1. Introduction
Endometriosis is characterized by the growth of
endometrial tissue outside the uterus. Endometriosis may result
from increased cellular proliferation or a reduction in apoptosis
(1). The balance between
proliferation and apoptosis is changed in eutopic endometrium from
endometriosis. Proposed hypothetical causes of endometriosis
include retrograde menstruation, coelomic metaplasia and embryonic
rest. Sampson’s theory of retrograde menstruation hypothesizes that
the mechanical transfer, invasive implantation and ectopic growth
of endometrial tissues causes endometriosis and it remains the most
widely accepted causal theory. However, no single theory is able to
completely explain the origin and all aspects of this disorder
(2). The mechanisms responsible
for the initial development and subsequent progression of
endometriosis are not clear.
Estrogen is involved in the development and
progression of endometriosis. Treatment for endometriosis is
expectant, medical or surgical. Medical treatment strategies focus
on the creation of states of pseudopregnancy or pseudomenopause.
However, the most crucial problem is that the current medical and
surgical treatments of endometriosis are associated with high rates
of relapse. Thus, a novel approach would be a beneficial
improvement for the treatment of endometriosis. The identification
of the pathogenesis of this condition is a crucial step for
developing novel strategies for the treatment.
The present study reviews the contemporary
literature on the infection and sterile inflammation that support
the pathogenesis of endometriosis.
2. Study methodology
The present study reviews the literature for
biological studies on the development of endometriosis. Data
pertaining to in vitro and in vivo studies were
included. A computerized literature search was performed to
identify relevant studies reported in the English language. All
abstracts obtained from Medline electronic database were reviewed
by two investigators (Y.H. and H.S.) to identify studies for
full-text review. The web-based databases were searched by
combining the keywords ‘TLR’, ‘PAMP’, ‘inflammation’ ‘iron’
‘oxidative stress’ ‘damage’ ‘DAMP’ ‘danger signal’ ‘NF-κB’ or
‘antiapoptosis’ with ‘endometriosis’. Additionally, references in
each report were searched to identify potentially missed studies.
Target publications were mainly studies on humans and animal
models, as well as basic studies in gene and protein expression
systems. Abstracts were not included, as they do not undergo a
stringent peer review process.
3. Innate immunity and inflammation in
endometriosis
Endometriosis is associated with angiogenesis,
lymphangiogenesis and neurogenesis, which may be induced through
inflammatory cell activation and contribute to the ectopic growth
of endometrial tissue. Mounting evidence suggests that
endometriosis is a common type of chronic inflammatory disease with
an immunological background (3).
Substantial numbers of immune cells, such as macrophages,
neutrophils, dendritic cells, natural killer cells and mast cells,
have been identified in peritoneal fluids associated with
endometriosis (4) and macrophage
activity may be fundamental in this disorder. However, these
immune-associated cells fail to detect and eliminate ectopic
endometrial cells, suggesting that they are dysfunctional (4).
The conditions for the development of endometriosis
are induction of angiogenesis and lymphangiogenesis, which comprise
a continuum of vascular development. Endometriotic lesions,
particularly deep infiltrating endometriosis, have lymphangiogenic
properties. Furthermore, mast cells are key mediators of allergic
reactions of the immune system (5)
and their immune function may extend far beyond this role (6). Endometriosis is a common cause of
pelvic pain, possibly by regulating the recruitment of their own
unique neural supplies through neurogenesis (7). Although the precise reason for the
endometriosis-associated increase in neurogenesis is unknown, there
is evidence of a closer proximity between mast cells and nerve
fibers, demonstrating that mast cells are able to contribute to the
development of pain (5,8).
There is increasing evidence to demonstrate marked
elevation of proinflammatory cytokines [interleukin (IL)-1β and
tumor necrosis factor (TNF)-α], angiogenic cytokines (leptin and
IL-8), angiogenic growth factors (vascular endothelial growth
factor and protein kinase CK2) and prostaglandin concentrations in
endometriosis (3,7,9).
IL-1β and TNF-α in peritoneal fluid are able to activate c-Jun
N-terminal kinase (JNK) in eutopic endometrial cells from women
with endometriosis, which in turn may upregulate inflammatory
cytokine expression (10). JNK is
also activated in response to cellular stress (10).
Endometriosis is often accompanied by marked changes
in the number and function of inflammatory products, including
human neutrophil peptides belonging to the α-defensin family
(11), macrophage migration
inhibitory factor (MIF) (12), C-C
chemokine monocyte chemoattractant protein-1 (MCP-1), serum amyloid
A (SAA), TNF-α, IL-1, IL-6, IL-8, chemokine (C-C motif) receptor 1
(CCR1) (13) and regulated on
activation, normal T cell expressed and secreted (RANTES) (14). Defensin is involved in innate
immunity against bacteria and kills microbes. MIF is a potent
proinflammatory and growth-promoting factor and acts on ectopic
endometrial cells to stimulate the production of COX-2 and
PGE2. MCP-1 activates monocytes and recruits into the
inflammation site. The hepatic biosynthesis of SAA is upregulated
by proinflammatory cytokines. CCR1 is a CC chemokine receptor with
high affinity for RANTES. TNF-α induces the expression of RANTES,
which in turn, stimulates recruitment of macrophages into the
endometriotic tissues. Angiogenic proinflammatory cytokines, leptin
and IL-8 are potentially involved in the pathophysiology of
endometriosis (3). Leptin produced
by the adipose tissue regulates innate and adaptive immune
responses and inflammation. IL-8 has been identified as a
chemotactic factor for leukocytes and is also produced following
inflammation. The levels of leptin and IL-8 are increased in
endometriosis, reflecting inflammation and dysregulated
immunomodulation (3).
Angiopoietins, ligands of the endothelial TEK (Tie2) tyrosine
kinase receptor, have been associated with angiogenesis (9). The Tie2-expressing macrophages
regulate angiogenesis and lymphangiogenesis to maintain the
viability of newly-formed vessels in endometriosis (9).
These data support the involvement of a chronic
inflammatory state in endometriotic cells growing in the
extra-uterine environment. Cytokines and growth factors may be
significant in endometriosis-associated inflammation (3,10,15).
Principal molecular factors of inflammation-associated
angiogenesis, lymphangiogenesis and neurogenesis should be
identified to develop novel therapeutic strategies for this
disorder.
4. Role of initial infection
Application of advances in genomic and proteomic
technologies has provided molecular insights into endometriosis.
Estrogen stimulates proliferation of endometrial and endometriotic
cells. In addition to estrogen, the proliferation of an
endometriotic lesion is regulated by the innate immune system.
Innate immunity is used as a first defense against pathogens. A
microbial infection of the upper genital tract may be critical for
the initiation of chronic pelvic inflammation. Although the immune
system is able to reject harmful pathogens, commensal microbes have
coexisted with cells at the cell surfaces in a symbiosis (16). Disturbances in the maintenance of
endometrial homeostasis and regulation of the host defense against
bacterial infection lead to a break in the endometrial barrier
function in genetically-susceptible hosts. Spontaneous
contamination of Escherichia coli in menstrual blood and
peritoneal fluid may promote Toll-like receptor (TLR) 4-mediated
growth of endometrial tissue originating from retrograde
menstruation (17). The TLR system
responds immediately to infectious agents. The proinflammatory
innate immune response leads to the activation of the slower
adaptive immune system. It has also been reported that the TLR4
A896G polymorphism (rs4986790) is a functional polymorphism
resulting in peritoneal inflammation (18). The initial development of
endometriosis, e.g., adhesion and growth of ectopic endometrial
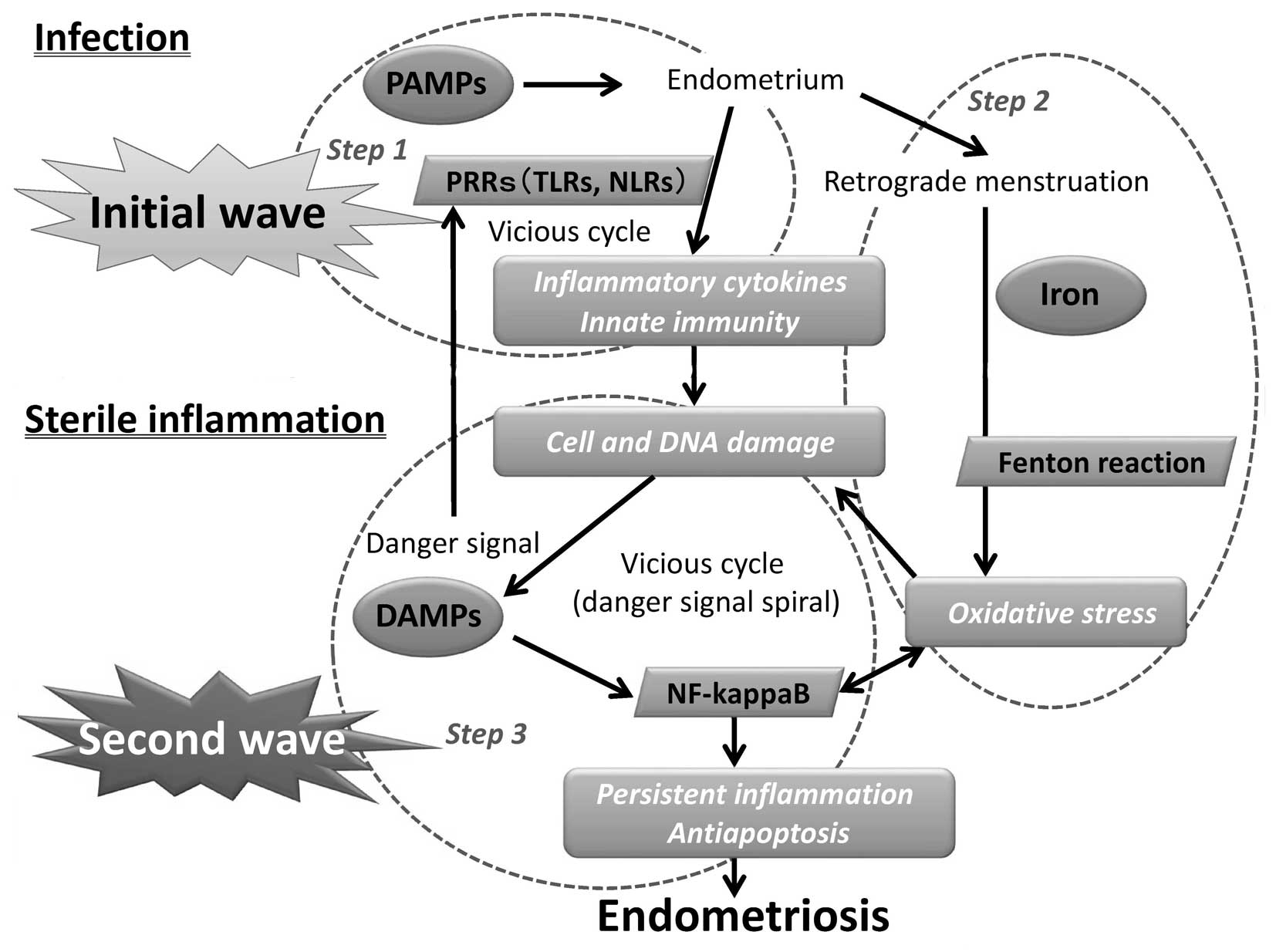
tissue, may be characteristically non-sterile (Fig. 1, Step 1).
Host pathogen recognition receptors (PRRs) recognize
microbial structures referred to as pathogen-associated molecular
patterns (PAMPs) (16). TLRs are
the first class of cell PRRs (16,19).
These PAMPs include lipopolysaccharide (LPS) and other components
such as flagellin, a bioactive TLR5 ligand (16). PAMPs trigger intracellular
signaling events, including ion influxes, cytosolic Ca2+
accumulation, production of reactive oxygen species (ROS) and
phosphorylation of specific proteins, and their signaling pathways
are involved in innate immunity through activation of TLRs. TLR
activation induces key inflammatory mechanisms such as nuclear
transcription factor-κB (NF-κB) activation and the synthesis of
IL-1β mRNA (19). A complex
network of pathways is targeted by immunosuppressive cytokines such
as IL-10 and transforming growth factor (TGF)-β.
TLR-mediated inflammation possibly occurs through
the exogenous PAMPs as well as the endogenous danger-associated
molecular pattern (DAMP) ligands (2). Nucleotide oligomerization domain-like
receptors (NLRs) and, to a lesser degree, the retinoic
acid-inducible gene I-like receptors are intracellular sensors
(16). Activation of PRR signaling
via members of the TLR and NLRs families initiates inflammatory
defense mechanisms that are required to protect the host. NLRs
respond to a variety of pathogen and intracellular danger signals
(20) and induce the transcription
of proinflammatory cytokines via the activation of NF-κB (19). NLRC5, expressed through the signal
transducers and activators of the transcription 1-mediated
signaling pathway, has been identified as a regulator of NF-κB,
type I interferon (IFN) and inflammasome signaling pathways (see
section 10) (20). NLRs also
trigger mitogen-activated protein kinase signaling pathways and
control the activation of inflammatory caspases and subsequent
activation of proinflammatory cytokines (16). Activation of caspase-1 is essential
for the processing of pro-IL-1β and pro-IL-18 (16). In the absence of the
anti-inflammatory feedback signals, physiological defense
mechanisms may turn into pathological responses.
Gene expression profiling studies may reveal factors
that explain variability in the development and progression of
endometriosis. Notably, TLR activation in eutopic endometrium may
be involved in the initial inflammatory response. The increased
expression levels of several factors, including inflammatory
cytokines and innate immunity, may be involved in the process of
TLR-dependent inflammation (2).
Khan et al studied the role of bacterial LPS and TLR4 in
endometriosis, suggesting the association between bacterial
infection and endometriotic proliferation (17). Bacterial LPS may have an initial
role in the development of endometriosis.
5. Role of subsequent sterile
inflammation
Inflammation is part of the non-specific immune
response. In certain cases, the inflammatory process becomes
continuous and the early endometriotic lesion may develop
subsequently. At least two waves of inflammation-induced gene
expression occurs in endometriotic tissues. The first wave includes
conserved infection and immune-associated early genes. The
second-wave genes encode oxidative stress and sterile inflammatory
factors required for proper regeneration. Therefore, the initial
wave of the LPS-dependent TLR activation in modulating immune
responses would be followed by a second wave of the mechanisms
responsible for enhancing the oxidative stress and sterile
inflammation.
6. Oxidative stress
Although the mechanism by which oxidative stress
induces inflammation remains unclear, one prominent and early
mediator for inflammation in endometriosis may be free iron
(21). Redox active metals such as
iron possess the ability to produce reactive radicals, for example
the superoxide anion radical. Iron-induced inflammation is mediated
through ROS production by driving the Fenton reaction (Fig. 1, Step 2). Excessive production of
ROS is secondary to peritoneal influx of pro-oxidants such as iron
during retrograde menstruation (22). Iron is able to induce oxidative
stress in endometriosis during the secondary persistent
inflammatory response (23). ROS
induce cellular and DNA damage and increased proinflammatory gene
expression through NF-κB activation (22). In mice deficient for the NF-κB
family, endometriosis development was reduced (24). Iron also induces IL-1β expression,
and the inhibition of IL-1β prevents development of endometriosis.
These data allow us to hypothesize that the iron overload affects
numerous mechanisms involved in the development of endometriosis
during sterile inflammation. Iron and NF-κB appear to be connected,
making these signaling pathways an attractive target for the future
treatment of this disease (23).
Sterile inflammation and oxidative stress by redox-active iron may
lead to a circulus vitiosus, resulting in the persistence of
inflammatory processes and the development of endometriosis when
the process becomes chronic (2).
7. DAMPs
TLRs are a critical environmental interface that
regulate infection and sterile injury by responding to a variety of
microbial and endogenous ligands (25). In addition to their response to
various exogenous PAMPs, TLRs recognize a wide range of endogenous
DAMPs, including high-mobility group box protein 1 (HMGB1), heat
shock protein 70 (HSP70), adenosine-5′-triphosphate (ATP), DNA,
urate crystals, asbestos, silica, aluminum hydroxide, S100,
neutrophil elastase, amyloid-β and soluble extracellular matrix
components, such as biglycan, hyaluronan, versican, fibrinogen,
heparan sulfate fragments and fibronectin extra domain A (2,26,27).
Intracellular contents released from damaged cells into the
extracellular space serve as DAMPs or alarmins that trigger
inflammation (Fig. 1, Step 3).
Infection and damage cause inflammation as PAMPs as well as DAMPs
are immunogenic. There is increasing evidence that this sterile
inflammatory response mediated through DAMPs is a key determinant
of further development of inflammation-associated diseases such as
atherosclerosis, gout, type II diabetes and pancreatitis (26). A number of DAMPS, including HMGB1,
DNA, ATP and HSP70, have been shown to be involved in endometriosis
(2). The TLR-mediated inflammation
persistently occurs possibly through the endogenous ligands.
Persistent sterile inflammatory insults and DAMPs released from
damaged cells are bidirectional, and they activate immunity and
further propagate tissue damage. Specific DAMP receptors, including
TLR4, TLR9 and P2X7, as well as downstream DAMP-sensing components,
including NLRP3, caspase-1, IL-1β, IL-18 and IL-1 receptor, are
required for full experimental endometriosis (26). These DAMP-mediated pathways may
provide novel therapeutic targets.
Mesothelial cells undergo injury and repair
themselves following retrograde menstruation-associated
inflammation. Peritoneal endometriosis is the result of ectopic
implantation and growth of endometrial tissue in women with a
deficient immune system, which is not able to defend against
regurgitated endometrial cells (28). Specific factors, including TNF-α,
α-enolase and hemoglobin, in menstrual effluent induce epithelial
to mesenchymal transitions (EMT) in mesothelial cells, resulting in
cell retraction and exposure of the submesothelial extracellular
matrix (ECM) (28). Structural
damage of the mesothelial layer attributable to the menstrual
effluent may facilitate regurgitated endometriotic cell adhesion
and growth. Fibrosis around the endometriotic foci is the
consequence of recurrent cell damage. Endometriotic cell migration
and the concomitant degradation of ECM are two essential steps in
the invasive process. ECM components such as biglycan, hyaluronan,
versican, fibrinogen, heparan sulfate fragments and fibronectin
extra domain A have now been demonstrated to act as signaling
molecules (29). They act as
fundamental danger signals or DAMPs, signifying tissue injury, and
they also potentiate the immune system (29). The expression of certain danger
signal proteins involved in the organization of the cytoskeleton,
signal transduction, regulation of the redox state and production
of ATP, has been demonstrated to be altered during the EMT
process.
ATP is a ubiquitous molecule in every cell and is
released into the extracellular milieu following tissue injury.
Extracellular ATP is a host-derived small-danger-molecule. ATP
synthase β subunit has been demonstrated to be differentially
expressed between women with and without endometriosis. ATP
synthase β-chain has been identified as phosphorylated and
activated in endometriosis. Thus, extracellular ATP released from
ectopic endometrial cells may constitute a major endogenous danger
signal that leads to IL-1β expression and fibrosis.
Although DAMPs such as endogenous DNA and nuclear
HMGB1 have been shown to be critical in sterile inflammation, the
role of nuclear histone proteins has not yet been investigated in
endometriosis. Numerous studies (26) have established that certain
non-histone proteins such as HMGB1 are released extracellularly and
induce innate/inflammatory and adaptive immune responses as a DAMP.
DNA binds histones to form nucleosomes. Ischemia/reperfusion (I/R)
injury of several organs enhances histone expression and selected
proinflammatory/profibrotic genes. Injection of exogenous histones
exacerbates injury through the TLR9 and MyD88-mediated cytotoxic
effects, and histone neutralization protects against injury
(30). Histones function as a DAMP
following I/R injury (30). We
hypothesize that histones may be a novel class of DAMP molecules
and serve as an activator of innate immunity during sterile
inflammation in endometriosis. These findings require additional
preclinical studies on DAMPs in endometriosis.
8. Antiapoptosis
The release of DAMPs from endometriotic-damaged
cells may engender a second wave of tissue damage during acute
processes and, if chronic, potentially trigger antiapoptotic
processes. Increased levels of cell proliferation and reduced
levels of apoptosis emerge as major mechanisms responsible for the
development of endometriosis. Attenuated susceptibility to
apoptosis may contribute to the pathogenesis of endometriosis
(31,32). Marked cell proliferation has been
attributed to a change in the expression levels of proteins such as
B-cell lymphoma-2 (Bcl-2) and Bcl-2-associated X proteins (Bcl-XL,
Bcl-XS). A number of other genes are associated with apoptosis in
endometriosis, including defender against cell death-1, P53,
survivin, caspase-1, calpain, proliferating cell nuclear antigen
and PGE2 synthesis genes (31,33–35).
PGE2 promotes cell survival through the EP2/EP4
receptor-dependent activation of ERK1/2, Akt, NF-κB, and β-catenin
signaling pathways, suppressing proapoptotic proteins (Bax and Bad)
and enhancing antiapoptotic proteins (Bcl-2/Bcl-XL) (33). The PGE2 signaling
components are abundantly expressed in endometriosis (33). COX-2 induces PGE2
expression through the cAMP/ERK pathways by activating EP2 and EP4
receptors. COX-2 also reduces apoptosis-associated caspase-3
expression (36). Selective COX-2
inhibition induces regression of endometrial grafts by stimulation
of apoptosis (36). Decreased
apoptosis is also associated with a pathway involving HOXA10,
calpain5 and caspase (37).
Expression levels of calpain5, a target of HOXA10 transcriptional
regulation, are reduced in endometriosis which is likely to be the
result of reduced HOXA10 expression levels (37).
9. Cytokines
Commonly upregulated cytokines in endometriosis
include IL-1β, TNF-α, IFN-γ, IL-18, IL-10 and TGF-β. IL-1β and
IL-18 are two powerful proinflammatory cytokines with pleiotropic
activities. IL-18 increases natural killer cell activity, and
stimulates IFN-γ production in T-helper type I cells. IL-18 is
involved in various immune diseases and induces COX-2 in peritoneal
monocytes (38). Although there
are a number of studies on the association between IL-18 and
endometriosis, their results appear to be contradictory (38).
Upregulation of IL-10 expression levels was also
observed in ovarian endometrioma when compared with a control group
(39). The biological actions of
IL-10 are inhibitory, including the inactivation of macrophages and
inhibition of proinflammatory cytokines, demonstrating that IL-10
functions as an immunoregulatory cytokine.
Host-derived TGF-β1 is a multifunctional cytokine
that is upregulated in endometriosis (40). TGF-β1 induces the secretion of IL-6
from endometrial stromal cells through an increase in
protease-activated receptor (PAR)-2 expression levels (41). TGF-β1 may enhance the development
of endometriosis (40).
NF-κB is involved in the transduction of
proinflammatory signals, upregulation of adhesion molecules and
suppression of apoptosis, thus enhancing the initial development of
endometriosis (43). NF-κB, as a
series of cascade events of an autocrine nature, regulates the
expression of proinflammatory cytokines and activation of NF-κB
itself, leading to the maintenance of autocrine self-amplifying
cycles (Fig. 1).
10. Inflammasome
Production of pro-IL-1β, an inactive precursor, is
induced by the activation of NF-κB in response to inflammatory
stimuli. In the generation of IL-1β, additional proteolytic
cleavage by cytosolic protein complexes termed ‘inflammasomes’ is
required for the activation of pro-IL-1β (16). Inflammasomes assemble in response
to endogenous danger signals. Caspase-1 is auto-activated within
inflammasomes that include NLR proteins, the adapter
apoptosis-associated speck-like protein containing a C-terminal
caspase recruitment domain and pro-caspase-1 (16). Accumulating evidence indicates that
several sterile inflammatory responses triggered by oxidative
stress or cell damage are mediated by inflammasomes. Subsequent
activation of inflammasomes leads to NF-κB activation and IL-1β
production, resulting in inflammatory responses. Therefore, we
hypothesize that the inflammasome is an initial sensor for danger
signals in endometriosis. However, regulation of the inflammasome
formation in endometriosis is poorly understood.
11. Novel treatment strategies
This review allows us to hypothesize that several
promising therapies that target DAMPs in antiapoptosis, sterile
inflammation and oxidative stress should be developed. Novel
treatments from genomic investigations may be identified due to the
accumulation of a therapeutic signature for endometriosis. A
high-throughput screening assay is capable of revealing a number of
compounds that specifically inhibit a target molecule activity.
12. Antiapoptosis as a target
Antiapoptosis may be involved in the pathophysiology
of endometriosis (32).
Accordingly, apoptosis of the nascent endometriotic lesion has
become an attractive target to use to inhibit the development of
endometriosis. An array of promising therapeutic agents, including
BAY 11–7085, β-hydroxyisovalerylshikonin (β-HIVS), bufalin and
rapamycin, have entered various stages of preclinical development
for endometriosis. Bay 11–7085 is reported to inhibit NF-κB
activation and this pharmacological inhibitor induces apoptosis
through the suppression of antiapoptotic proteins such as
caspase-3, -8, and -9 (43).
Extract of the roots of Onosma paniculata, β-HIVS, induces
apoptosis in cancer cells (44).
β-HIVS is an ATP non-competitive inhibitor of protein-tyrosine
kinases. It induces G0/G1 phase cell-cycle arrest and apoptosis via
the downregulation of Bcl-2 expression with the activation of
caspase-3, -8 and -9, leading to the inhibition of endometriotic
cell proliferation (44). Bufalin,
an apoptosis-inducing agent, is a major digoxin-like compound
isolated from the skin and parotid venom glands of toads (35). Rapamycin is a drug with antifungal,
immunosuppressant, antiapoptotic and antiangiogenic effects
(45). The apoptotic activities of
these agents may be sufficient for additional clinical
investigation on the development of endometriosis.
13. Inhibitors of DAMPs
Molecules of DAMPs are stimulated under conditions
of stress, such as injury, infection, ischemia or oxidative stress.
They induce immune tolerance or various chronic diseases such as
arthritis, atherosclerosis, cancer, systemic lupus erythematosus
and possibly endometriosis. These data suggest a role for DAMP
molecules in the enhanced and persistent activation of
endometriosis. HMGB1 functions as a major DAMP and inhibits
apoptosis, promoting Bcl-2 expression and inhibiting Bax
translocation. Gabexate mesilate, a synthetic protease inhibitor
with a degree of anti-inflammatory action, inhibits PAI-1 and
PAR-2, thereby indirectly inhibiting HMGB1. HSP70 is a molecular
chaperone that protects cells from damage in response to various
stress stimuli (46). Preclinical
studies suggest that the use of specific inhibitors for HSP70 is an
effective strategy to treat Alzheimer’s disease. Statin, a HMG-CoA
reductase inhibitor, also inhibits the expression of HSP70 through
the inactivation of NF-κB. The present findings indicate that DAMP
molecules released from damaged endometriotic cells may serve as a
link between the initial cell damage induced by bacterial infection
and the subsequent activation of innate immune cells by sterile
inflammation and oxidative stress. Direct or indirect inhibitors of
DAMP molecules may be a potential therapeutic target to prevent or
minimize endometriosis.
14. Iron chelators
Hemolysis occurring during the development of
endometriosis results in high levels of free iron. Redox-active
iron participates in the generation of highly toxic free radicals
and oxidatively leads to cell and DNA damage. A previous study
demonstrated the molecular basis for the initial inflammatory
response following iron-induced oxidative stress and suggested that
iron is a potential novel therapeutic target for preventing the
development of endometriosis (21). Iron chelator treatment may be
beneficial in endometriosis via the suppression of cell
proliferation (47).
15. Conclusion
Based on epidemiological and experimental data, it
is possible to a certain extent to hypothesize that retrograde
menstruation promotes implantation and the development of
endometrial tissue, in accordance with Sampson’s hypothesis. The
mechanism of the initial development and subsequent progression of
endometriosis is largely unknown. There may be at least two waves
of development of endometriosis; the first wave due to bacterial
infection and the second wave from sterile inflammation (Fig. 1). The initial stage of a
non-specific bacterial infection, such as E. coli, is
believed to occur in the uterine endometrium and peritoneal fluid
(Step 1). Oxidative stress is secondary to the influx of iron
during retrograde menstruation (Step 2). Redox-active
iron-dependent oxidative stress and PAMP/DAMP-receptor signaling
(Step 3) provide the combination of antiapoptosis and persistent
inflammation. The release of DAMPs from damaged cells may engender
a second big wave of tissue damage during chronic processes and
potentially trigger sterile inflammation-induced antiapoptotic and
oxidative stress processes. In conclusion, initial infection and
subsequent sterile inflammation are closely associated with the
development of endometriosis.
Acknowledgements
This study was supported by a Grant-in-Aid for
Scientific Research to H.K. from the Ministry of Education,
Culture, Sports, Science and Technology (Tokyo, Japan).
References
|
1
|
Agic A, Djalali S, Diedrich K and Hornung
D: Apoptosis in endometriosis. Gynecol Obstet Invest. 68:217–223.
2009. View Article : Google Scholar
|
|
2
|
Kajihara H, Yamada Y, Kanayama S, Furukawa
N, Noguchi T, Haruta S, Yoshida S, Sado T, Oi H and Kobayashi H:
New insights into the pathophysiology of endometriosis: from
chronic inflammation to danger signal. Gynecol Endocrinol.
27:73–79. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Malhotra N, Karmakar D, Tripathi V, Luthra
K and Kumar S: Correlation of angiogenic cytokines-leptin and IL-8
in stage, type and presentation of endometriosis. Gynecol
Endocrinol. 28:224–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Khoufache K, Michaud N, Harir N, Kibangou
Bondza P and Akoum A: Anomalies in the inflammatory response in
endometriosis and possible consequences: a review. Minerva
Endocrinol. 37:75–92. 2012.PubMed/NCBI
|
|
5
|
Kirchhoff D, Kaulfuss S, Fuhrmann U,
Maurer M and Zollner TM: Mast cells in endometriosis: guilty or
innocent bystanders? Expert Opin Ther Targets. 16:237–241. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Menzies FM, Shepherd MC, Nibbs RJ and
Nelson SM: The role of mast cells and their mediators in
reproduction, pregnancy and labour. Hum Reprod Update. 17:383–396.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Asante A and Taylor RN: Endometriosis: the
role of neuroangiogenesis. Annu Rev Physiol. 73:163–182. 2011.
View Article : Google Scholar
|
|
8
|
D’Cruz OJ and Uckun FM: Targeting mast
cells in endometriosis with janus kinase 3 inhibitor, JANEX-1. Am J
Reprod Immunol. 58:75–97. 2007.PubMed/NCBI
|
|
9
|
Capobianco A, Monno A, Cottone L, Venneri
MA, Biziato D, Di Puppo F, Ferrari S, De Palma M, Manfredi AA and
Rovere-Querini P: Proangiogenic Tie2(+) macrophages infiltrate
human and murine endometriotic lesions and dictate their growth in
a mouse model of the disease. Am J Pathol. 179:2651–2659. 2011.
|
|
10
|
Uz YH, Murk W, Bozkurt I, Kizilay G, Arici
A and Kayisli UA: Increased c-Jun N-terminal kinase activation in
human endometriotic endothelial cells. Histochem Cell Biol.
135:83–91. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Milewski Ł, Dziunycz P, Barcz E, Radomski
D, Roszkowski PI, Korczak-Kowalska G, Kamiński P and Malejczyk J:
Increased levels of human neutrophil peptides 1, 2, and 3 in
peritoneal fluid of patients with endometriosis: association with
neutrophils, T cells and IL-8. J Reprod Immunol. 91:64–70.
2011.PubMed/NCBI
|
|
12
|
Carli C, Metz CN, Al-Abed Y, Naccache PH
and Akoum A: Up-regulation of cyclooxygenase-2 expression and
prostaglandin E2 production in human endometriotic cells by
macrophage migration inhibitory factor: involvement of novel kinase
signaling pathways. Endocrinology. 150:3128–3137. 2009. View Article : Google Scholar
|
|
13
|
Agic A, Xu H, Finas D, Banz C, Diedrich K
and Hornung D: Is endometriosis associated with systemic
subclinical inflammation? Gynecol Obstet Invest. 62:139–147. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lebovic DI, Chao VA and Taylor RN:
Peritoneal macrophages induce RANTES (regulated on activation,
normal T cell expressed and secreted) chemokine gene transcription
in endometrial stromal cells. J Clin Endocrinol Metab.
89:1397–1401. 2004. View Article : Google Scholar
|
|
15
|
Michaud N, Al-Akoum M, Gagnon G, Girard K,
Blanchet P, Rousseau JA and Akoum A: Decreased concentrations of
soluble interleukin-1 receptor accessory protein levels in the
peritoneal fluid of women with endometriosis. J Reprod Immunol.
92:68–73. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Franchi L, Warner N, Viani K and Nuñez G:
Function of Nod-like receptors in microbial recognition and host
defense. Immunol Rev. 227:106–128. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Khan KN, Kitajima M, Hiraki K, Yamaguchi
N, Katamine S, Matsuyama T, Nakashima M, Fujishita A, Ishimaru T
and Masuzaki H: Escherichia coli contamination of menstrual
blood and effect of bacterial endotoxin on endometriosis. Fertil
Steril. 94:2860–2863. 2010. View Article : Google Scholar
|
|
18
|
Latha M, Vaidya S, Movva S, Chava S,
Govindan S, Govatati S, Banoori M, Hasan Q and Kodati VL: Molecular
pathogenesis of endometriosis; Toll-like receptor-4 A896G (D299G)
polymorphism: a novel explanation. Genet Test Mol Biomarkers.
15:181–184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brown J, Wang H, Hajishengallis GN and
Martin M: TLR-signaling networks: an integration of adaptor
molecules, kinases, and cross-talk. J Dent Res. 90:417–427. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tong Y, Cui J, Li Q, Zou J, Wang HY and
Wang RF: Enhanced TLR-induced NF-κB signaling and type I interferon
responses in NLRC5 deficient mice. Cell Res. 22:822–835. 2012.
|
|
21
|
Kobayashi H, Yamada Y, Kanayama S,
Furukawa N, Noguchi T, Haruta S, Yoshida S, Sakata M, Sado T and Oi
H: The role of iron in the pathogenesis of endometriosis. Gynecol
Endocrinol. 25:39–52. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lousse JC, Van Langendonckt A, Defrere S,
Ramos RG, Colette S and Donnez J: Peritoneal endometriosis is an
inflammatory disease. Front Biosci (Elite Ed). 4:23–40. 2012.
View Article : Google Scholar
|
|
23
|
Defrère S, González-Ramos R, Lousse JC,
Colette S, Donnez O, Donnez J and Van Langendonckt A: Insights into
iron and nuclear factor-kappa B (NF-kappaB) involvement in chronic
inflammatory processes in peritoneal endometriosis. Histol
Histopathol. 26:1083–1092. 2011.PubMed/NCBI
|
|
24
|
Lu Y, Sun Q, Zheng Y, Liu X, Geng JG and
Guo SW: The role of nuclear factor-kappa-B p50 subunit in the
development of endometriosis. Front Biosci (Elite Ed). 591–603.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mollen KP, Anand RJ, Tsung A, Prince JM,
Levy RM and Billiar TR: Emerging paradigm: toll-like receptor
4-sentinel for the detection of tissue damage. Shock. 26:430–437.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hoque R, Malik AF, Gorelick F and Mehal
WZ: Sterile inflammatory response in acute pancreatitis. Pancreas.
41:353–357. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zeiser R, Penack O, Holler E and Idzko M:
Danger signals activating innate immunity in graft-versus-host
disease. J Mol Med (Berl). 89:833–845. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Demir AY, Demol H, Puype M, de Goeij AF,
Dunselman GA, Herrler A, Evers JL, Vandekerckhove J and Groothuis
PG: Proteome analysis of human mesothelial cells during epithelial
to mesenchymal transitions induced by shed menstrual effluent.
Proteomics. 4:2608–2623. 2004. View Article : Google Scholar
|
|
29
|
Schaefer L: Extracellular matrix
molecules: endogenous danger signals as new drug targets in kidney
diseases. Curr Opin Pharmacol. 10:185–190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang H, Evankovich J, Yan W, Nace G,
Zhang L, Ross M, Liao X, Billiar T, Xu J, Esmon CT and Tsung A:
Endogenous histones function as alarmins in sterile inflammatory
liver injury through Toll-like receptor 9 in mice. Hepatology.
54:999–1008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Watanabe A, Taniguchi F, Izawa M, Suou K,
Uegaki T, Takai E, Terakawa N and Harada T: The role of survivin in
the resistance of endometriotic stromal cells to drug-induced
apoptosis. Hum Reprod. 24:3172–3179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Izawa M, Harada T, Deura I, Taniguchi F,
Iwabe T and Terakawa N: Drug-induced apoptosis was markedly
attenuated in endometriotic stromal cells. Hum Reprod. 21:600–604.
2006. View Article : Google Scholar
|
|
33
|
Banu SK, Lee J, Speights VO Jr,
Starzinski-Powitz A and Arosh JA: Selective inhibition of
prostaglandin E2 receptors EP2 and EP4 induces apoptosis of human
endometriotic cells through suppression of ERK1/2, AKT, NFkappaB,
and beta-catenin pathways and activation of intrinsic apoptotic
mechanisms. Mol Endocrinol. 23:1291–1305. 2009. View Article : Google Scholar
|
|
34
|
Braun DP, Ding J, Shaheen F, Willey JC,
Rana N and Dmowski WP: Quantitative expression of
apoptosis-regulating genes in endometrium from women with and
without endometriosis. Fertil Steril. 87:263–268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nasu K, Nishida M, Ueda T, Takai N, Bing
S, Narahara H and Miyakawa I: Bufalin induces apoptosis and the
G0/G1 cell cycle arrest of endometriotic stromal cells: a promising
agent for the treatment of endometriosis. Mol Hum Reprod.
11:817–823. 2005. View Article : Google Scholar
|
|
36
|
Laschke MW, Elitzsch A, Scheuer C, Vollmar
B and Menger MD: Selective cyclo-oxygenase-2 inhibition induces
regression of autologous endometrial grafts by down-regulation of
vascular endothelial growth factor-mediated angiogenesis and
stimulation of caspase-3-dependent apoptosis. Fertil Steril.
87:163–171. 2007. View Article : Google Scholar
|
|
37
|
Penna I, Du H, Ferriani R and Taylor HS:
Calpain5 expression is decreased in endometriosis and regulated by
HOXA10 in human endometrial cells. Mol Hum Reprod. 14:613–618.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Oku H, Tsuji Y, Kashiwamura SI, Adachi S,
Kubota A, Okamura H and Koyama K: Role of IL-18 in pathogenesis of
endometriosis. Hum Reprod. 19:709–714. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Podgaec S, Dias JA Junior, Chapron C,
Oliveira RM, Baracat EC and Abrão MS: Th1 and Th2 immune responses
related to pelvic endometriosis. Rev Assoc Med Bras. 56:92–98.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hull ML, Johan MZ, Hodge WL, Robertson SA
and Ingman WV: Host-derived TGFB1 deficiency suppresses lesion
development in a mouse model of endometriosis. Am J Pathol.
180:880–887. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Saito A, Osuga Y, Yoshino O, Takamura M,
Hirata T, Hirota Y, Koga K, Harada M, Takemura Y, Yano T and
Taketani Y: TGF-β1 induces proteinase-activated receptor 2 (PAR2)
expression in endometriotic stromal cells and stimulates PAR2
activation-induced secretion of IL-6. Hum Reprod. 26:1892–1898.
2011.
|
|
42
|
González-Ramos R, Van Langendonckt A,
Defrère S, Lousse JC, Mettlen M, Guillet A and Donnez J: Agents
blocking the nuclear factor-kappaB pathway are effective inhibitors
of endometriosis in an in vivo experimental model. Gynecol Obstet
Invest. 65:174–186. 2008.PubMed/NCBI
|
|
43
|
Nasu K, Nishida M, Ueda T, Yuge A, Takai N
and Narahara H: Application of the nuclear factor-kappaB inhibitor
BAY 11–7085 for the treatment of endometriosis: an in vitro study.
Am J Physiol Endocrinol Metab. 293:E16–E23. 2007.
|
|
44
|
Nishida M, Nasu K, Ueda T, Yuge A, Takai N
and Narahara H: Beta-hydroxyisovalerylshikonin induces apoptosis
and G0/G1 cell-cycle arrest of endometriotic stromal cells: a
preliminary in vitro study. Hum Reprod. 21:2850–2856. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Laschke MW, Elitzsch A, Scheuer C,
Holstein JH, Vollmar B and Menger MD: Rapamycin induces regression
of endometriotic lesions by inhibiting neovascularization and cell
proliferation. Br J Pharmacol. 149:137–144. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Khan KN, Kitajima M, Imamura T, Hiraki K,
Fujishita A, Sekine I, Ishimaru T and Masuzaki H: Toll-like
receptor 4-mediated growth of endometriosis by human heat-shock
protein 70. Hum Reprod. 23:2210–2219. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Defrère S, Van Langendonckt A, Vaesen S,
Jouret M, González Ramos R, Gonzalez D and Donnez J: Iron overload
enhances epithelial cell proliferation in endometriotic lesions
induced in a murine model. Hum Reprod. 21:2810–2816.
2006.PubMed/NCBI
|