenance of
homeostasis? Stroke. 39:e177–e178. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang X, Liu J, Zhu H, et al: Effects of
neuroglobin overexpression on acute brain injury and long-term
outcomes after focal cerebral ischemia. Stroke. 39:1869–1874. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dong Y, Zhao R, Chen XQ and Yu AC:
14-3-3gamma and neuroglobin are new intrinsic protective factors
for cerebral ischemia. Mol Neurobiol. 41:218–231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu Z, Liu J, Guo S, et al:
Neuroglobin-overexpression alters hypoxic response gene expression
in primary neuron culture following oxygen glucose deprivation.
Neuroscience. 162:396–403. 2009. View Article : Google Scholar
|
|
5
|
Haines B, Demaria M, Mao X, et al:
Hypoxia-inducible factor-1 and neuroglobin expression. Neurosci
Lett. 514:137–140. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jin K, Mao Y, Mao X, Xie L and Greenberg
DA: Neuroglobin expression in ischemic stroke. Stroke. 41:557–559.
2010. View Article : Google Scholar
|
|
7
|
Wakasugi K, Nakano T and Morishima I:
Oxidized human neuroglobin acts as a heterotrimeric Galpha protein
guanine nucleotide dissociation inhibitor. J Biol Chem.
278:36505–36512. 2003. View Article : Google Scholar
|
|
8
|
Hota KB, Hota SK, Srivastava RB and Singh
SB: Neuroglobin regulates hypoxic response of neuronal cells
through Hif-1α- and Nrf2-mediated mechanism. J Cereb Blood Flow
Metab. 32:1046–1060. 2012.PubMed/NCBI
|
|
9
|
Brittain T, Skommer J, Henty K, Birch N
and Raychaudhuri S: A role for human neuroglobin in apoptosis.
IUBMB Life. 62:878–885. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brittain T and Skommer J: Does a redox
cycle provide a mechanism for setting the capacity of neuroglobin
to protect cells from apoptosis? IUBMB Life. 64:419–422. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin K, Mao X, Xie L and Greenberg DA:
Neuroglobin expression in human arteriovenous malformation and
intracerebral hemorrhage. Acta Neurochir. 111:315–319. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cai B, Lin Y, Xue XH, Fang L, Wang N and
Wu ZY: TAT-mediated delivery of neuroglobin protects against focal
cerebral ischemia in mice. Exp Neurol. 227:224–231. 2011.
View Article : Google Scholar
|
|
13
|
Sun Y, Jin K, Peel A, Mao XO, Xie L and
Greenberg DA: Neuroglobin protects the brain from experimental
stroke in vivo. Proc Natl Acad Sci USA. 100:3497–3500. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dudek H, Datta SR, Franke TF, et al:
Regulation of neuronal, survival by the serine-threonine protein
kinase Akt. Science. 275:661–665. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu EH and Wong YH: Activation of
muscarinic M4 receptor augments NGF-induced pro-survival Akt
signaling in PC12 cells. Cell Signal. 18:285–293. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li X, Lu Y, Jin W, Liang K, Mills GB and
Fan Z: Autophosphorylation of Akt at threonine 72 and serine 246. A
potential mechanism of regulation of Akt kinase activity. J Biol
Chem. 281:13837–13843. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wystub S, Laufs T, Schmidt M, et al:
Localization of neuroglobin protein in the mouse brain. Neurosci
Lett. 346:114–116. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu Y, Sun Y, Jin K and Greenberg DA:
Hemin induces neuroglobin expression in neural cells. Blood.
100:2494–2498. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jin K, Mao XO, Xie L, Khan AA and
Greenberg DA: Neuroglobin protects against nitric oxide toxicity.
Neurosci Lett. 430:135–137. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Herold S, Fago A, Weber RE, Dewilde S and
Moens L: Reactivity studies of the Fe(III) and Fe(II)NO forms of
human neuroglobin reveal a potential role against oxidative stress.
J Biol Chem. 279:22841–22847. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chaudhry K, Rogers R, Guo M, et al: Matrix
metalloproteinase-9 (MMP-9) expression and extracellular
signal-regulated kinase 1 and 2 (ERK1/2) activation in
exercise-reduced neuronal apoptosis after stroke. Neurosci Lett.
474:109–114. 2010. View Article : Google Scholar
|
|
23
|
Jover-Mengual T, Miyawaki T, Latuszek A,
Alborch E, Zukin RS and Etgen AM: Acute estradiol protects CA1
neurons from ischemia-induced apoptotic cell death via the PI3K/Akt
pathway. Brain Res. 1321:1–12. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nayak GH, Prentice HM and Milton SL:
Neuroprotective signaling pathways are modulated by adenosine in
the anoxia tolerant turtle. J Cereb Blood Flow Metab. 31:467–475.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao H, Shimohata T, Wang JQ, et al: Akt
contributes to neuroprotection by hypothermia against cerebral
ischemia in rats. J Neurosci. 25:9794–9806. 2005. View Article : Google Scholar : PubMed/NCBI
|