1. Introduction
Limb-girdle muscular dystrophies (LGMD) are a group
of muscular dystrophies, that until the late 1980s were identified
in patients by ‘diagnosis by exclusion’. Revolutionary advances in
molecular biology in the last several decades have allowed the
scientific community to understand and recognize this disease more
clearly. Currently, there are >25 LGMD types that have been
linked to specific gene loci, and they are now estimated to
constitute one third of all Duchenne muscular dystrophy cases
(1).
The review is constructed to cover LGMD from a
variety of viewpoints and is established on the authors’ own
experience and investigations, as well as inclusive MEDLINE
searches on the topics of ‘limb-girdle muscular dystrophies’,
‘review’, ‘genotype-phenotype correlations’, ‘prevention’ and
‘surveillance’. In this review, we focused on peer-reviewed studies
(in English) published in key scientific journals from 1995 to
date, combined with several historical articles. All available
articles were reviewed in depth. In consideration of the summary
and usefulness for clinicians in the field of myology and for
future citation, the information and references were summarized
into tables.
2. Historical background
The term LGMD was, among hereditary muscle
disorders, one of the most difficult to establish as a clinical
entity. Early definitions were described by Erb where he designated
a type of juvenile, scapulohumeral progressive muscular atrophy
known as ‘a juvenile form of progressive muscular dystrophy’. In
1891, Erb (2) proposed to include
with his cases the previous observations of Leyden (3) and Mobius (4). The theory was generally well
accepted. Bell (5) was the first
to differentiate this type of dystrophy from X-linked Duchenne
muscular dystrophy and from autosomal dominant facioscapulohumeral
muscular dystrophy. In their archetypal paper, Walton and Nattrass
(6) first devised the name
‘limb-girdle muscular dystrophies’ to comprise cases of both sexes,
beginning usually within the first three decades, with major
involvement of scapular, pelvic girdle and trunk muscles, with
sparing of facial muscles and infrequent pseudo hypertrophy,
moderately severe progression and usually an autosomal recessive
mode of inheritance.
With the development of physiological and
histopathological means to assess muscular disorders, it rapidly
seemed that a number of patients considered to be suffering from
LGMD were, in fact, affected by other conditions, including spinal
muscular atrophies, congenital myopathies or metabolic disorders.
Hence, the clinicopathological consistency of this suggested entity
was, nevertheless, again promptly disputed.
In the early development of immunostaining methods
in the late 1980s, a precise distinction between LGMD and other
conditions such as Becker’s dystrophy characterized by dystrophin
abnormality was established (7).
In 1995, the European Neuromuscular Centre Workshop
established more precise criteria for the diagnosis and
classification of LGMD. More specifically, different subtypes of
LGMD were grouped according to their genetic characteristics
(8).
The abbreviation of the autosomal-dominant type is
now LGMD1, whereas autosomal-recessive types are LGMD2. Each
separate gene locus has a unique classification (Table I) (9–35).
 | Table ILGMD classification. |
Table I
LGMD classification.
| Form | Locus | Gene |
Proteinopathies | Key references |
|---|
| Autosomal
dominant |
| LGMD1A | 5q31 | MYOTM |
Myotilinopathies | (9) |
| LGMD1B | 1q11-q21 | LMNA | Lamin A/C
opathies | (10) |
| LGMD1C | 3p25 | CAV3 |
Caveolinopathies | (11) |
| LGMD1D | 2q35 | DES | Desminopathies | (12) |
| LGMD1E | 7q36 | DNAJB6 | HSP40/DNAJ | (13,14) |
| LGMD1F | 7q32.1-q32.2 | - | - | (15) |
| LGMD1G | 4p21 | - | - | (16) |
| LGMD1H | 3p23-p25 | - | - | (17) |
| Autosomal
recessive |
| LGMD2A | 15q15.1 | CAPN3 | Calpainopathy | (18) |
| LGMD2B | 2p13 | DYSF |
Dysferlinopathies | (19) |
| LGMD2Ca | 13q12 | SGCG |
γ-sarcoglycanopathy | (20) |
| LGMD2Da | 17q12-q21.33 | SGCA |
α-sarcoglycanopathy | (21) |
| LGMD2Ea | 4q12 | SGCB |
β-sarcoglycanopathy | (22) |
| LGMD2Fa | 5q33 | SGCD |
δ-sarcoglycanopathy | (23) |
| LGMD2G | 17q12 | TCAP |
Telethoninopathy | (24) |
| LGMD2H | 9q31-q34 | TRIM32 | E3-ubiquitin
ligase | (25) |
| LGMD2Ib | 19q13 | FKRP | Fukutin-related
protein | (26) |
| LGMD2J | 2q31 | TTN | Titinopathies | (27) |
| LGMD2Kb | 9q34.1 | POMT1 | POMT1 | (28) |
| LGMD2L | 11p14.3 | ANO5 |
Anoctaminopathies | (29) |
| LGMD2Mb | 9p3 | FKTN |
Fukutinopathies | (30) |
| LGMD2Nb | 14q10-q24 | POMT2 | POMT2 | (31) |
| LGMD2Ob | 1p34-33 | POMGnT1 | POMGnT1 | (32) |
| LGMD2Pb | 3p21 | DAG1 | Dystroglycan | (33,34) |
| LGMD2Q | 8q24.3 | PLEC |
Plectinopathies | (35) |
As is evident from its long nosological history,
LGMD is not an homogeneous disease. LGMD can be considered an
‘umbrella’ term under which >24 gene defects have been
recognized, where single gene defects encode numerous phenotypes
and vice versa.
3. Mechanism of action
The mechanisms of action of LGMD-involved proteins
are diverse. Emphasis is moving away from the identification of
structural proteins and their implications in muscular dystrophies
towards investigating proteins involved in muscle fiber maintenance
in response to repeated injury (dysferlin, caveolin 3 and anoctamin
5), fiber remodeling under stressful conditions (calpain 3), and
post-translational modifications of proteins and the enzymes
involved in these processes (POMT1, POMT2, POMTGnT1). Despite the
advances in modern biochemical techniques, certain proteins still
exist without defined functions (Table II) (13,35–51).
| Table IILGMD subtype: Proteins and putative
functions. |
Table II
LGMD subtype: Proteins and putative
functions.
| Form | Protein | Site | Anticipated
function (ref.) |
|---|
| LGMD1A | Myotilin | Sarcomere | Z-disc structure
protection, anchorage of thin filaments to the Z-disc (36) |
| LGMD1B | Lamin A/C | Nuclear
membrane | Nuclear membrane
stabilization, cell signaling, differentiation (37) |
| LGMD1C | Caveolin 3 | Sarcolemma | Membrane
trafficking, signal transduction (38) |
| LGMD1D | Desmin | Sarcomere | Assembly and the
formation of the extra-sarcomeric cytoskeleton (39) |
| LGMD1E | HSP40/DNAJ | Ubiquitous | Protecting client
proteins from irreversible aggregation (13) |
| LGMD1F | - | - | |
| LGMD1G | - | - | |
| LGMD1H | - | - | |
| LGMD2A | Calpain3 | Cytosol,
sarcomere | Sarcomeric
remodeling; zygomatic and structural function (40,41) |
| LGMD2B | Dysferlin | Sarcolemma | Membrane repair and
vesicle trafficking (42) |
| LGMD2C | γ-sarcoglycan | Sarcolemma | Part of DGC,
involved in membrane integrity, cell signaling (43) |
| LGMD2D | α-sarcoglycan | Sarcolemma | Part of DGC,
involved in membrane integrity, cell signaling |
| LGMD2E | β-sarcoglycan | Sarcolemma | Part of DGC,
involved in membrane integrity, cell signaling |
| LGMD2F | δ-sarcoglycan | Sarcolemma | Part of DGC,
involved in membrane integrity, cell signaling |
| LGMD2G | Telethonin | Sarcomere | Sarcomeric
assembly, titin anchor (44) |
| LGMD2H | E3-ubiquitin
ligase | Cytosol | Involved in
ubiquitin-proteasome pathway (45) |
| LGMD2I | Fukutin-related
protein | Extracellular | Unknown,
glycosylation of α-dystroglycan (46) |
| LGMD2J | Titin | Sarcomere | Sarcomeric
scaffold, elasticity, force bearing mechanism, cell signaling
(47) |
| LGMD2K | POMT1 | Extracellular | Catalyze the first
step in O-mannosylation of α-DG (48) |
| LGMD2L | Anoctamin 5 | Sarcolemma | Calcium-activated
chloride channel function, reseal mechanism (29) |
| LGMD2M | Fukutin | Extracellular | Unknown, putative
phospholigand transferase (49) |
| LGMD2N | POMT2 | Extracellular | Catalyze the first
step in O-mannosylation of α-DG (48) |
| LGMD2O | POMGnT1 | Extracellular | Catalyze the second
step in O-mannosylation of α-DG (50) |
| LGMD2P | Dystroglycan | Sarcolemma | Connect
extracellular medium to intracellular scaffold (51) |
| LGMD2Q | Plectin | Sarcomere | Cytoskeleton system
linker (35) |
4. Pathophysiology
The majority of the developments in LGMD
therapeutics have derived from insight gained in animal model
investigations, created through the reversal of etiopathogenic
agents prompting the disease or by disrupting certain steps
believed to be downstream of the defect gene.
When reviewing all mutations in MYOT through the
Leiden muscular dystrophy page (http://www.dmd.nl), it was evident that no null
mutation has been reported to date in the MYOT gene, hence all
mutations are of missense type. This evidence supports the theory
that myotilin missense, but not nonsense, mutations are pathogenic
in humans and there must be other proteins, such as palladin and
myopalladin, which reimburse the structural and functional
properties of myotilin (52). This
hypothesis is further supported by the fact that the myotilin-null
mouse demonstrates normal development, histology and performance,
yet myotilin transgenic mice show dystrophic processes and double
transgenic mice report a more severe phenotype (52,53).
Authors hypothesize that genetic or pharmaceutical interference
with mutant myotilin translation, may serve as promising
therapeutic approaches in the treatment of LGMD. The LMNA gene
encodes the lamin A/C protein, which is important in its function
as a scaffold for nuclear lamina and as a vital component of cell
signaling and differentiation. Currently, three pathways are
implicated in the pathophysiology of LMNA-mediated cardiac and
musculoskeletal dystrophies; retinoblastoma protein (pRb), mitogen
activated protein kinases-extracellular signal-regulated kinases
(MAPK-ERK) and transforming growth factor beta ligands (TGF-β).
MAPK-ERK and pRb signaling mediate regulation of the cell cycle,
while the TGF-β pathway is involved in the transduction of
extracellular signals to trigger downstream TGF-β gene
transcription in the nucleus (37). Several in vivo studies have
obtained promising results by targeting these pathways. For
example, Muchir et al despite identifying that inhibition of
the ERK pathway had a minimal effect on cardiomyopathy, the impact
of therapy on cardiac rhythm was not examined; the most common
cause of mortality in humans (54). Further studies are essential to
elucidate the precise mechanism involved in cardiac arrhythmias and
skeletal muscle weakness. Myostatin or TGF-β receptor inhibition by
various techniques is evolving therapeutic rationales for caveolin
3 mutations. Yet, these treatments were applied to a particular
mutation (55). Whether these
therapies are effective on other types of mutations or other models
remains to be explored.
Accumulating lines of evidence have demonstrated
that gene transfer of CAPN3 and mutated myostatin propeptide
delivery were efficient therapies in investigations utilizing the
calpainopathy murine animal model. However in this study, the
duration for gene persistence in muscles was relatively short (9
days), the mode of gene delivery (intramuscular) was not sufficient
to improve lifestyle, and the parameters used for assessing
improvement were poor, such as the contractile force, which was
measured ex vivo (56).
Several novel therapeutic strategies have been piloted to treat
DYSF gene (DYSF) deficiency. The most well characterized study was
that performed by Han et al, who disrupted the complement
component C3 (C3), that is considered to be a crucial mediator of
inflammatory cascades in DYSF deficient mice (57). Nonetheless, several queries were
raised against the proposed model, including the limitation that
the contractile force was not examined. In addition, the authors
hypothesized that it is complement activation rather than
contraction-induced injuries that is responsible for muscle
weakness in the DYSF deficient mouse, however some dysferlinopathic
patients have no infiltrates on their biopsies (58). It is unlikely that membrane attack
complexes (MAC) underlie muscle weakness, given that the same study
identified that C5 ablation (a terminal component of the MAC
pathway) had minimal effects. This issue should be addressed in
future trials when utilizing inflammatory and non-inflammatory
models. Whether complement C3 inhibitors (easily administered and
more convenient for human trials) will have a similar effect as C3
ablation remains to be clarified. In sarcoglycanopthies, various
therapeutic stratgies have been investigated in vitro and
in vivo, including gene transfer, stem cell grafting,
myostatin blockade and calcium channel blockers. Notably,
sarcoglycanopathies conveyed more success of gene therapy than
other LGMD subtypes due to small size genes. Lastly, calpain 3
inhibition provided a rationale for the treatment of
titinopathy.
Though numerous therapies have proved efficacious
and safe in several different murine models, caution should be
exercised when considering the applicability of these treatments to
humans, given the evident differences in size, life span, genetic
variants and immunological complexity between the two species.
Table III (54–57,59–82)
summarizes the majority of murine models constructed and the
interventions applied with their results.
| Table IIIMurine models with assumed
therapy. |
Table III
Murine models with assumed
therapy.
| LGMD | Animal model | Description | Comment (ref.) | Intervention
(ref.) | Result (ref.) |
|---|
| LGMD1A |
Myo−/− | Knockout mouse | Normal | | |
|
Myo+/T57I | Transgenic
mouse | Phenotype similar
to myotilinopathy | | |
| DTg | Double
transgenic | More severe
phenotype than TgT57I, Tg WT | | |
| LGMD1B |
LmnaH222P/H222P | Knockin mouse | More relevant to
human laminopathy | ERK inhibition
(PD98059) (54) | Reverse DCM in
mice |
|
Lmna−/− | Knockout mouse | Less relevant to
human laminopathy | | |
| LGMD1C |
Cav-3−/− | Knockout mouse | Muscle disease,
HCM, defect in T-tubules | | |
|
Cav-3+/P104L | Transgenic
mouse | | Myostatin (M)
inhibition (55) | Revere atrophy,
weakness |
| | | | TGF-β receptor
inhibitor (59) | Revere atrophy,
weakness |
| LGMD2A |
Capn3C129S/C129S | Knockin mouse | Proteolytically
inactive, structurally intact | AAV delivery
mutated (M) (56) | Revese atrophy,
weakness |
|
Capn3−/− | Knockout mouse | No calpain-3 | AAV delivery
calpain 3 (60) | Revese atrophy,
weakness |
| Capn3 Tg | Transgenic
mouse | Normal | | |
| LGMD2B | A/J | Spontaneous | Retrotransposon
insertion in intron 4 | Diltiazem (61), pyridostigmine (62) | Improved
contractile function |
| B6.A/J | By breeding | Retrotransposon
insertion in intron 4 | Dual AAV gene
transfer (63) | Clinical,
biochemical imp. |
| SJL/J | Spontaneous | Splice site
mutation at exon 45 | HUCB i.v.
administration (64) | Biochemical
imp. |
| | | | Q10 and resveratrol
(65) | Histological
imp |
| B10.SJL | By breeding | Splice site
mutation at exon 45 | | |
|
Dysf−/− | Knockout mouse | Deletion of exon
45 | Genetic disruption
of C3 (57) | Improved muscle
pathology |
| LGMD2C |
gsg−/− | Knockout mouse | Exon 2
disruption | AAV gene transfer
(66) | Biochemical
imp. |
| | | | Myostatin blockade
(67) | Improve function
not histology |
| gxi | Double
knockout | Lacking both
integrin α7 and γ-sarcoglycan | Integrin α7β1
(68) | Compensate
γ-sarcoglycan |
| LGMD2D |
Sgca−/− | Knockout mouse | | AAV gene transfer
(69) | Biochemical
imp. |
| | | | Mesoangioblasts
i.a. (70) | Biochemical
imp. |
| | | | Deacetylase
inhibitors (71) | Reverse morphology,
function |
|
SGCAH77C/H77C | Knockin mouse | Normal | | |
| LGMD2E |
Sgcb−/− | Knockout mouse | Exon 2
disruption | AAV gene transfer
(72) | Biochemical
imp. |
| LGMD2F | BIO14.6 | Spontaneous | | AAV gene transfer
(73) | Reverse morphology,
function |
| | | | Tranilast,
diltiazem (74) | Biochemical
imp. |
| TO-2 | By breeding | | AAV gene transfer
(75) | Biochemical and
functional |
| | | | HUCB i.myo.
administration (76) | Short-term
imp. |
|
Sgcd−/− | Knockdown | Exon 2 targeted
replacement | Hematopoietic stem
cells (77) | No imp. |
| | | | Myosphere-derived
progenitors (78) | Improved heart
function |
| | | | Myostatin blockade
(79) | Early-stage
imp. |
| | | | AAV gene transfer
(80) | Heart but not
muscle imp. |
| LGMD2G |
TCap−/− | Knockout mouse | | | |
| LGMD2H |
Trim32−/− | Knockout mouse | | | |
| T32KI | Knockin mouse | Carries
c.1459G>A, p. D487N mutation | | |
| LGMD2J |
TTN+/c.43628insAT | Knockin mouse
(het) | Wild allele
compensate mutated one | | |
|
TTNhom | Knockin mouse
(hom) | Lethal at E9.5 | | |
|
PEVK−/− | Knockout mouse | PEVK part of TTN
highly phosphorylated | | |
|
N2B−/− | Knockout mouse | Calcium-sensitive
area of TTN | | |
|
FINmaj | Knockout mouse | C-terminal area of
TTN (homo. Het.) | FINmaj
(Het.) X capn3−/−mice (81) | Improve muscle,
heart |
| LGMD2L | No model yet | | | | |
| Dystroglycan | | | | | |
|
LARGEmyd | Myodystrophy | LARGE gene
mutated | LARGE gene transfer
(82) | Improve structure
and function |
|
LARGE++/++ | Transgenic
mouse | | | Late-onset loss of
force |
| LGMD2I |
FKRP-NeoTyr307Asn | Knockin mouse | Lethal soon after
birth | | |
|
FKRPTyr307Asn | Knockin mouse | Normal | | |
| | FKRP−/− | Knockout mouse | Lethal at
E12.5 | |
| FKRPP448L/
P448L | Knockin mouse | Structural
anomalies reminiscent to human | | |
| LGMD2K |
POMT1−/− | Knockout | Lethal between E7.5
and E9.5 | | |
| LGMD2M |
FKTN+ETN | Transgenic
mouse | Retrotransposon
insertion in FKTN gene | | |
| LGMD2N |
POMT2−/− | Knockout mouse | Lethal at E9.5 | | |
| LGMD2O |
POMGnT1−/− | Knockout mouse | Muscle-eye-brain
model | | |
| LGMD2P | DGS654A | Transgenic
mice | Inhibit
dystroglycan cleavage | | |
| DAG1T192M/
T192M | Knockin mouse | Neuromuscular
abnormalities | | |
| LGMD2Q | No model yet | | | | |
5. Disease markers
With recent advances in therapeutic trials, more
light has been shed on the clinical outcome measures that can be
utilized to predict disease activity, regression and treatment
efficacy without requiring multiple muscle biopsies.
There are multiple disease markers for LGMD, some of
which are well known, and others which have only been identified in
recent years. For example, high creatine kinase (CK) levels
indicate disease activity; while low levels may indicate fibrosis
or therapeutic responsiveness. In cases of local delivery of target
therapy, CK level is not helpful. Hand grip strength, Medical
Research Council (MRC) scale, Gardner-Medwin and Walton scales or
other clinical scales are beneficial for assessing clinical
outcome, however inter and intra examiner variability and
false-negative results in depressed patients reduces the efficacy
and reliability of this method. Another measure utilizes contrast
agent-enhanced MRI scanning. Typically, albumin-targeted contrast
agent, (MS-325) does not enter into myocytes. In membranopathies,
the dye is usually observed in the sarcoplasm. The technique is
considered a robust noninvasive disease follow-up tool in the
therapeutic trials of the sarcoglycanopathies (83). Measuring the secreted alkaline
phosphatase levels (Se AP) in blood (succeeded in murine models),
involves insertion of the secreted alkaline phosphatase gene with
the gene of interest into a viral vector. Following this, simple
blood-based assay of Se AP can predict gene expression in specific
muscles. The assay overcomes limitations of CK level identification
that is nonspecific and minimally affected in cases of local
delivery of the gene (84).
Another approach measures dysferlin and calpain 3 expression levels
using circulating monocytes that reflect expression level sin
muscles given that the target therapy is delivered systemically
(61).
Luciferase assay
Luciferase assay is considered as ‘regeneration
reporter’ and its level mirrors the number of centrally nucleated
fibers and embryonic heavy chain myosin positive cells (61).
Neutralizing antibodies and interferon
(INF)-γ to r AAV
Measuring antibody titer or mononuclear cell
mediated INF-γ secretion, by enzyme linked immunosorbent assay
(ELISA) and enzyme-linked immunsorboent spot (ELI Spot) assays,
respectively, were used to detect degree of immune rejection to
inserted vector gene (85).
6. Disease prevalence
With the exception of countries such as Norway,
Denmark and Finland, where the founder effect was determined,
LGMD2A is the most frequent type of LGMD worldwide, followed either
by dysferlinopathies in some areas or by sarcoglycanopthies in
others (Fig. 1). The relative
frequencies of the subtypes that exist among various ethnicities
are described in Table IV
(86,87,89–92,94–107). LGMD is considered the second most
common muscular dystrophy in England, Mexico and Turkey, after
dystrophinopathies, with a disease prevalence of up to 1/14,500 and
a carrier frequency of up to 1/150 (86–88).
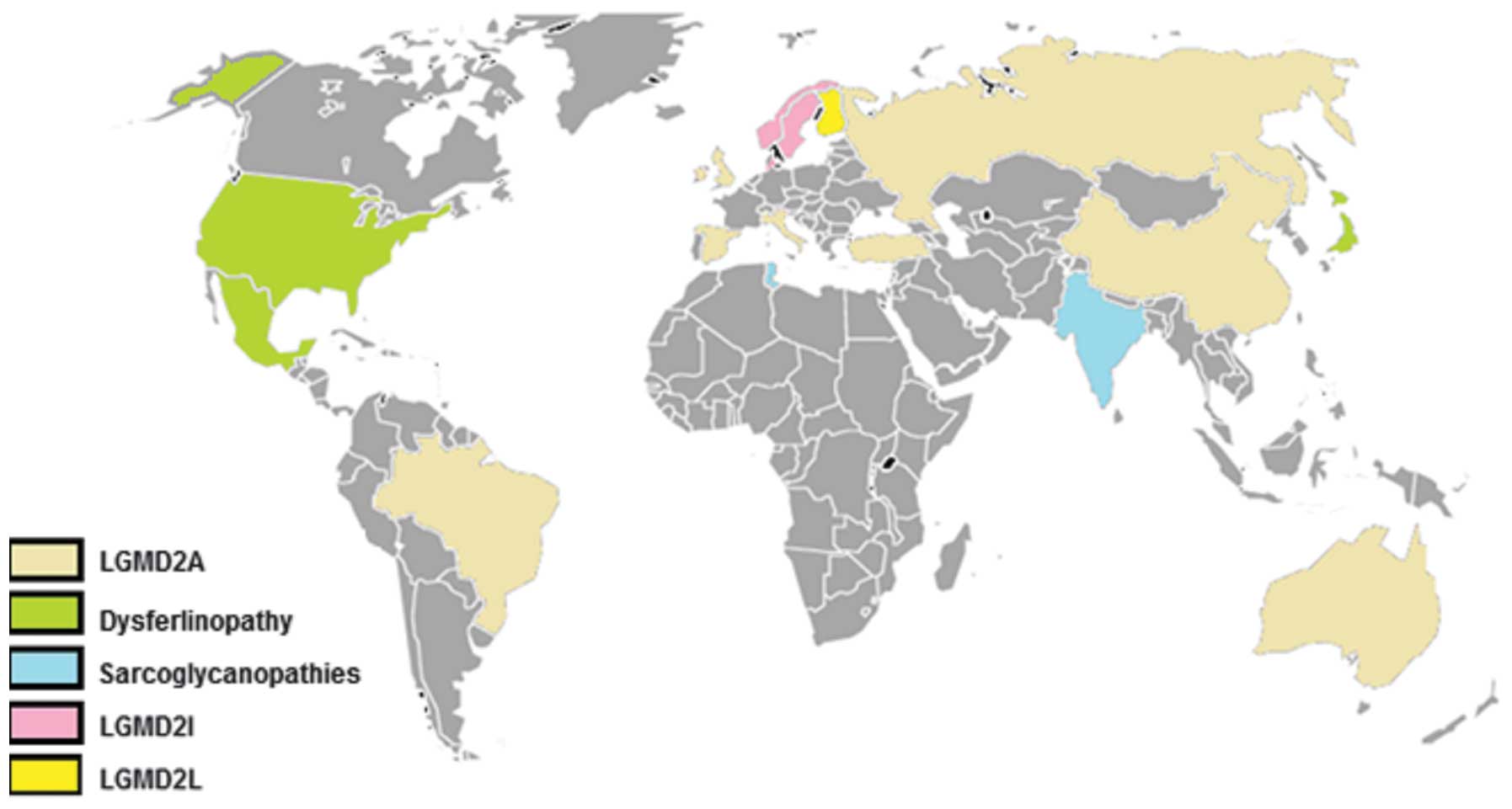 | Figure 1World map with the most common LGMD
forms represented in colors. Grey color indicates countries with no
known cohort. In Spain, Italy, England, Turkey, Russia, China,
Brazil and Australia, the most common type was calpainopathy.
LGMD2I was more frequent than other forms in the Scandinavian
Peninsula. However, dysferlinopathy was the most frequent in US,
Japan and Mexico. In India, sarcoglycanopathies had the highest
incidence, whereas in Finland, anoctaminopathy ranked the first
(25%) amongst other forms. |
| Table IVRelative % of different LGMD forms in
different countries. |
Table IV
Relative % of different LGMD forms in
different countries.
| | LGMD |
|---|
| |
|
|---|
| Country (ref.) | No. of
patients | 2A | 2B | 2C-F | 2I | 2L | 1B | 1C |
|---|
| Italy (89) | 228 | 37% | 27% | 23% | 9% | 2% | - | 2% |
| Italy (90) | 181 | 28.4% | 18.7% | 18.1% | 6.4% | - | - | 1.3% |
| Italy (91) | 346 | 25.1% | 11.2% | 15% | 4.3% | - | 1.4% | 1.4% |
| Spain (92) | - | 80% | - | - | - | - | - | - |
| German (89) | 124 | - | - | - | 16% | - | - | - |
| UK (86,94) | 68 | 26.5% | 5.9% | 11.8% | 19.1% | 11.8% | 8.8% | - |
| Norway (95) | 326 | - | - | - | 27% | - | - | - |
| Denmark (96) | 118 | 10.2% | 1.7% | 19% | 32.2% | - | - | - |
| Finland (97) | 101 | - | - | - | - | 25% | - | - |
| Australia (98) | 76 | 8% | 5% | 2% | 3% | - | 1% | 3% |
| USA (99) | 226 | 12% | 18% | 15% | 15% | - | - | 1.5% |
| Mexico (87) | - | 25% | 40.6% | 31.2% | - | - | - | 3.1% |
| Turkey (100) | 20 | 50% | 5% | 40% | - | - | - | - |
| Russia (101) | 19 | 75% | - | - | - | - | - | - |
| Brazil (102) | - | 32% | 22% | 32% | 11% | - | - | - |
| China (In
press) | 68 | 17% | 15% | 3% | - | - | - | 3% |
| Japan (103,104) | 80 | 26% | Most | 9% | - | - | - | - |
| India (105) | 26 | - | - | 53.8% | - | - | - | - |
| India (106) | 171 | 47% | - | - | - | - | - | |
| India (107) | 30 | 21% | - | - | - | - | - | - |
7. Genotype-phenotype correlation
Clinical presentations of LGMD disorders vary among
patients of the same subtype, or even within the same family
(108). Genotype-phenotype
correlational analysis is however, difficult to predict and it
represents one of the most challenging obstacles in the field of
genetic disorders. Trials to elucidate the specific clinical
picture for individual genetic subtypes were futile (108).
Currently, two null mutations in the CAPN3 gene have
been associated with severe phenotype, early onset and risk of
being crippled (90). Natural exon
32 skipping of the DYSF gene notably reduced the severity of
symptoms in a mother of two severely affected daughters by
homozygous mutation (109). Mild
features are most commonly reported in relation to the common Asian
mutation c.2997G>T, p.W999C). However, the mutation has also
been described in association with a variety of phenotypes
(110–112). In cases of LGMD2H, it has been
identified that mutations gathered in the NHL (named after the
proteins NCL1, HT2A and LIN-41) domain result in
LGMD2H/sarcotubular myopathy, whereas in Bardet-Biedl Syndrome,
mutations are most commonly located in the B-box region of the
gene. This may suggest that mutations in the NHL area render
individuals more susceptible to muscular disorders (113). Late disease onset, mild
phenotype, less susceptibility to loss of ambulation and more
liability to myoglobinuria, are consistent features observed in
patients homozygous to c.826C>A (p.L276I) of the FKRP gene
compared with patients heterozygous to the same mutation (96). While several LGMD forms are
phenotypically heterogeneous, it appears that ‘hot spot’ c.191dupA
mutation in the ANO5 gene is associated with a more homogeneous
phenotype (94). Of note, no
LGMD2M patients to date have exhibited a 3 kb retrotransposal
insertion in the FKTN gene, a founder mutation accounted for 87% of
Fukuyama type congenital muscular dystrophy (114). Finally, it has been identified
that mutations clustered in immunoglobulin-like fold and coil 2 of
the LMNA gene are inconsistently correlated with the autosomal
dominant form, LGMD1B (115).
Matching gene expression profiles of normal and
affected muscles, identifying the crystalline structure of the
protein of interest and recognizing the precise function of each
protein domain, are approaches that will improve our understanding
of the associations between various pathogenic mutations and
disease presentation in LGMD (116).
8. Diagnostic strategy
Clinical, electrophysiological, imaging, biochemical
and genetic testing techniques collectively should be utilized and
tailored according to specific LGMD patient cases. Not only will
this facilitate diagnosis and provide individualized genetic
counseling to proband and relatives, but will also enhance the
understanding of the underlying pathophysiology, to allow delivery
of a therapeutic strategy that targets the precise pathways that
are specific to that patient.
Clinical
Historical analysis and clinical examination are
commonly used approaches to identify specific LGMD subtypes.
Age of onset
The vast majority of autosomal recessive LGMD cases
have teenage onset-progressive muscle weakness, however, there are
exceptions to this rule. In certain dystroglycanopathies, sporadic
dysferlinopathy cases may start exhibiting perinatal ‘floppiness’
and mild weakness as late as 70 years-old (117,118). While calpainopathy and
dysferlinopathies tend to manifest in late childhood to late teens,
early childhood onset indicates the diagnosis will be a
sarcoglycanopathy or dystroglycanopathy. The majority of, but not
all, autosomal dominant LGMD patients begin experiencing symptoms
following their twenties.
Pattern of distribution of muscles
weakness
LGMD is associated with marked clinical disparity.
As the rule of thumb, a defect in membrane scaffolds cause
predominant proximal myopathies, whereas sarcomeric protein
deficiencies typically result in initial distal myopathies. The
exceptions to this include distal Miyoshi myopathies (MMs), which
are associated with the membrane patch proteins dysferlinopathy and
anoctaminopathy. Scapulohumeral muscle weakness or ‘Erb phenotype’
is most commonly observed in sarcoglycanopathies, calpainopathies
and Duchene’s type of LGMD2I, while pelvic muscle weakness or
‘Leyden-Mobius phenotype’ (LM) is the leading indicator for the
majority of LGMD subtypes. While calf, thigh and tongue
hypertrophies are mostly encountered in sarcoglycanopthies and
dystroglycanopathies, deltoid hypertrophy appears to be restricted
to dysferlinopahies (119,120,121).
Skeletal manifestations
Contracture is reported in numerous types of LGMD,
however predilection is given to laminopathy, calpainopathy,
dystroglycanopathies and anoctminopathy. While kyphoscoliosis and
lordosis manifest in calpainopathies, dystroglycanopathies and
plectinopathies, features such as bent and rigid spines, appear to
be limited to dysferlinopathies (112,122).
LGMD and heart
With the exception of myotilinopathy, laminopathies,
telethoninopathy and LGMD2I, cardiac muscles are spared in the
majority of LGMD subtypes or seldom involved in others (LGMD2A, 2B
and 2C-2F). Patients with LGMD may present with wide range of
cardiac abnormalities e.g., atrial fibrillations, flutters,
atrio-ventricular conduction blocks, supraventricular, ventricular
ectopic beats, ventricular tachycardia and sudden death commonly
seen in the laminopathy patients; on the other side, dilated,
hypertrophic and restrictive cardiomyopathy are noticed in some
sarcoglycanopathies and a third of LGMD2I cases. Cardiac problems
may precede, overlap with or follow skeletal muscle weakness.
Periodical cardiac monitoring and pacemaker or defibrillator
implantation are warranted in certain cases (123).
LGMD and pulmonary function
Respiratory muscle weakness is a rare manifestation
and predominantly indicates diagnosis of LGMD2I, myotilinopathy and
occasionally, LGMD2A and LGMD2C-2F. Patients with LGMD2I may also
develop respiratory failure while ambulant. Pulmonary function
tests should be part of routine examination in suspected LGMD
subtypes.
Extra muscular features
Detailed neurological exams often provide an
implication of the specific LGMD form. While neuropathy,
paresthesia, nasal speech and dysarthria appear to commonly occur
in myotilinopathies; opthalmoparesis, foot drop, and paresthesias
have been sporadically reported in LGMD1C, 2G and 2H respectively
(124,125). In dystroglycanopathies, with the
exception of LGMD2I, muscle weakness is usually coupled with
cognitive impairment, which has serious psychosocial consequences
for the patient and their family. Patients with LGMD2I usually have
normal intellect, nonetheless there may be some difficulties in
visiospatial planning and memory function (126).
Clinical course
LGMD subtypes have a characteristically slow
progression, with weakness most commonly beginning in the proximal
lower limbs and sometimes the distal limbs. This is followed by
weakness in all other limbs and then permanent disability is
established two to three decades after disease onset. The subtypes
LGMD1D, LGMD2L and LGMD2M represent slowly progressive diseases and
patients are mildly affected and remain ambulant (94). Rapid progression is a classical
feature of dysferlinopathies, sarcoglycanopathies and
plectinopathies. In some LGMD forms there may be inconsistencies in
the progression of the disease between male and female-affected
patients. In female mild-onset patients, estrogen is considered to
impact disease progression. LGMD2G and LGMD2L are examples of LGMD
groups with inconsistent disease progression between male and
female patients. The study discerned another type of LGMD that is
also characterized by a two-peak onset; rapid progressive Duchene
like childhood onset and milder Becker-like adolescent to adulthood
onset. LGMD2I is representative of this set (96). Fig.
2 denotes disease spectrum for the rapid recall of clinical
features of LGMD subtypes.
Clinical constellations
LGMD remains an expanding group of diseases, and
approximately one third of cases have not yet been associated with
a specific subtype. Certain clinical constellations often favor
specific LGMD subtypes, and eliminate a wide array of unnecessary
genetic tests. In patients presenting with distal muscular
weakness, contractures and cardiac problems in adulthood,
myotilinopathy should be considered. Limb-girdle muscle weakness
with a family history of sudden death and late onset contractures,
most commonly indicate a laminopathy, whereas myalgia, rippling
phenomenon, in association with proximal limb muscle weakness in
the first four decades of life should raise suspicion of
caveolinopathy. Proximal limb muscle weakness with contractures and
calf atrophy in teens are features consistently associated with
LGMD2A. Early childhood onset muscle weakness with cognitive
impairment is a pathognomonic feature of dystroglycanopathies. In
patients with cardiopulmonary involvement in association with
myopathy and myoglobinuria, LGMD2I should be suspected.
Biochemical, imaging and
electrophysiological studies
Muscle enzymes (CK)
CK levels are either normal or mildly elevated in
the majority of autosomal dominant LGMDs, whereas in autosomal
recessive types, they are highly elevated. While high CK levels in
dysferlinopathies, dystroglycanopathies and sarcoglycanopathies are
usually correlated with disease activity, calpain 3 deficient
patients typically manifest mild to moderate CK level elevation.
However, normal and high levels of CK have been sporadically
described amongst the disease subtypes. Although CK levels are not
specific measurements for the diagnosis of muscular diseases, they
often act as a useful tool for guiding physicians to a specific
disease process and exclude metabolic or acquired myopathic
diseases (127).
Magnetic resonance imaging (MRI)
Novel diagnostic tools have been recently introduced
to the field of neuromuscular disorders. Studies have utilized
modern imaging facilities to facilitate in the diagnosis of highly
complicated genetic diseases and to allow physicians to direct
patients for further protein and gene testing. The distinction
between the most common subtypes (LGMD2A and LGMD2I) that account
for up to 80% of recessive LGMD disorders (in some ethnic groups)
is now possible using imaging techniques (128). LGMD2A is characterized by the
involvement of gluteus maximus, the posteromedial thigh area and
the selective involvement of medial calf muscles. This is in marked
contrast to LGMD2I where calf muscles are non-selectively involved.
In addition, winging of scapulae and calf atrophy are more evident
in calpainopathies than in LGMD2I. The anterior thigh muscles are
more affected than the posterior ones in α-sarcoglycanopathy, where
the calf muscles are relatively spared. This is in contrast to
dystrophinopathies, where early and striking changes in the
gastrocnemii muscles are prominent (128,129). The specific pattern of affected
muscles was eventually delineated in anoctaminopathy, as
demonstrated by high signal intensities in posterior thigh muscles,
partial atrophy of quadriceps and posteromedial calf affection
(89). In myotilinopathy, distal
muscle groups represented by the calves are more affected than
proximal peroneal muscles and medial pelvic, thigh and lower leg
muscles are more involved than lateral sets. The opposite is true
for desminopathy (130).
Electrophysiological studies; nerve
conduction velocity (NCV) study and electromyography (EMG)
Electrophysiological studies are highly important in
differentiating myopathic diseases from neurogenic ones,
particularly if the patient presents with distal myopathy or
contracture deformity where other differential diagnoses, including
Charcot-Marie-Tooth syndrome, may be considered. However,
neurogenic damages have been sporadically reported in calpainopathy
(131) and dysferlinopathy
(unpublished observations).
Muscle biopsy: optical and electron
microscopy (EM)
Muscle biopsy is a transitional step in the
diagnostic algorithm of muscle disorders. Several studies are
ongoing to simplify the diagnosis of muscular disorders using more
superficial tissues like skin (132).
A wide spectrum of optical microscopic alterations,
ranging from mild to severe degenerative muscle changes, has been
described in LGMD biopsy specimens. Aside from the myopathic
features (fiber size variation, internal myonuclei and fiber
splitting), inflammatory components appear frequently in biopsy
analysis of specific LGMD subtypes like dysferlinopathies (133), anoctaminopathies (94) and dystroglycanopathies and rarely
in others, including LGMD2A (134), LGMD2C (135) and LGMD2D (manuscript in press).
While rimmed and non-rimmed vacuoles are non-classical features in
LGMD disorders (observed mainly in autosomal dominant patients),
amyloid deposits are frequently encountered in dysferlinopathies
and anoctaminopathies. Features of chronicity, including lobulated
fibers, cytochrome C oxidase (COX) negative fibers and fibers with
focal areas of reduced or absent nicotinamide adenine dinucleotide
tetrazolium reductase (NADH-TR) are frequently detected in
calpainopathy. Desminopathy should be considered in cases with
menadione-linked nitro blue tetrazolium (M-NTB) positive
cytoplasmic inclusions (12). In
cases with only trivial muscle pathology, biopsy analyses are
usually non-specific or even normal. However, many genetically
confirmed LGMD cases have been mistakably diagnosed as acquired
myopathies (136). Hence, EM and
immunohistochemistry are essential to establish the diagnosis of
LGMD.
EM has a crucial role in diagnostic analyses of
certain LGMD subtypes particularly those associataed with
myofibrillar proteinopathies. Cytoplasmic filamentous inclusions,
spheroid bodies, myofibrillar protein aggregates and Z-disc
streaming are features that are commonly diagnosed as
myotilinopathies, whereas excess subsarcolemmal granulofilamentous
material is in keeping with the characteristics of desminopathy
(137). With the exception of
laminopathy and myotilinopathies, myonuclei are usually spared in
LGMD, which is a feature that facilitates its distinction from
sporadic and hereditary inclusion body myopathy. Recently, EMs has
been used to explore etiopathogenic mechanism underlying some
dystrophic processes. These studies identified plasma membrane
defects, basal lamina duplications and submembranous flocculations
detected in LGMD2B and LGMD2L as the key indicators of a membrane
reseal defect. Of note, some features e.g., vacuolar structures,
dilated T tubules and myelin bodies, are non-specific and are
shared with a variety of muscular disorders.
Immunohistochemistry (IHC) and western
blot (WB) assays
With respect to their diagnostic significance, WB
and immunostaining of muscle sections with antibodies against
dysferlin (138), sarcoglycans
(139), caveolin 3 (140) and telethonin (141) are now the ‘gold standards’ owing
to their high specificities and cost-effectiveness.
In LGMD2A, this approach is often hindered by
incomplete sensitivity and specificity. The process of staining
muscle sections with available antibodies against calpain 3 is
generally disputed as different staining patterns have been
detected in abnormal LGMD2A biopsies; however, the specificity of
western blotting has been improved by assessing calpain 3 autolytic
activity (142,143). In addition, quantitative analyses
of calpain 3 bands offer high diagnostic yield ranging from 84% in
certain studies to 100% in others (144–147).
Other specific antibodies are those raised against
the C-terminus of the giant titin protein and against truncated
TRIM32 protein due to compound heterozygous mutations (148,149). A putative broken linkage between
the sarcolemma and sarcomere, due to plectin 1f isoform mutation,
most commonly results in absent sarcolemma immunostaining that is
highly suggestive of plectinopathy. Whereas, reduced expression of
dystroglycan-α and laminin-α2 overlay is a sensitive diagnostic
indicator for dystroglycanopathies. However, this approach should
be interpreted in association with clinical data, to facilitate
selecting a specific gene testing method for the patient.
Proper sample handling, freezing and homogenization
usually solve the limitation of denatured proteins, when certain
antibodies cannot identify and may not be appropriate for
biochemical assays. On the other hand, the masking effect of the
epitope of an antibody may provide an inaccurate signal and some
cases can be easily be overlooked (150).
High expression of calpain 3 and dysferlin in
monocytes and the skin has reduced the necessity for muscle
biopsies (132,151). Furthermore, multiplex blot
analyses technique allows the investigator to envisage protein
interaction and secondary reductions more clearly. Also, ‘reverse
protein array’ confers high sensitivity to minimal protein changes
that make it suitable to follow-up markers and predict drug
responsiveness in upcoming trials (152).
Genetic diagnosis
Careful analysis of clinical and pathological
findings and physiological and biochemical data, often provides
crucial clues for the diagnosis of a distinct LGMD form. Currently,
the documentation of a pathogenic mutation is currently warranted
as a tool for identifying the diagnosis of a hereditary muscular
disorder.
Genetic analyses are presumed to offer diagnosis for
~99% of cases with known gene loci. However in some types, such as
LGMD 2A, virtually 25% of cases have no defects in the CAPN3 gene
and ~22% have only one affected allele (153). It was estimated ~10–15 % of
mutations are either intronic or subtle exonic splice sites.
Therefore, muscle flesh, skin or in special cases (LGMD2A, 2B)
blood monocytes are necessary to obtain mRNA (153,154). With the exception of selected
centers in USA and certain European countries that offer genetic
analyses for LGMD patients, genetic diagnosis is only affordable on
a research basis. Nonetheless, the common hot spot mutations in
certain ethnicities can be targeted prior to running whole gene
sequencing (Table V) (13,14,25,27,35,90,92,
94,95,96,101,104,141,148,155–162).
| Table VLGMD: Common mutations with founder
effects. |
Table V
LGMD: Common mutations with founder
effects.
| Type | Gene | Exon | Hot-spot mutations
(exon no.) | Populations that
express mutations (ref.) | Predicted
phenotype |
|---|
| LGMD1A | MYOT | 10 | Exon 2 | - | |
| LGMD1B | LMNA | 12 | - | - | |
| LGMD1C | CAV3 | 2 | - | - | |
| LGMD1D | DES | 9 | - | - | |
| LGMD1E | DNAJB6 | 10 | c.279C>G
(E5) | Finland, Americans
(13,14) | |
| LGMD2A | CAPN3 | 24 | c.550delA (E4) | Russia, Czech,
Turkey (40%), Italy, UK (101,155,156) | |
| | |
c.2362_2363delinsTCATCT (E22) | Spain (30%), Brazil
(Hispanics) (92) | |
| | | c.1469G4A, p.R490Q
(E11) | Italy, Turkey (10%)
(155,156) | |
| LGMD2B | DYSF | 55 | c.937+1G>A
(E10) | Japan (104) | |
| | | c.1566C>G,
p.Y522X (E18) | Japan (104,157) | |
| | | c.2997G>T,
p.W999C (E28) | Japan, China, S.
Korea (104) | Homozygous,
mild |
| | | c.3373delG,
p.E1125KfsX1134 (E1) | Japan (104) | |
| | | c.2494C>T,
p.Q832X (E24) | S. Korea (158) | |
| | | c.663+1G>C,
splicing defect (E6) | S. Korea (158) | |
| | | c.2372C>G,
p.p791R (E24) | Canada (natives)
(159) | |
| | | c.2875C>T,
p.R959W (E27) | Italy (159) | |
| | | c.5713C>T,
p.R1905X (E51) | Spain (159) | |
| | | c.2779delG.,
p.A927LfsX21 (E26) | Caucasian Jewish
population (159) | |
| | |
c.4872_4876delinsCCCC (E44) | Libyan Jewish
population (159) | |
| LGMD2C | SGCG | 8 | - | - | |
| LGMD2D | SGCA | 10 | c.229C>T,
p.R77C(E3) | Europe, Finland,
Brazil (160) | Homozygous,
mild |
| LGMD2E | SGCB | 6 | - | - | |
| LGMD2F | SGCD | 9 | - | - | |
| LGMD2G | TCAP | 2 | c.172C>T, p.Q53X
(E2) | Brazil (141) | |
| LGMD2H | TRIM32 | 2 | c.1459G>A,
p.D487N (E2) | Hutterites (USA,
Canada, Germany) (25,161) | |
| LGMD2I | FKRP | 4 | c.826C>A,
p.L276I (E4) | Europe, American
(90,95,96,162) | Homozygous, Becker
like
Heterozygous, Duchenne-like |
| LGMD2J | TTN | 363 | Mex6( 11-bp
change) | Finland (148) | |
| LGMD2K | POMT1 | 20 | c.598G>C,
p.A200P (E7) | Turkey (27) | |
| LGMD2L | ANO5 | 22 | c.191dupA,
p.Asn64Lysfs*15 (E5) | Northern europeans
(94) | Homogeneous
phenotype |
| LGMD2M | FKTN | 11 | - | - | |
| LGMD2N | POMT2 | 21 | - | - | |
| LGMD2O | POMGnT1 | 23 | - | - | |
| LGMD2P | DAG1 | 6 | - | - | |
| LGMD2Q | PLEC | 33 | c.1_9del,
p.0(E2i) | Turkey (35) | |
As delineated in this review, LGMD is still an
underdiagnosed entity and families with no identifiable gene locus
may benefit from approaches like linkage analysis (34). Furthermore, gene chips, exome and
wide genome sequencing are promising diagnostic tools for the
diagnosis of de novo mutations, yet due to the high cost and
extensive number of sequence variants, they are limited by
challenging monetary and interpretation tasks (163).
9. Prevention and surveillance
The optimum time for determination of genetic risk,
elucidation of carrier status, and discussion of the availability
of prenatal testing, is prior to pregnancy. It is appropriate and
necessary to offer genetic counseling to young adults who are
affected, are carriers, or are at risk of being carriers. The
identification of certain LGMD subtypes has led to changed advice
e.g., certain sarcoglycanopathies (autosomal recessive trait) had
previously been diagnosed as Becker muscular dystrophy (X-linked
recessive trait that mainly transmitted to males) (164).
In certain LGMD disorders, joint contracture,
cardiac or respiratory muscle dysfunctions arise earlier and later
in the disease course. Regular surveillance with early
physiotherapy, orthotics and stretching exercises facilitate joint
deformities and delays disabilities for approximately two years
(165). Consistent monitoring of
cardiac function with early pacemaker or improved implantable
cardioverter-defibrillator (ICD) instruments, may rescue the lives
of certain laminopathy patients. Consistent monitoring of
respiratory function, using of annual influenza vaccines, early
physiotherapy, nocturnal ventilation with use of mucolytic and
antibiotics if necessary, are all strategies that will reduce the
rate of hospitalization and delay the need for tracheostomy and
mechanical ventilation for several years. The majority of LGMD
patients suffer from depression, social isolation, low self-esteem
and culpability (166), so often
pyschiatric therapy is of much benefit too.
The patient may be monitored for cardiac and
respiratory function in the outpatient neurology clinic,
nonetheless, a multi-disciplinary team is recommended to improve
outcome and to confirm the optimal timing for intervention.
Finally, in cases with unidentified gene defects, should they
develop cardiopulmonary or skeletal complications then the above
mentioned principles will apply.
10. Management
Like other hereditary disorders, despite extensive
research, there are currently no therapeutic strategies to treat
LGMD. The existing management techniques include emotion and
physical support, such as in the use of canes, walkers, splinters,
surgical intervention in case of contracture deformities, and
support of cardiac and respiratory functions in cases of
myotilinopathy, laminopathy and dystroglycanopathy.
Several clinical trials have been completed in
humans and others are actively recruiting. These trials in humans
have been initiated due to the trials in murine models which
successfully demonstrated gene, cell transfer and pharmaceutical
therapy in prinicipal (54–82).
While ‘replacement therapy’ of the defect using
gene- or cell-based therapy is the gold standard, pharmaceutical
therapy seems to be a more favorable approach, due to the fact the
medicine can be evenly distributed to the whole body and is
suitable for both modes of inheritance. The proposed pharmaceutical
approaches act by targeting specific pathophysiological pathways in
the disease. Increasing muscle mass by enhancing positive
regulators or by inhibition of negative regulators of muscle
growth, such as neutralizing myostatin antibodies, have been
utilized in clincal trials, however unfortunately, despite proving
safe and tolerable, demonstrated negative results at the endpoints
(167). Another therapeutic
target is calcium channels, which are more permeable in
sarcoglycanopthies, caveolinopathies and other LGMD forms. These
observations have been confirmed by reports that anecdotal
improvement of CK level was demonstrated in an MM Japanese patient
treated with dantrolene to reduce muscle pain, which is a drug that
can block calcium channels. Furthermore, a combination of
lisinopril (a calcium channel blocker agent) and Co Q10 are
included in the upcoming trials in the treatment of LGMD. In
dysferlinopathies, treatment startegies differ, because
inflammatory mechanisms are often active in the DYSF mutant
muscles. These approaches include, the use monoclonal antibodies
like rituximab to block B cell activation or the use of intravenous
immunoglobulin to prevent complement attack complex activations.
Recently, vitamin D3 has been shown to possess DYSF promoter
properties and has improved dysferlin expression in muscles and
monocytes of DYSF mutation carriers (Table VI) (30, 85,167–174). The beneficial effect of steroids
in the treatment of sarcoglycanopthies and dystroglycanopathies is
a matter of interest, but the mechanism underlying how they improve
muscle strength remains elusive. Similarly, this also applies to
creatine monohydrate and Co Q10. Another encouraging potential
strategy involves the use of calpain inhibitors to stop ubiquitous
degradation of misfolded proteins in the Golgi apparatus, however
there is no evidence of their effect in humans. Lastly, identifying
drugs that upregulate surrogate proteins like ɛ-sarcoglycan in
cases of α-sarcoglycanopthies, Integrin α7β1 in cases of
γ-sarcoglycanopathies, or LARGE in cases of dystroglycanopathies,
may also be of potential interest.
| Table VIUpdate of therapeutic trials in
humans. |
Table VI
Update of therapeutic trials in
humans.
| Therapeutic
option | Mechanism | LGMD form
(ref.) | Comment |
|---|
| Gene therapy
(rAAV) | I.M. Intact gene
transfer | LGMD2D (85) | Application on
larger and more functional muscle is required
Intra-arterial delivery to whole-body muscles is
warranted
The study represents histological but not functional
improvement |
| Rituximab (I.V)
(monoclonal AB) | Against
CD20-positive B cells 375 mg/m2/week (4 doses) | Miyoshi M (168) | Small number of
patients, female responsiveness is requested
Muscle adaptation to specific exercise should be
considered
Effect of treatment on quality of life is doubtful |
| Dantrolene (25
mg/day) | Ca2+ ion
blocker in ER (ryanodine receptor binding) | Miyoshi M (169) | Query effect on
weakness
Hepatopathy side effect in up to 91% |
| Vitamin D3 (1/week
for 1 year) | MEK/ERK
pathway
D3 receptor to DYSF promoter | DYSF carriers
(170) | The study
represents cohort of asymptomatic carriers |
| Deflazacort | Steroids (1
mg/kg/day) | DYSF-opathy
(171) | Worsening of muscle
strength |
| LGMD2D (172) | Mildly symptomatic
female patient |
| Prednisone | Steroids (1–2
mg/kg/day) (0.35 mg/kg/day) | LGMD2M (30) | Partial
responsiveness, multiple fractures and susceptibility to
infections |
| LGMD2I (173) | Growth arrest,
vertebral fractures and susceptibility to infection |
| Creatine MH | Helps to supply
energy | Sarcoglycans
(174) | Mild improvement
(3%) |
| MYO-029 | Neutralizing AB to
myostatin | MD (167) | No improvements at
end point |
| CoQ10 +
lisinopril |
Vitamin-like+ Ca2+
blocker | LGMD | Recruiting |
11. Conclusion
In recent years, our understanding of LGMD has
advanced, with regards to disease occurrence, founder effect in
some locations, certain aspects of pathophysiology and
phenotype-genotype correlations. In addition, elucidation of hot
spot mutations, disease biomarkers, general strategies of the
diagnosis and treatments would point toward efficient and safe
follow up, and intervention. However, more emphasis should be
placed on pathogenesis, updating of diagnostic guidelines, with
regular assessment and close follow up of disease progression to
better elucidate the history of the disease and to enrich
translational research. Universal patient registry and collaborated
multicenter LGMD disease projects should be encouraged, to provide
invaluable insight into its common and unique features and to
settle standardized patient care.
12. Future perspectives
In the past six decades, advances in the field of
molecular biology has opened new avenues to understand the LGMD
clinical diagnosis, classification, pathogenesis and treatment
possibilities. However, our understanding of the pathophysiology of
the majority of LGMD forms is still in its infancy.
The majority of of autosomal dominant disorder
mutations behave in a ‘dominant negative fashion’. It remains
unresolved why single amino acid substitution results in negative
adverse function. What is equally intriguing, is whether all
mutations act by same mechanism and whether protein interaction
with other known and yet undisclosed proteins affect the phenotypes
of these diseases. Multiple strategies have been devised to
overcome dominant negative cytotoxicity in animal models, including
RNA interference, however their safety and efficacy needs to be
proved in forthcoming clinical trials.
While the pathogenicity of most autosomal recessive
disorders remains to be elucidated, a combination of biochemical
tests and the availability of variable animal models have provided
invaluable clues for physiological functions of key proteins
involved in LGMD. However, it remains unclear how protein
deficiencies result in muscle fiber degeneration. A parallel
question is why some ubiquitous proteins are involved specifically
in muscle diseases. What is equally unclear, is how congenital and
adult muscular diseases are produced by the same mutation. The use
of a variety of animal models with different types of mutations
within the same gene and observing their effects may feasibly solve
the ‘paradox of single gene and multiple phenotypes’.
Acknowledgements
This study was supported by the Jilin University
Award. The authors would like to thank Mr. Ming Chang and Mr. Yu G.
Ma for their technical support.
References
|
1
|
Danièle N, Richard I and Bartoli M: Ins
and outs of therapy in limb girdle muscular dystrophies. Int J
Biochem Cell Biol. 39:1608–1624. 2007.PubMed/NCBI
|
|
2
|
Erb W: Dystrophia muscularis progressiva.
Dtsch Z Nervenheilkd. 1:13–94. 173–261. 1891.(In German).
|
|
3
|
Leyden E: Klinik Der
Rückenmarks-Krankheiten. 2. Hirschwald; Berlin: pp. 531–540. 1875,
(In German).
|
|
4
|
Möbius PJ: Ueber die hereditären
nervenkrankheiten. Samml Klin Votr 171. Breitkopf und Härtel;
Leipzig: pp. 1505–1531. 1879, (In German).
|
|
5
|
Bell J: On pseudohypertrophic and allied
types of progressive Muscular dystrophy. The Treasury of Human
Inheritance. Fischer RA: 4(Part 4)Cambridge University Press;
London: pp. 283–342. 1943
|
|
6
|
Walton JN and Nattrass FJ: On the
classification, natural history and treatment of the myopathies.
Brain. 77:169–231. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bushby KM and Gardner-Medwin D: The
clinical, genetic and dystrophin characteristics of Becker muscular
dystrophy. I. Natural history. J Neurol. 240:98–104. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bushby KM: Diagnostic criteria for the
limb-girdle muscular dystrophies: report of the ENMC Consortium on
Limb-Girdle Dystrophies. Neuromuscul Disord. 5:71–74. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hauser MA, Horrigan SK, Salmikangas P, et
al: Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum
Mol Genet. 9:2141–2147. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Muchir A, Bonne G, van der Kooi AJ, et al:
Identification of mutations in the gene encoding lamins A/C in
autosomal dominant limb girdle muscular dystrophy with
atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet.
9:1453–1459. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Minetti C, Sotgia F, Bruno C, et al:
Mutations in the caveolin-3 gene cause autosomal dominant
limb-girdle muscular dystrophy. Nat Genet. 18:365–368. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Greenberg SA, Salajegheh M, Judge DP, et
al: Etiology of limb girdle muscular dystrophy 1D/1E determined by
laser capture microdissection proteomics. Ann Neurol. 71:141–145.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harms MB, Sommerville RB, Allred P, et al:
Exome sequencing reveals DNAJB6 mutations in dominantly-inherited
myopathy. Ann Neurol. 71:407–416. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sarparanta J, Jonson PH, Golzio C, et al:
Mutations affecting the cytoplasmic functions of the co-chaperone
DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet. 44:450–455.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Palenzuela L, Andreu AL, Gàmez J, et al: A
novel autosomal dominant limb-girdle muscular dystrophy (LGMD 1F)
maps to 7q32.1-32.2. Neurology. 61:404–406. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Starling A, Kok F, Passos-Bueno MR,
Vainzof M and Zatz M: A new form of autosomal dominant limb-girdle
muscular dystrophy (LGMD1G) with progressive fingers and toes
flexion limitation maps to chromosome 4p21. Eur J Hum Genet.
12:1033–1040. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bisceglia L, Zoccolella S, Torraco A, et
al: A new locus on 3p23-p25 for an autosomal-dominant limb-girdle
muscular dystrophy, LGMD1H. Eur J Hum Genet. 18:636–641. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Richard I, Broux O, Allamand V, et al:
Mutations in the proteolytic enzyme calpain 3 cause limb-girdle
muscular dystrophy type 2A. Cell. 81:27–40. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu J, Aoki M, Illa I, et al: Dysferlin, a
novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb
girdle muscular dystrophy. Nat Genet. 20:31–36. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Noguchi S, McNally EM, Ben Othmane K, et
al: Mutations in the dystrophin-associated protein
gamma-sarcoglycan in chromosome 13 muscular dystrophy. Science.
270:819–822. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roberds SL, Leturcq F, Allamand V, et al:
Missense mutations in the adhalin gene linked to autosomal
recessive muscular dystrophy. Cell. 78:625–633. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lim LE, Duclos F, Broux O, et al:
Beta-sarcoglycan: characterization and role in limb-girdle muscular
dystrophy linked to 4q12. Nat Genet. 1:257–265. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nigro V, de Sá Moreira E, Piluso G, et al:
Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is
caused by a mutation in the delta-sarcoglycan gene. Nat Genet.
14:195–198. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moreira ES, Wiltshire TJ, Faulkner G, et
al: Limb-girdle muscular dystrophy type 2G is caused by mutations
in the gene encoding the sarcomeric protein telethonin. Nat Genet.
24:163–166. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Frosk P, Weiler T, Nylen E, et al:
Limb-girdle muscular dystrophy type 2H associated with mutation in
TRIM32, a putative E3-ubiquitin-ligase gene. Am J Hum Genet.
70:663–672. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brockington M, Yuva Y, Prandini P, et al:
Mutations in the fukutin-related protein gene (FKRP) identify limb
girdle muscular dystrophy 2I as a milder allelic variant of
congenital muscular dystrophy MDC1C. Hum Mol Genet. 10:2851–2859.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Haravuori H, Vihola A, Straub V, et al:
Secondary calpain3 deficiency in 2q-linked muscular dystrophy:
titin is the candidate gene. Neurology. 56:869–877. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Balci B, Uyanik G, Dincer P, et al: An
autosomal recessive limb girdle muscular dystrophy (LGMD2) with
mild mental retardation is allelic to Walker-Warburg syndrome (WWS)
caused by a mutation in the POMT1 gene. Neuromuscul Disord.
15:271–275. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bolduc V, Marlow G, Boycott KM, et al:
Recessive mutations in the putative calcium-activated chloride
channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular
dystrophies. Am J Hum Genet. 86:213–221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Godfrey C, Escolar D, Brockington M, et
al: Fukutin gene mutations in steroid-responsive limb girdle
muscular dystrophy. Ann Neurol. 60:603–610. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Biancheri R, Falace A, Tessa A, et al:
POMT2 gene mutation in limb-girdle muscular dystrophy with
inflammatory changes. Biochem Biophys Res Commun. 363:1033–1037.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Clement EM, Godfrey C, Tan J, et al: Mild
POMGnT1 mutations underlie a novel limb-girdle muscular dystrophy
variant. Arch Neurol. 65:137–141. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hara Y, Balci-Hayta B, Yoshida-Moriguchi
T, et al: A dystroglycan mutation associated with limb-girdle
muscular dystrophy. N Engl J Med. 364:939–946. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nigro V, Aurino S and Piluso G: Limb
girdle muscular dystrophies: update on genetic diagnosis and
therapeutic approaches. Curr Opin Neurol. 24:429–436. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gundesli H, Talim B, Korkusuz P, et al:
Mutation in exon 1f of PLEC, leading to disruption of plectin
isoform 1f, causes autosomal-recessive limb-girdle muscular
dystrophy. Am J Hum Genet. 87:834–841. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
von Nandelstadh P, Grönholm M, Moza M,
Lamberg A, Savilahti H and Carpén O: Actin-organising properties of
the muscular dystrophy protein myotilin. Exp Cell Res. 310:131–139.
2005.PubMed/NCBI
|
|
37
|
Maraldi NM, Capanni C, Cenni V, Fini M and
Lattanzi G: Laminopathies and lamin-associated signaling pathways.
J Cell Biochem. 112:979–992. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gazzerro E, Sotgia F, Bruno C, Lisanti MP
and Minetti C: Caveolinopathies: from the biology of caveolin-3 to
human diseases. Eur J Hum Genet. 18:137–145. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schröder R and Schoser B: Myofibrillar
myopathies: a clinical and myopathological guide. Brain Pathol.
19:483–492. 2009.
|
|
40
|
Ojima K, Ono Y, Ottenheijm C, et al:
Non-proteolytic functions of calpain-3 in sarcoplasmic reticulum in
skeletal muscles. J Mol Biol. 407:439–449. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ojima K, Kawabata Y, Nakao H, et al:
Dynamic distribution of muscle-specific calpain in mice has a key
role in physical-stress adaptation and is impaired in muscular
dystrophy. J Clin Invest. 120:2672–2683. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bansal D, Miyake K, Vogel SS, et al:
Defective membrane repair in dysferlin-deficient muscular
dystrophy. Nature. 423:168–172. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen YW, Zhao P, Borup R and Hoffman EP:
Expression profiling in the muscular dystrophies: identification of
novel aspects of molecular pathophysiology. J Cell Biol.
151:1321–1336. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zou P, Pinotsis N, Lange S, et al:
Palindromic assembly of the giant muscle protein titin in the
sarcomeric Z-disk. Nature. 439:229–233. 2006. View Article : Google Scholar
|
|
45
|
Shieh PB, Kudryashova E and Spencer MJ:
Limb-girdle muscular dystrophy 2H and the role of TRIM32. Handb
Clin Neurol. 101:125–133. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Brockington M, Blake DJ, Prandini P, et
al: Mutations in the fukutin-related protein gene (FKRP) cause a
form of congenital muscular dystrophy with secondary laminin alpha2
deficiency and abnormal glycosylation of alpha-dystroglycan. Am J
Hum Genet. 69:1198–1209. 2001. View
Article : Google Scholar
|
|
47
|
Isralewitz B, Gao M and Schulten K:
Steered molecular dynamics and mechanical functions of proteins.
Curr Opin Struct Biol. 11:224–230. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Akasaka-Manya K, Manya H, Nakajima A,
Kawakita M and Endo T: Physical and functional association of human
protein O-mannosyltransferases 1 and 2. J Biol Chem.
281:19339–19345. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yamamoto T, Shibata N, Saito Y, Osawa M
and Kobayashi M: Functions of fukutin, a gene responsible for
Fukuyama type congenital muscular dystrophy, in neuromuscular
system and other somatic organs. Cent Nerv Syst Agents Med Chem.
10:169–179. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yoshida A, Kobayashi K, Manya H, et al:
Muscular dystrophy and neuronal migration disorder caused by
mutations in a glycosyltransferase, POMGnT1. Dev Cell. 1:717–724.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Barresi R and Campbell KP: Dystroglycan:
from biosynthesis to pathogenesis of human disease. J Cell Sci.
119:199–207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Moza M, Mologni L, Trokovic R, Faulkner G,
Partanen J and Carpén O: Targeted deletion of the muscular
dystrophy gene myotilin does not perturb muscle structure or
function in mice. Mol Cell Biol. 27:244–252. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Garvey SM, Liu Y, Miller SE and Hauser MA:
Myotilin overexpression enhances myopathology in the LGMD1A mouse
model. Muscle Nerve. 37:663–667. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Muchir A, Shan J, Bonne G, Lehnart SE and
Worman HJ: Inhibition of extracellular signal-regulated kinase
signaling to prevent cardiomyopathy caused by mutation in the gene
encoding A-type lamins. Hum Mol Genet. 18:241–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kawakami E, Kinouchi N, Adachi T, et al:
Atelocollagen-mediated systemic administration of
myostatin-targeting siRNA improves muscular atrophy in
caveolin-3-deficient mice. Dev Growth Differ. 53:48–54. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bartoli M, Poupiot J, Vulin A, et al:
AAV-mediated delivery of a mutated myostatin propeptide ameliorates
calpain 3 but not alpha-sarcoglycan deficiency. Gene Ther.
14:733–740. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Han R, Frett EM, Levy JR, et al: Genetic
ablation of complement C3 attenuates muscle pathology in
dysferlin-deficient mice. J Clin Invest. 120:4366–4374. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gallardo E, Rojas-García R, de Luna N, Pou
A, Brown RH Jr and Illa I: Inflammation in dysferlin myopathy:
immunohistochemical characterization of 13 patients. Neurology.
57:2136–2138. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ohsawa Y, Okada T, Nishimatsu S, et al: An
inhibitor of transforming growth factor beta type I receptor
ameliorates muscle atrophy in a mouse model of caveolin 3-deficient
muscular dystrophy. Lab Invest. 92:1100–1114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bartoli M, Roudaut C, Martin S, et al:
Safety and efficacy of AAV-mediated calpain 3 gene transfer in a
mouse model of limb-girdle muscular dystrophy type 2A. Mol Ther.
13:250–259. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Albrecht DE, Rufibach LE, Williams BA,
Monnier N, Hwang E and Mittal P: 5th Annual Dysferlin Conference;
11–14, July 2011; Chicago, Illinois, USA. Neuromuscul Disord. 22.
pp. 471–477. 2012, View Article : Google Scholar
|
|
62
|
Albrecht DE, Garg N, Rufibach LE, et al:
3rd Annual Dysferlin Conference; 2–5 June, 2009; Boston,
Massachusetts, USA. Neuromuscul Disord. 19. pp. 867–873. 2009,
View Article : Google Scholar
|
|
63
|
Lostal W, Bartoli M, Bourg N, et al:
Efficient recovery of dysferlin deficiency by dual adeno-associated
vector-mediated gene transfer. Hum Mol Genet. 19:1897–1907. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kong KY, Ren J, Kraus M, Finklestein SP
and Brown RH Jr: Human umbilical cord blood cells differentiate
into muscle in sjl muscular dystrophy mice. Stem Cells. 22:981–993.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Potgieter M, Pretorius E, Van der Merwe
CF, et al: Histological assessment of SJL/J mice treated with the
antioxidants coenzyme Q10 and resveratrol. Micron. 42:275–282.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Cordier L, Hack AA, Scott MO, et al:
Rescue of skeletal muscles of gamma-sarcoglycan-deficient mice with
adeno-associated virus-mediated gene transfer. Mol Ther. 1:119–129.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bogdanovich S, McNally EM and Khurana TS:
Myostatin blockade improves function but not histopathology in a
murine model of limb-girdle muscular dystrophy 2C. Muscle Nerve.
37:308–316. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Allikian MJ, Hack AA, Mewborn S, Mayer U
and McNally EM: Genetic compensation for sarcoglycan loss by
integrin alpha7beta1 in muscle. J Cell Sci. 117:3821–3830. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Allamand V, Donahue KM, Straub V, Davisson
RL, Davidson BL and Campbell KP: Early adenovirus-mediated gene
transfer effectively prevents muscular dystrophy in
alpha-sarcoglycan-deficient mice. Gene Ther. 7:1385–1391. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Galvez BG, Sampaolesi M, Brunelli S, et
al: Complete repair of dystrophic skeletal muscle by
mesoangioblasts with enhanced migration ability. J Cell Biol.
174:231–243. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Minetti GC, Colussi C, Adami R, et al:
Functional and morphological recovery of dystrophic muscles in mice
treated with deacetylase inhibitors. Nat Med. 12:1147–1150. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Dressman D, Araishi K, Imamura M, et al:
Delivery of alpha- and beta-sarcoglycan by recombinant
adeno-associated virus: efficient rescue of muscle, but
differential toxicity. Hum Gene Ther. 13:1631–1646. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hoshijima M, Hayashi T, Jeon YE, et al:
Delta-sarcoglycan gene therapy halts progression of cardiac
dysfunction, improves respiratory failure, and prolongs life in
myopathic hamsters. Circ Heart Fail. 4:89–97. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Iwata Y, Katanosaka Y, Shijun Z, et al:
Protective effects of Ca2+ handling drugs against abnormal Ca2+
homeostasis and cell damage in myopathic skeletal muscle cells.
Biochem Pharmacol. 70:740–751. 2005.
|
|
75
|
Zhu T, Zhou L, Mori S, et al: Sustained
whole-body functional rescue in congestive heart failure and
muscular dystrophy hamsters by systemic gene transfer. Circulation.
112:2650–2659. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Henning RJ, Aufman J, Shariff M, et al:
Human umbilical cord blood mononuclear cells decrease fibrosis and
increase cardiac function in cardiomyopathy. Regen Med. 5:45–54.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lapidos KA, Chen YE, Earley JU, et al:
Transplanted hematopoietic stem cells demonstrate impaired
sarcoglycan expression after engraftment into cardiac and skeletal
muscle. J Clin Invest. 114:1577–1585. 2004. View Article : Google Scholar
|
|
78
|
Nomura T, Ashihara E, Tateishi K, et al:
Skeletal myosphere-derived progenitor cell transplantation promotes
neovascularization in delta-sarcoglycan knockdown cardiomyopathy.
Biochem Biophys Res Commun. 352:668–674. 2007. View Article : Google Scholar
|
|
79
|
Parsons SA, Millay DP, Sargent MA, McNally
EM and Molkentin JD: Age-dependent effect of myostatin blockade on
disease severity in a murine model of limb-girdle muscular
dystrophy. Am J Pathol. 168:1975–1985. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Goehringer C, Rutschow D, Bauer R, et al:
Prevention of cardiomyopathy in delta-sarcoglycan knockout mice
after systemic transfer of targeted adeno-associated viral vectors.
Cardiovasc Res. 82:404–410. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Charton K, Danièle N, Vihola A, et al:
Removal of the calpain 3 protease reverses the myopathology in a
mouse model for titinopathies. Hum Mol Genet. 19:4608–4624. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Barresi R, Michele DE, Kanagawa M, et al:
LARGE can functionally bypass alpha-dystroglycan glycosylation
defects in distinct congenital muscular dystrophies. Nat Med.
10:696–703. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
83
|
Straub V, Donahue KM, Allamand V, Davisson
RL, Kim YR and Campbell KP: Contrast agent-enhanced magnetic
resonance imaging of skeletal muscle damage in animal models of
muscular dystrophy. Magn Reson Med. 44:655–659. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Bartoli M, Poupiot J, Goyenvalle A, et al:
Noninvasive monitoring of therapeutic gene transfer in animal
models of muscular dystrophies. Gene Ther. 13:20–28. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Mendell JR, Rodino-Klapac LR, Rosales XQ,
et al: Sustained alpha-sarcoglycan gene expression after gene
transfer in limb-girdle muscular dystrophy, type 2D. Ann Neurol.
68:629–638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Norwood FL, Harling C, Chinnery PF, Eagle
M, Bushby K and Straub V: Prevalence of genetic muscle disease in
Northern England: in-depth analysis of a muscle clinic population.
Brain. 132:3175–3186. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Gómez-Díaz B, Rosas-Vargas H,
Roque-Ramírez B, et al: Immunodetection analysis of muscular
dystrophies in Mexico. Muscle Nerve. 45:338–345. 2012.PubMed/NCBI
|
|
88
|
Diniz G, Eryaşar G, Türe S, et al: A
regional panorama of dysferlinopathies. Turk Patoloji Derg.
28:259–265. 2012.PubMed/NCBI
|
|
89
|
Magri F, Bo RD, D’Angelo MG, et al:
Frequency and characterisation of anoctamin 5 mutations in a cohort
of Italian limb-girdle muscular dystrophy patients. Neuromuscul
Disord. 22:934–943. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Guglieri M, Magri F, D’Angelo MG, et al:
Clinical, molecular, and protein correlations in a large sample of
genetically diagnosed Italian limb girdle muscular dystrophy
patients. Hum Mutat. 29:258–266. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Fanin M, Nascimbeni AC, Aurino S, et al:
Frequency of LGMD gene mutations in Italian patients with distinct
clinical phenotypes. Neurology. 72:1432–1435. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Urtasun M, Sáenz A, Roudaut C, et al:
Limb-girdle muscular dystrophy in Guipúzcoa (Basque Country,
Spain). Brain. 121:1735–1747. 1998.
|
|
93
|
Walter MC, Petersen JA, Stucka R, et al:
FKRP (826C>A) frequently causes limb-girdle muscular dystrophy
in German patients. J Med Genet. 41:e502004.
|
|
94
|
Hicks D, Sarkozy A, Muelas N, et al: A
founder mutation in Anoctamin 5 is a major cause of limb-girdle
muscular dystrophy. Brain. 134:171–182. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Stensland E, Lindal S, Jonsrud C, et al:
Prevalence, mutation spectrum and phenotypic variability in
Norwegian patients with Limb Girdle Muscular Dystrophy 2I.
Neuromuscul Disord. 21:41–46. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Sveen ML, Schwartz M and Vissing J: High
prevalence and phenotype-genotype correlations of limb girdle
muscular dystrophy type 2I in Denmark. Ann Neurol. 59:808–815.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Penttila S, Palmio J, Suominen T, et al:
Eight new mutations and the expanding phenotype variability in
muscular dystrophy caused by ANO5. Neurology. 78:897–903. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Lo HP, Cooper ST, Evesson FJ, et al:
Limb-girdle muscular dystrophy: diagnostic evaluation, frequency
and clues to pathogenesis. Neuromuscul Disord. 18:34–44. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Moore SA, Shilling CJ, Westra S, et al:
Limb-girdle muscular dystrophy in the United States. J Neuropathol
Exp Neurol. 65:995–1003. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Dinçer PP, Leturcq F, Richard I, et al: A
biochemical, genetic, and clinical survey of autosomal recessive
limb girdle muscular dystrophies in Turkey. Ann Neurol. 42:222–229.
1997.PubMed/NCBI
|
|
101
|
Pogoda TV, Krakhmaleva IN, Lipatova NA,
Shakhovskaya NI, Shishkin SS and Limborska SA: High incidence of
550delA mutation of CAPN3 in LGMD2 patients from Russia. Hum Mutat.
15:2952000. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Zatz M, de Paula F, Starling A and Vainzof
M: The 10 autosomal recessive limb-girdle muscular dystrophies.
Neuromuscul Disord. 13:532–544. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Chae J, Minami N, Jin Y, et al: Calpain 3
gene mutations: genetic and clinico-pathologic findings in
limb-girdle muscular dystrophy. Neuromuscul Disord. 11:547–555.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Hayashi S, Ohsawa Y, Takahashi T, et al:
Rapid screening for Japanese dysferlinopathy by fluorescent primer
extension. Intern Med. 49:2693–2696. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Meena AK, Sreenivas D, Sundaram C, et al:
Sarcoglycanopathies: a clinico-pathological study. Neurol India.
55:117–121. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Pathak P, Sharma MC, Sarkar C, et al: Limb
girdle muscular dystrophy type 2A in India: a study based on
semi-quantitative protein analysis, with clinical and
histopathological correlation. Neurol India. 58:549–554. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Gayathri N, Alefia R, Nalini A, et al:
Dysferlinopathy: spectrum of pathological changes in skeletal
muscle tissue. Indian J Pathol Microbiol. 54:350–354. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Hadj Salem I, Kamoun F, Louhichi N, Trigui
M, Triki C and Fakhfakh F: Impact of single-nucleotide
polymorphisms at the TP53-binding and responsive promoter region of
BCL2 gene in modulating the phenotypic variability of LGMD2C
patients. Mol Biol Rep. 39:7479–7486. 2012.PubMed/NCBI
|
|
109
|
Sinnreich M, Therrien C and Karpati G:
Lariat branch point mutation in the dysferlin gene with mild
limb-girdle muscular dystrophy. Neurology. 66:1114–1116. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Tagawa K, Ogawa M, Kawabe K, et al:
Protein and gene analyses of dysferlinopathy in a large group of
Japanese muscular dystrophy patients. J Neurol Sci. 211:23–28.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Takahashi T, Aoki M, Tateyama M, et al:
Dysferlin mutations in Japanese Miyoshi myopathy: relationship to
phenotype. Neurology. 60:1799–1804. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Nagashima T, Chuma T, Mano Y, et al:
Dysferlinopathy associated with rigid spine syndrome.
Neuropathology. 24:341–346. 2004. View Article : Google Scholar
|
|
113
|
Saccone V, Palmieri M, Passamano L, et al:
Mutations that impair interaction properties of TRIM32 associated
with limb-girdle muscular dystrophy 2H. Hum Mutat. 29:240–247.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Kondo-lida E, Kobayashi K, Watanabe M, et
al: Novel mutations and genotype-phenotype relationships in 107
families with Fukuyama-type congenital muscular dystrophy (FCMD).
Hum Mol Genet. 8:2303–2309. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Scharner J, Gnocchi VF, Ellis JA and
Zammit PS: Genotype-phenotype correlations in laminopathies: how
does fate translate? Biochem Soc Trans. 38:257–262. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhang Y, Ye J, Chen D, et al: Differential
expression profiling between the relative normal and dystrophic
muscle tissues from the same LGMD patient. J Transl Med. 4:532006.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Godfrey C, Clement E, Mein R, et al:
Refining genotype phenotype correlations in muscular dystrophies
with defective glycosylation of dystroglycan. Brain. 130:2725–2735.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Klinge L, Dean AF, Kress W, et al: Late
onset in dysferlinopathy widens the clinical spectrum. Neuromuscul
Disord. 18:288–290. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Rosales XQ, Gastier-Foster JM, Lewis S, et
al: Novel diagnostic features of dysferlinopathies. Muscle Nerve.
42:14–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Broglio L, Tentorio M, Cotelli MS, et al:
Limb-girdle muscular dystrophy-associated protein diseases.
Neurologist. 16:340–352. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Mercuri E, Brockington M, Straub V, et al:
Phenotypic spectrum associated with mutations in the
fukutin-related protein gene. Ann Neurol. 53:537–542. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Gáti I, Danielsson O, Gunnarsson C, et al:
Bent spine syndrome: a phenotype of dysferlinopathy or a
symptomatic DYSF gene mutation carrier. Eur Neurol. 67:300–302.
2012.PubMed/NCBI
|
|
123
|
Hermans MC, Pinto YM, Merkies IS, de
Die-Smulders CE, Crijns HJ and Faber CG: Hereditary muscular
dystrophies and the heart. Neuromuscul Disord. 20:479–492. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Filosto M, Tonin P, Vattemi G, et al:
Chronic ophthalmoparesis in limb girdle muscular dystrophy 1C. J
Neurol Neurosurg Psychiatry. 80:448–449. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Selcen D: Myofibrillar myopathies.
Neuromuscul Disord. 21:161–171. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Palmieri A, Manara R, Bello L, et al:
Cognitive profile and MRI findings in limb-girdle muscular
dystrophy 2I. J Neurol. 258:1312–1320. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zhang Y, Huang JJ, Wang ZQ, Wang N and Wu
ZY: Value of muscle enzyme measurement in evaluating different
neuromuscular diseases. Clin Chim Acta. 413:520–524. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Wattjes MP, Kley RA and Fischer D:
Neuromuscular imaging in inherited muscle diseases. Eur Radiol.
20:2447–2460. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Fischer D, Walter MC, Kesper K, et al:
Diagnostic value of muscle MRI in differentiating LGMD2I from other
LGMDs. J Neurol. 252:538–547. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Fischer D, Kley RA, Strach K, et al:
Distinct muscle imaging patterns in myofibrillar myopathies.
Neurology. 71:758–765. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Starling A, de Paula F, Silva H, Vainzof M
and Zatz M: Calpainopathy: how broad is the spectrum of clinical
variability? J Mol Neurosci. 21:233–236. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Santoro L, Nolano M, Faraso S, et al:
Perioral skin biopsy to study skeletal muscle protein expression.
Muscle Nerve. 41:392–398. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Nagaraju K, Rawat R, Veszelovszky E, et
al: Dysferlin deficiency enhances monocyte phagocytosis: a model
for the inflammatory onset of limb-girdle muscular dystrophy 2B. Am
J Pathol. 172:774–785. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Brown RH Jr and Amato A: Calpainopathy and
eosinophilic myositis. Ann Neurol. 59:875–877. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Baumeister SK, Todorovic S, Milić-Rasić V,
Dekomien G, Lochmüller H and Walter MC: Eosinophilic myositis as
presenting symptom in gamma-sarcoglycanopathy. Neuromuscul Disord.
19:167–171. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Vinit J, Samson M Jr, Gaultier JB, et al:
Dysferlin deficiency treated like refractory polymyositis. Clin
Rheumatol. 29:103–106. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Claeys KG, Fardeau M, Schröder R, et al:
Electron microscopy in myofibrillar myopathies reveals clues to the
mutated gene. Neuromuscul Disord. 18:656–666. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Cacciottolo M, Numitone G, Aurino S, et
al: Muscular dystrophy with marked Dysferlin deficiency is
consistently caused by primary dysferlin gene mutations. Eur J Hum
Genet. 19:974–980. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Trabelsi M, Kavian N, Daoud F, et al:
Revised spectrum of mutations in sarcoglycanopathies. Eur J Hum
Genet. 16:793–803. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Herrmann R, Straub V, Blank M, et al:
Dissociation of the dystroglycan complex in caveolin-3-deficient
limb girdle muscular dystrophy. Hum Mol Genet. 9:2335–2340. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Vainzof M, Moreira ES, Suzuki OT, et al:
Telethonin protein expression in neuromuscular disorders. Biochim
Biophys Acta. 1588:33–40. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Charlton R, Henderson M, Richards J, et
al: Immunohistochemical analysis of calpain 3: advantages and
limitations in diagnosing LGMD2A. Neuromuscul Disord. 19:449–457.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Fanin M, Nascimbeni AC, Fulizio L,
Trevisan CP, Meznaric-Petrusa M and Angelini C: Loss of calpain-3
autocatalytic activity in LGMD2A patients with normal protein
expression. Am J Pathol. 163:1929–1936. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Fanin M, Nascimbeni AC, Tasca E and
Angelini C: How to tackle the diagnosis of limb-girdle muscular
dystrophy 2A. Eur J Hum Genet. 17:598–603. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Sáenz A, Leturcq F, Cobo AM, et al:
LGMD2A: genotype-phenotype correlations based on a large mutational
survey on the calpain 3 gene. Brain. 128:732–742. 2005.PubMed/NCBI
|
|
146
|
Groen EJ, Charlton R, Barresi R, et al:
Analysis of the UK diagnostic strategy for limb girdle muscular
dystrophy 2A. Brain. 130:3237–3249. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Fanin M, Fulizio L, Nascimbeni AC, et al:
Molecular diagnosis in LGMD2A: mutation analysis or protein
testing? Hum Mutat. 24:52–62. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Hackman P, Vihola A, Haravuori H, et al:
Tibial muscular dystrophy is a titinopathy caused by mutations in
TTN, the gene encoding the giant skeletal-muscle protein titin. Am
J Hum Genet. 71:492–500. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
149
|
Borg K, Stucka R, Locke M, et al:
Intragenic deletion of TRIM32 in compound heterozygotes with
sarcotubular myopathy/LGMD2H. Hum Mutat. 30:E831–844. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Sewry CA: Muscular dystrophies: an update
on pathology and diagnosis. Acta Neuropathol. 120:343–358. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Ho M, Gallardo E, McKenna-Yasek D, De Luna
N, Illa I and Brown RH Jr: A novel, blood-based diagnostic assay
for limb girdle muscular dystrophy 2B and Miyoshi myopathy. Ann
Neurol. 51:129–133. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Escher C, Lochmüller H, Fischer D, et al:
Reverse protein arrays as novel approach for protein quantification
in muscular dystrophies. Neuromuscul Disord. 20:302–309. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Blázquez L, Azpitarte M, Sáenz A, et al:
Characterization of novel CAPN3 isoforms in white blood cells: an
alternative approach for limb-girdle muscular dystrophy 2A
diagnosis. Neurogenetics. 9:173–182. 2008.PubMed/NCBI
|
|
154
|
De Luna N, Freixas A, Gallano P, et al:
Dysferlin expression in monocytes: a source of mRNA for mutation
analysis. Neuromuscul Disord. 17:69–76. 2007.PubMed/NCBI
|
|
155
|
Teer JK and Mullikin JC: Exome sequencing:
the sweet spot before whole genomes. Hum Mol Genet. 19:R145–151.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Balci B, Aurino S, Haliloglu G, et al:
Calpain-3 mutations in Turkey. Eur J Pediatr. 165:293–298. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Fanin M, Nascimbeni AC, Fulizio L and
Angelini C: The frequency of limb girdle muscular dystrophy 2A in
northeastern Italy. Neuromuscul Disord. 15:218–224. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Takahashi T, Aoki M, Suzuki N, et al:
Clinical features and a mutation with late onset of limb girdle
muscular dystrophy 2B. J Neurol Neurosurg Psychiatry. 84:433–440.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Park YE, Kim HS, Lee CH, Nam TS, Choi YC
and Kim DS: Two common mutations (p.Gln832X and c.663+1G>C)
account for about a third of the DYSF mutations in Korean patients
with dysferlinopathy. Neuromuscul Disord. 22:505–510.
2012.PubMed/NCBI
|
|
160
|
Barthélémy F, Wein N, Krahn M, Lévy N and
Bartoli M: Translational research and therapeutic perspectives in
dysferlinopathies. Mol Med. 17:875–882. 2011.PubMed/NCBI
|
|
161
|
Hackman P, Juvonen V, Sarparanta J, et al:
Enrichment of the R77C alpha-sarcoglycan gene mutation in Finnish
LGMD2D patients. Muscle Nerve. 31:199–204. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Schoser BG, Frosk P, Engel AG, Klutzny U,
Lochmüller H and Wrogemann K: Commonality of TRIM32 mutation in
causing sarcotubular myopathy and LGMD2H. Ann Neurol. 57:591–595.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Kang PB, Feener CA, Estrella E, et al:
LGMD2I in a North American population. BMC Musculoskelet Disord.
8:1152007. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Norwood F, de Visser M, Eymard B,
Lochmüller H and Bushby K: EFNS guideline on diagnosis and
management of limb girdle muscular dystrophies. Eur J Neurol.
14:1305–1312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Eagle M: Report on the muscular dystrophy
campaign workshop: exercise in neuromuscular diseases; Newcastle.
January 2002; Neuromuscul Disord. 12. pp. 975–983. 2002, View Article : Google Scholar
|
|
166
|
Miladi N, Bourguignon JP and Hentati F:
Cognitive and psychological profile of a Tunisian population of
limb girdle muscular dystrophy. Neuromuscul Disord. 9:352–354.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Wagner KR, Fleckenstein JL, Amato AA, et
al: A phase I/II trial of MYO-029 in adult subjects with muscular
dystrophy. Ann Neurol. 63:561–571. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Lerario A, Cogiamanian F, Marchesi C, et
al: Effects of rituximab in two patients with dysferlin-deficient
muscular dystrophy. BMC Musculoskelet Disord. 11:1572010.
View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Hattori H, Nagata E, Oya Y, et al: A novel
compound heterozygous dysferlin mutation in Miyoshi myopathy
siblings responding to dantrolene. Eur J Neurol. 14:1288–1291.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Luna ND, Díaz-Manera J, Paradas C, et al:
1α,25(OH)(2)-Vitamin D3 increases dysferlin expression in vitro and
in a human clinical trial. Mol Ther. 20:1988–1997. 2012.
|
|
171
|
Walter MC, Reilich P, Thiele S, et al:
Treatment of dysferlinopathy with deflazacort: a double-blind,
placebo-controlled clinical trial. Orphanet J Rare Dis. 8:262013.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Angelini C, Fanin M, Menegazzo E, Freda
MP, Duggan DJ and Hoffman EP: Homozygous alpha-sarcoglycan mutation
in two siblings: one asymptomatic and one steroid-responsive mild
limb-girdle muscular dystrophy patient. Muscle Nerve. 21:769–775.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Darin N, Kroksmark AK, Ahlander AC,
Moslemi AR, Oldfors A and Tulinius M: Inflammation and response to
steroid treatment in limb-girdle muscular dystrophy 2I. Eur J
Paediatr Neurol. 11:353–357. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Walter MC, Lochmüller H, Reilich P, et al:
Creatine monohydrate in muscular dystrophies: A double-blind,
placebo-controlled clinical study. Neurology. 54:1848–1850. 2000.
View Article : Google Scholar : PubMed/NCBI
|















