Introduction
The inhibitor of growth 1 (ING1) gene was previously
identified and characterized as a type-II tumor suppressor gene.
The ING1 gene is located on chromosome 13q34 and encodes a minimum
of 4 protein isoforms (p47ING1a, p33ING1b,
p24ING1c, and p27ING1d), as a result of the
effects of various promoters, exons and alternative splicing
(1). The ING1b gene exists in
numerous species, including humans, mice, rats, frogs and yeast
(2). The ING1b proteins have
significant roles in the regulation of chromatin structure,
inhibition of cell proliferation (3),
cell cycle regulation (4), apoptosis
(5), damaged DNA repair (6), regulation of gene transcription and
other biological functions (7). Of
these four variants, p33ING1b is the isoform which is
most extensively expressed in human tissues, and has therefore been
the most intensively analyzed. Studies have indicated that
p33ING1b is expressed in the majority of normal cells
and tissues, suggesting that it may have fundamental functions
(8–10). The expression of p33ING1b
is significantly downregulated in multiple human malignancies,
including breast, esophageal, gastric and brain cancer, as well as
leukemia (11–15). Blocking ING1 expression has been shown
to enhance cell proliferation in vitro and tumor formation
in vivo (16). Additionally,
adenovirus-mediated ING1b gene transfer has been demonstrated to
significantly suppress growth and increase apoptosis in glioma
(17) and gastric adenocarcinoma
cells (18). These results suggest
that ING1b may be a universal tumor suppressor gene.
Colorectal cancer (CRC) is the third most common
human malignancy amongst males and the second most common amongst
females, with >1.2 million novel cases diagnosed and 608,700
associated mortalities worldwide, in 2008 (19). It has been generally accepted that CRC
develops through step-wise genetic alterations (20). Analysis of the molecular mechanism of
carcinogenesis facilitates the development of novel approaches for
the prevention and treatment of a particular cancer (21). However, the carcinogenic mechanisms of
CRC remain far from being fully elucidated. Although previous
reports have demonstrated that the expression of
p33ING1b is downregulated at the mRNA level in CRC
(22), the role of
p33ING1b in colorectal carcinogenesis has yet to be
investigated.
In the present study, the expression of
p33ING1b in CRC tissues was evaluated, and the effects
of adenovirus-mediated p33ING1b ectopic expression in
colorectal adenocarcinoma cells in vitro were
investigated.
Materials and methods
Patients and tissue preparation
CRC samples and adjacent non-malignant tissues,
which were ≥2 cm from the tumor, were collected endoscopically from
10 patients with histologically verified colorectal adenocarcinomas
at the First Affiliated Hospital, School of Medicine, Xi'an Jiaotong
University (Xi'an, China). The samples were immediately snap-frozen
in liquid nitrogen and stored at −80°C. This study was approved by
the ethics committee of The First Affiliated Hospital, School of
Medicine, Xi'an Jiaotong University (Xi'an, China). Written
informed consent was obtained from all patients.
Cell culture and adenoviral
infection
The human colorectal adenocarcinoma cell lines
SW480, HT29 and LoVo were obtained from the American Type Culture
Collection (Manassas, VA, USA). The cells were maintained in
Dulbecco's modified Eagle's medium (DMEM; Gibco-BRL, Grand Island,
NY, USA) supplemented with 10% fetal calf serum (FCS; Gibco-BRL),
100 U/ml penicillin [Runze Pharmaceutical (Suzhou) Co., Ltd.,
Suzhou, China] and 100 µg/ml streptomycin [Runze Pharmaceutical
(Suzhou) Co., Ltd.] at 37°C in a humidified 5% CO2
atmosphere.
Recombinant adenoviruses
Recombinant adenoviruses (type 5) encoding ING1b
(Ad-ING1b) or green fluorescent protein (Ad-GFP), were constructed
as previously reported (18).
Briefly, p33ING1b was cloned into the adenoviral shuttle plasmid,
pShuttle-CMV (Stratagene, La Jolla, CA, USA). The recombinant
pShuttle-CMV-p33ING1b plasmid was digested by PmeI (New England
Biolabs, Ipswich, MA, USA) to linearize, followed by homologous
recombination with adenoviral bone plasmid pAdEasy-1 (Stratagene)
in E.coli BJ5183. The positive recombinant adenoviral
plasmid, pAd-p33ING1b, was identified by PacI digestion (New
England Biolabs) and the recombinant adenovirus, Ad-p33ING1b, was
obtained following transfection in HEK293 cells (Xi'an Huaguang
Biological Engineering. Co., Ltd., Xi'an, China). The
cytopathogenic effect was observed under a fluorescent microscope
(IX-50; Olympus Corporation, Tokyo, Japan). The titers of
adenoviral stocks were determined by polymerase chain reaction
(Qiagen, Hilden, Germany) performed under the following conditions:
Initial denaturation at 95°C for 10 sec followed by 40 cycles of
denaturation at 95°C for 5 sec, annealing at 50°C for 15 sec and
elongation at 72°C for 23 sec. The viral stocks were aliquoted and
stored in 10% glycerol at −80°C prior to use.
Adenovirus-mediated gene transfer
The extent of the adenovirus-mediated gene transfer
in the three colorectal adenocarcinoma cell lines was determined by
measuring the expression of GFP 72 h post-infection with Ad-GFP.
Briefly, when the cells reached ~70–90% confluence, the medium was
aspirated and the cell monolayer was washed with pre-warmed sterile
phosphate-buffed saline (PBS; Gibco-BRL). The cells were incubated
with Ad-GFP at various multiplicity of infections (MOIs: 0, 10, 20,
40, 80 and 160) at 37°C. Two hours later, 2 ml of fresh growth
medium was added and the cells were incubated for an additional 72
h at 37°C. Subsequently, the transduction efficiency was evaluated
under a fluorescence microscope (BX61; Olympus Corporation). When
the cells were infected with Ad-GFP at an MOI of 40, ~70% of the
cells were identified to be GFP-positive, without exhibiting marked
toxic effects. Higher MOIs did not result in higher transduction
efficiencies but did result in toxicity. Therefore, the subsequent
experiments were performed using Ad-ING1b and Ad-GFP at an MOI of
40.
Western blot analysis
Colonic tissues and cultured cells were homogenized
in radioimmunoprecipitation lysis buffer (50 mM Tris-HCl, 150 mM
NaCl, 0.1% SDS, 0.02% sodium azide, 1% Nonidet P-40, 0.5% sodium
deoxycholate and protease inhibitor cocktail; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) in an ice-bath for 30
min. The extracts were clarified by centrifugation at 17,465 × for
10 min at 4°C. Following measurement of the protein concentrations
using the Bradford assay (23), 100
µg total protein was subjected to 10% SDS-PAGE (Santa Cruz
Biotechnology, Inc.). Following electrophoresis, the proteins were
electrophoretically transferred onto a nitrocellulose membrane (EMD
Millipore, Bedford, MA, USA), and blocked with 5% nonfat dry milk
in PBS with 0.2% Tween-20 for 1 h. Subsequently, the membrane was
incubated overnight at 4°C with primary mouse anti-human antibodies
against p33ING1b (polyclonal antibody prepared in the
Reproductive Medical Laboratory of Xi'an Jiaotong University; 1:400
dilution) or β-actin (1:1,000 dilution; Sigma-Aldrich, St. Louis,
MO, USA). Following three 15-min washes with Tris-buffered saline
with 0.1% Tween-20 (Sigma-Aldrich), the membrane was incubated with
horseradish peroxidase-conjugated goat anti-mouse Immunoglobulin G
as a secondary antibody (1:2,000 dilution; Sigma-Aldrich) for 1 h
at room temperature. Visualization of the blots was accomplished
using enhanced chemiluminescence solution (Pierce, Rockford, IL,
USA).
MTT assay
An MTT assay was conducted to assess the number of
viable cells pre- and post-infection at 24 h intervals. The cells
were seeded in 96-well plates at a density of 1×103
cells/well and cultured for 24 h at 37°C with 5% CO2.
Subsequently, the cells were infected with Ad-GFP or Ad-ING1b,or
treated with PBS, as described above. At the indicated time-points
(day 1, 2, 3, 4, 5, 6, 7 and 8), 5 mg/ml MTT (Sigma-Aldrich) was
added to each well, and the cells were incubated at 37°C for 4 h
prior to removal of the supernatants. The crystals were dissolved
in 150 µl dimethyl sulfoxide (Sigma-Aldrich). The absorbance was
examined using an automated microplate reader (BioTek Instruments,
Inc., Winooski, VT, USA) at an absorption wavelength of 490 nm.
Each sample was evaluated in triplicate, and three independent
experiments were conducted.
Cell cycle analysis
The cells were cultured in 6-well plates (Corning
Inc., Corning, NY, USA) with growth medium for 24 h prior to
adenoviral infection. Twenty-four hours post-infection, the cells
were cultured with DMEM without bovine serum (serum starvation) for
24 h to achieve cell cycle synchronization. The cells were then
cultured with complete growth medium (Gibco-BRL) for 48 h. The
harvested cells were washed twice with ice-cold PBS and fixed with
70% ethanol at 4°C overnight. Following RNase A (20 mg/ml;
Sigma-Aldrich) digestion at 37°C for 30 min, the cells were stained
with 50 mg/ml propidium iodide (PI; Sigma-Aldrich) at 4°C for 30
min in the dark. The cell cycle distribution was analyzed by flow
cytometry (FCM; FACSCalibur; BD Biosciences, San Jose, CA,
USA).
Apoptosis detection
Apoptosis was determined by staining cells with
Annexin V (ApoScreen Annexin V; Southern-Biotech, Birmingham, AL,
USA) and PI (Sigma-Aldrich) according to the manufacturer's
instructions. Briefly, the cells were cultured in 6-well plates
(Corning Inc.) and infected with adenoviruses as described.
Seventy-two hours post-infection, the cells were harvested and
resuspended in binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl,
2.5 mM CaCl2 and 0.5% bovine serum albumin;
Southern-Biotech) at a concentration of 1×106 cells/ml.
Following incubation with Annexin V-fluorescein isothiocyanate
(Southern-Biotech) in an ice bath in the dark for 15 min, binding
buffer and PI were added to the cells. The cells were then analyzed
immediately using a FACScan flow cytometer (BD Biosciences).
Statistical analysis
All statistical analyses were performed using SPSS
software version 19.0 (SPSS, Inc., Chicago, IL, USA). All values
are presented as the mean ± standard deviation. Statistical
analyses of the data were performed using one-way analysis of
variance followed by the Student-Newman-Keuls method. P<0.05 was
considered to indicate a statistically significant difference.
Results
p33ING1b expression is
downregulated in CRC tissues
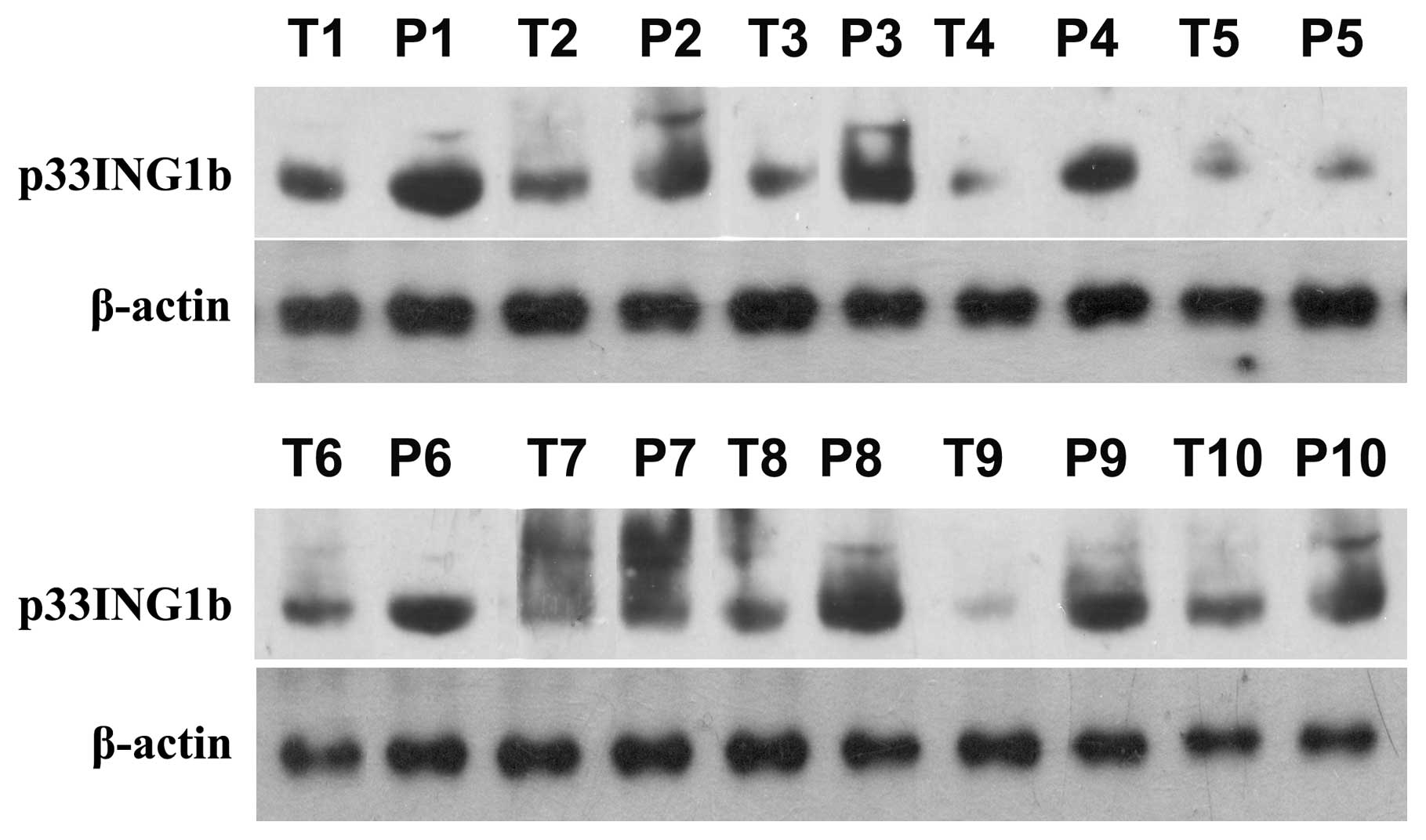
Western blot analysis of p33ING1b
expression in CRC tissues demonstrated that p33ING1b was
steadily expressed in the non-tumorous colonic tissues. However, in
the colonic adenocarcinoma tissues, the expression of
p33ING1b was markedly lower than that in the adjacent
non-tumorous colonic tissues. The results indicated that the
expression of p33ING1b was downregulated in CRC tissues
(Fig. 1; P<0.05).
Recombinant adenovirus-mediated
p33ING1b expression in colorectal adenocarcinoma
cells
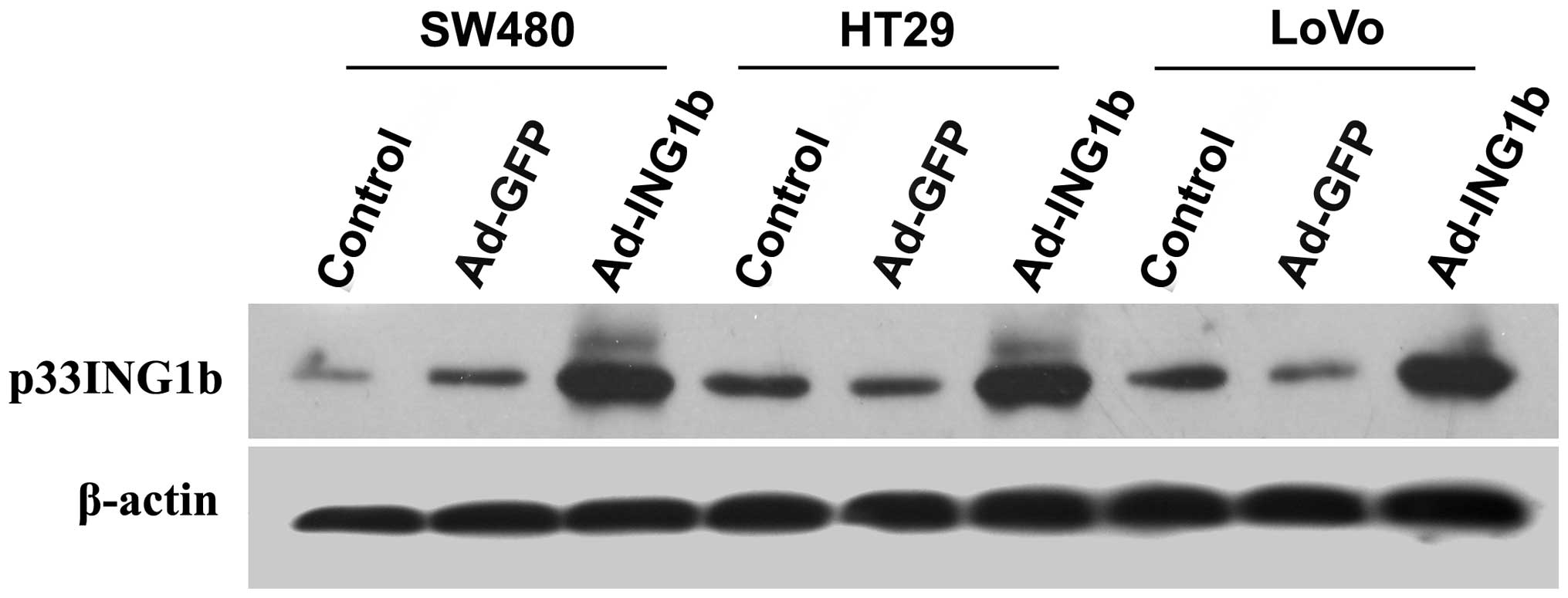
To verify the ectopic expression of
p33ING1b mediated by adenoviral infection, western blot
analysis with a specific antibody against p33ING1b was
performed. As shown in Fig. 2, low
levels of p33ING1b expression were detected in all three
CRC cell lines following infection with Ad-GFP, which had no
notable effect on the expression of p33ING1b protein.
Ad-ING1b-infected cells demonstrated markedly higher
p33ING1b expression levels than those of the
Ad-GFP-infected or blank control cells (P<0.05).
p33ING1b suppresses
proliferation of colorectal adenocarcinoma cells
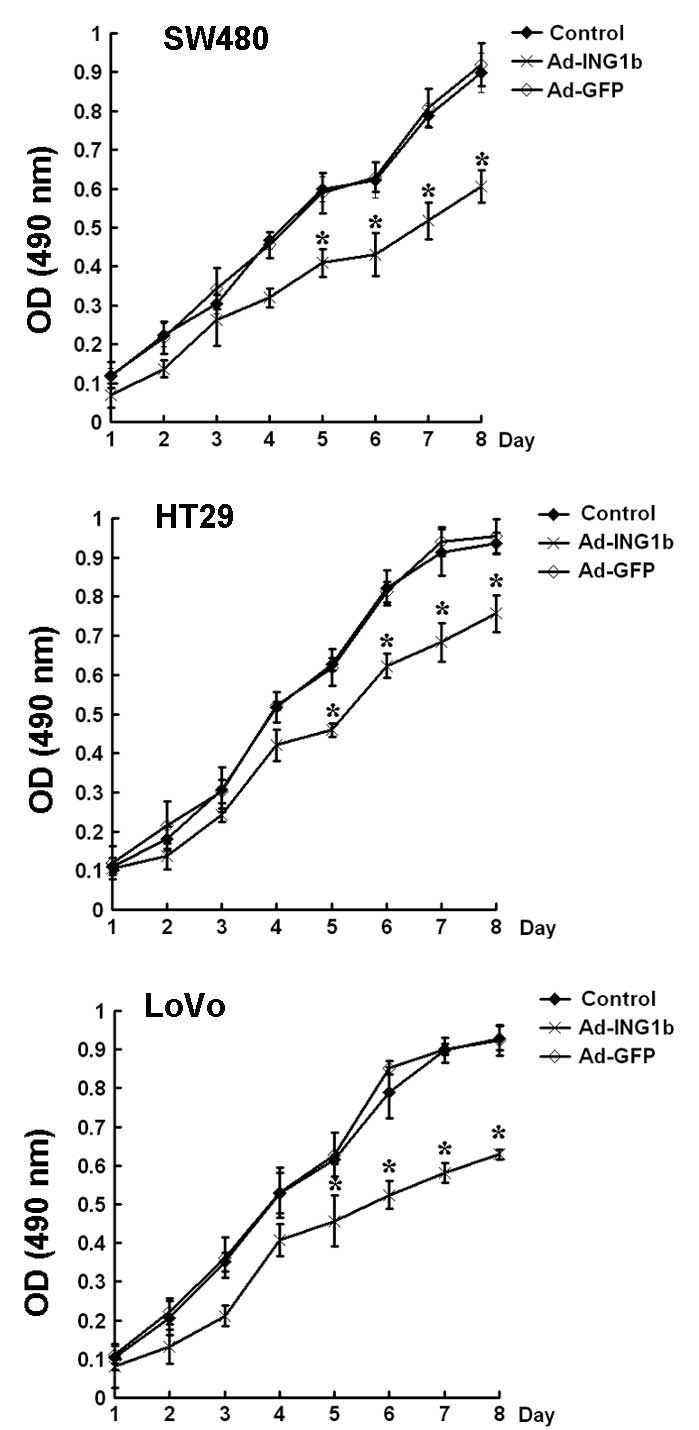
Subsequently, the effect of p33ING1b on
the proliferation of colorectal cancer cells in vitro was
examined using an MTT assay. The results demonstrated that the
three colorectal cancer cell lines exhibited slower growth rates
following Ad-ING1b infection than those of cells infected with
Ad-GFP at the same MOI or the blank control (P<0.05). No
significant difference in growth rate was observed between the
Ad-GFP and blank control groups (Fig.
3).
Ectopic expression of
p33ING1b induces apoptosis in colorectal adenocarcinoma
cells
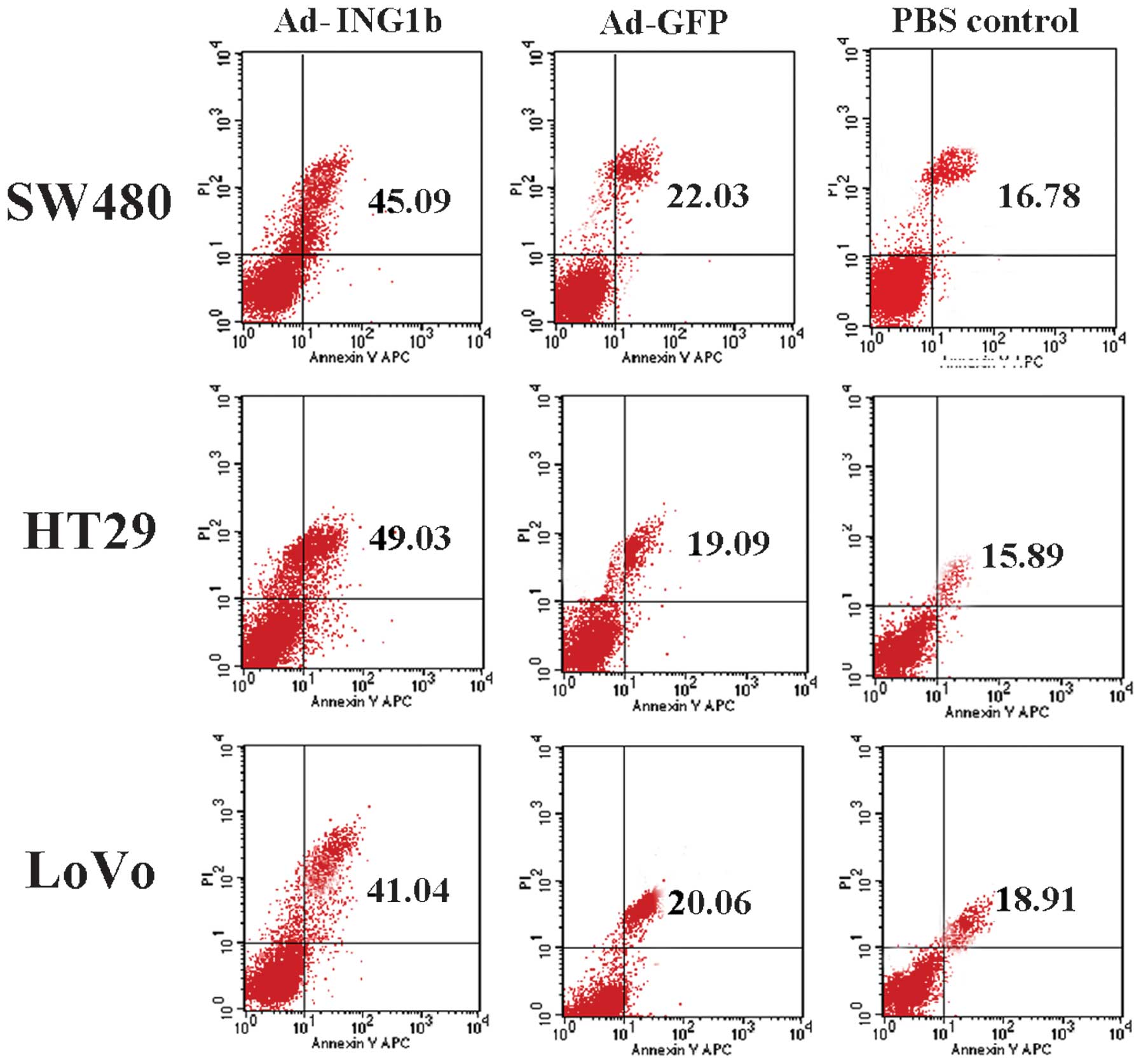
Apoptosis, or programmed cell death, has a
significant role in the development of CRC. To examine the role of
p33ING1b in the apoptosis of CRC cells, apoptotic cells
were evaluated following Ad-ING1b infection by Annexin V-PI
staining followed by FCM analysis. For all three cell lines,
Ad-ING1b transduction resulted in an increase in apoptosis,
compared with that of cells infected with Ad-GFP or the blank
control. The Ad-GFP-infected cells demonstrated a slight increase
in apoptotic ratio compared with that of the blank control group
(Fig. 4). These results demonstrated
that ectopic expression of p33ING1b induces apoptosis in
colorectal adenocarcinoma cells.
p33ING1b induces G1 phase
cell cycle arrest in colorectal adenocarcinoma cells
To further study the potential mechanisms underlying
the growth-inhibitory and pro-apoptotic effects of
p33ING1b in colorectal adenocarcinoma cells, alterations
in the cell cycle distribution following adenoviral infection were
analyzed. As shown in Fig. 5, the
Ad-GFP and blank control groups exhibited similar cell cycle
profiles, indicating that the expression of Ad-GFP exerted little
effect on the cell cycle. However, Ad-ING1b infection resulted in
an enlarged proportion of cells in G1 phase in all three cell
lines. These results suggested that ectopic p33ING1b may
inhibit proliferation and induce apoptosis in colorectal
adenocarcinoma cells by arresting the cells in G1 phase.
In accordance with the results of Annexin V-PI
staining and FCM analysis of apoptotic cells (Fig. 4), analysis of the cell cycle
distribution by FCM also indicated that Ad-ING1b infection induced
an increase in the number of apoptotic cells (Fig. 5).
Discussion
It has previously been demonstrated that the
expression of ING1b is downregulated in malignancies derived from
various organs and tissues, and that downregulation of
p33ING1b has a significant role in carcinogenesis
(24–28). The function of p33ING1b in
colorectal carcinogenesis has not been extensively studied;
however, a previous study has demonstrated that the mRNA expression
of ING1b was downregulated in human sporadic colorectal cancer
(29). In the present study, the
expression of p33ING1b in CRC specimens was detected
using western blot analysis. The results indicated that the
expression of p33ING1b in CRC tissues was markedly
reduced compared with that of their paired peritumoral mucosa
tissues. This result, along with the data obtained by Chen et
al (24), indicated that the
expression of p33ING1b was downregulated in CRC, as
previously described for malignancies derived from other tissues
(11,13–15,24). This
evidence suggests that the downregulation of p33ING1b
may be involved in the development and progression of CRC.
Subsequently, the effects of adenovirus-mediated
ectopic expression of p33ING1b on the proliferation of
three colorectal adenocarcinoma cell lines were evaluated. A
recombinant adenovirus was used to deliver ING1b into the in
vitro cultured colorectal adenocarcinoma cells, as this
approach facilitated guaranteed transduction efficiency, whilst
avoiding the potential adoptive alterations that may occur during
long-term selection following transfection with plasmids. The
results revealed that infection with the reporter adenovirus,
Ad-GFP, at an MOI of 40, resulted in GFP expression in ~70% of the
cultured colorectal adenocarcinoma cells and exerted no notable
effects on p33ING1b expression, proliferation, cell
cycle distribution or apoptosis. However, the cells infected with
Ad-ING1b exhibited significantly decreased growth rates due to the
ectopic expression of p33ING1b. This result was in
agreement with previous reports using adenoviral transduction in
gastric adenocarcinoma cells (18)
and using plasmid transfection in human fibroblast cells and breast
cancer cell lines (22,30).
Early studies revealed that one of the major
biological functions of p33ING1b is to promote
apoptosis, via a p53-dependent pathway (31–35). In
the present study, three colorectal adenocarcinoma cell lines with
different p53 statuses were used, in order to observe the effects
of p33ING1b on apoptosis in vitro. The three cell
lines comprised the LoVo cell line, which has wild-type p53, the
HT29 cell line, which has a heterozygous mutation of p53 and the
SW480 cell line, which has two copies of mutated p53 (36,37). The
results revealed that, despite the varying p53 statuses, the
ectopic expression of p33ING1b exerted similar
pro-apoptotic effects in all three adenocarcinoma cell lines,
suggesting that the pro-apoptosis effect of p33ING1b may
not be fully dependent on the presence of wild-type, functional
p53.
Cell cycle progression is correlated with
proliferation and apoptosis. G1 phase of the cell cycle involves a
critical DNA-damage checkpoint, which functions as a safeguard
against genomic instability (38).
Cells that arrest in G1 phase may undergo apoptosis, or may recover
from the G1 phase arrest and enter into S phase. In a previous
study, the overexpression of p33ING1b in human diploid
fibroblasts was shown to induce a 50% increase in the number of
cells in the G0/G1 phase of the cell cycle,
whereas knockdown of p33ING1b in these cells resulted in
abrogation of this arrest and the entry of the cells into S phase
(39). These results indicate that
p33ING1b may have a role in mediating the
G1-S phase transition (2).
Accordingly, in the present study, cell cycle distribution analysis
revealed that cells infected with Ad-ING1b were arrested in G1
phase, whereas the normal control and Ad-GFP-infected cells
proliferated normally, suggesting that the inhibitory effect on
proliferation and the pro-apoptotic effects of p33ING1b
may be, at least partially, attributed to its influence on cell
cycle progression.
In conclusion, the results of the present study
provide substantial evidence indicating that the downregulation of
p33ING1b has a significant role in CRC development.
Furthermore, gene therapy based on the ectopic expression of
p33ING1b may provide a promising therapeutic approach
for the treatment of CRC.
Acknowledgements
The authors would like to thank Dr K. T. Riabowol,
University of Calgary (Calgary, AB, Canada), for providing the
pCI-ING1b plasmid.
References
|
1
|
Garkavtsev I, Demetrick D and Riabowol K:
Cellular localization and chromosome mapping of a novel candidate
tumor suppressor gene (ING1). Cytogenet Cell Genet. 76:176–178.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cheung KJ Jr and Li G: The tumor
suppressor ING1: Structure and function. Exp Cell Res. 268:1–6.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Garkavtsev I, Grigorian IA, Ossovskaya VS,
et al: The candidate tumour suppressor p33ING1 cooperates with p53
in cell growth control. Nature. 391:295–298. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gunduz M, Ouchida M, Fukushima K, et al:
Genomic structure of the human ING1 gene and tumor-specific
mutations detected in head and neck squamous cell carcinomas.
Cancer Res. 60:3143–3146. 2000.PubMed/NCBI
|
|
5
|
Vieyra D, Toyama T, Hara Y, et al: ING1
isoforms differentially affect apoptosis in a cell age-dependent
manner. Cancer Res. 62:4445–4452. 2002.PubMed/NCBI
|
|
6
|
Kataoka H, Bonnefin P, Vieyra D, et al:
ING1 represses transcription by direct DNA binding and through
effects on p53. Cancer Res. 63:5785–5792. 2003.PubMed/NCBI
|
|
7
|
Ythier D, Larrieu D, Brambilla C, et al:
The new tumor suppressor genes ING: Genomic structure and status in
cancer. Int J Cancer. 123:1483–1490. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jäger D, Stockert E, Scanlan MJ, et al:
Cancer-testis antigens and ING1 tumor suppressor gene product are
breast cancer antigens: Characterization of tissue-specific ING1
transcripts and a homologue gene. Cancer Res. 59:6197–6204.
1999.PubMed/NCBI
|
|
9
|
Soliman MA and Riabowol K: After a decade
of study-ING, a PHD for a versatile family of proteins. Trends
Biochem Sci. 32:509–519. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nouman GS, Angus B, Lunec J, et al:
Comparative assessment expression of the inhibitor of growth 1 gene
(ING1) in normal and neoplastic tissues. Hybrid Hybridomics.
21:1–10. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Toyama T, Iwase H, Watson P, et al:
Suppression of ING1 expression in sporadic breast cancer. Oncogene.
18:5187–5193. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cheung KJ Jr, Mitchell D, Lin P and Li G:
The tumor suppressor candidate p33(ING1) mediates repair of
UV-damaged DNA. Cancer Res. 61:4974–4977. 2001.PubMed/NCBI
|
|
13
|
Oki E, Maehara Y, Tokunaga E, Kakeji Y and
Sugimachi K: Reduced expression of p33(ING1) and the relationship
with p53 expression in human gastric cancer. Cancer Lett.
147:157–162. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ito K, Kinjo K, Nakazato T, Ikeda Y and
Kizaki M: Expression and sequence analyses of p33(ING1) gene in
myeloid leukemia. Am J Hematol. 69:141–143. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tallen G, Riabowol K and Wolff JE:
Expression of p33ING1 mRNA and chemosensitivity in brain tumor
cells. Anticancer Res. 23:1631–1635. 2003.PubMed/NCBI
|
|
16
|
Garkavtsev I, Kazarov A, Gudkov A and
Riabowol K: Suppression of the novel growth inhibitor p33ING1
promotes neoplastic transformation. Nat Genet. 14:415–420. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shinoura N, Muramatsu Y, Nishimura M,
Yoshida Y, Saito A, Yokoyama T, Furukawa T, Horii A, Hashimoto M,
Asai A, et al: Adenovirus-mediated transfer of p33ING1 with p53
drastically augments apoptosis in gliomas. Cancer Res.
59:5521–5528. 1999.PubMed/NCBI
|
|
18
|
Lv Y, Purbey BK, Huang Y, Li S, Radha G
and Hao Z: Adenovirus-mediated expression of p33(ING1b) induces
apoptosis and inhibits proliferation in gastric adenocarcinoma
cells in vitro. Gastric Cancer. 15:355–362. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Morán A, Ortega P, de Juan C,
Fernández-Marcelo T, Frías C, Sánchez-Pernaute A, Torres AJ,
Díaz-Rubio E, Iniesta P and Benito M: Differential colorectal
carcinogenesis: Molecular basis and clinical relevance. World J
Gastrointest Oncol. 2:151–158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Center MM, Jemal A, Smith RA and Ward E:
Worldwide variations in colorectal cancer. CA Cancer J Clin.
59:366–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen LS, Wei JB, Zhou YC, Zhang S, Liang
JL, Cao YF, Tang ZJ, Zhang XL and Gao F: Genetic alterations and
expression of inhibitor of growth 1 in human sporadic colorectal
cancer. World J Gastroenterol. 11:6120–6124. 2005.PubMed/NCBI
|
|
23
|
Rhoads TW, Williams JR, Lopez NI, et al:
Using theoretical protein isotopic distributions to parse
small-mass-difference post-translational modifications via mass
spectrometry. J Am Soc Mass Spectrom. 24:115–124. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen L, Matsubara N, Yoshino T, Nagasaka
T, Hoshizima N, Shirakawa Y, Naomoto Y, Isozaki H, Riabowol K and
Tanaka N: Genetic alterations of candidate tumor suppressor ING1 in
human esophageal squamous cell cancer. Cancer Res. 61:4345–4349.
2001.PubMed/NCBI
|
|
25
|
Russell M, Berardi P, Gong W and Riabowol
K: Grow-ING, Age-ING and Die-ING: ING proteins link cancer,
senescence and apoptosis. Exp Cell Res. 312:951–961. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nouman GS, Anderson JJ, Mathers ME, et al:
Nuclear to cytoplasmic compartment shift of the p33ING1b tumour
suppressor protein is associated with malignancy in melanocytic
lesions. Histopathology. 40:360–366. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nouman GS, Anderson JJ, Wood KM, et al:
Loss of nuclear expression of the p33(ING1b) inhibitor of growth
protein in childhood acute lymphoblastic leukaemia. J Clin Pathol.
55:596–601. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nouman GS, Anderson JJ, Crosier S, et al:
Downregulation of nuclear expression of the p33(ING1b) inhibitor of
growth protein in invasive carcinoma of the breast. J Clin Pathol.
56:507–511. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen XY, Zhu TH and Li Y: Advances in the
research on ING gene family. J Mol Med. 2:83–92. 2005.
|
|
30
|
Ohgi T, Masaki T, Nakai S, Morishita A,
Yukimasa S, Nagai M, Miyauchi Y, Funaki T, Kurokohchi K, Watanabe S
and Kuriyama S: Expression of p33(ING1) in hepatocellular
carcinoma: Relationships to tumour differentiation and cyclin E
kinase activity. Scand J Gastroenterol. 37:1440–1448. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shimada H, Liu TL, Ochiai T, et al:
Facilitation of adenoviral wild-type p53-induced apoptotic cell
death by overexpression of p33(ING1) in T.Tn human esophageal
carcinoma cells. Oncogene. 21:1208–1216. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Skowyra D, Zeremski M, Neznanov N, et al:
Differential association of products of alternative transcripts of
the candidate tumor suppressor ING1 with the mSin3/HDAC1
transcriptional corepressor complex. J Biol Chem. 276:8734–8739.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nagashima M, Shiseki M, Miura K, et al:
DNA damage-inducible gene p331NG2 negatively regulates cell
proliferation through acetylation of p53. Proc Natl Acad Sci USA.
98:9671–9676. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tsang FC, Po LS, Leung KM, et al: ING1b
decreases cell proliferation through p53-dependent and -independent
mechanisms. FEBS Lett. 553:277–285. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Coles AH, Liang H, Zhu Z, et al: Deletion
of p37Ing1 in mice reveals a p53-independent role for Ing1 in the
suppression of cell proliferation, apoptosis, and tumorigenesis.
Cancer Res. 67:2054–2061. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rochette PJ, Bastien N, Lavoie J, Guérin
SL and Drouin R: SW480, a p53 double-mutant cell line retains
proficiency for some p53 functions. J Mol Biol. 352:44–57. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Haupt S, di Agostino S, Mizrahi I,
Alsheich-Bartok O, Voorhoeve M, Damalas A, Blandino G and Haupt Y:
Promyelocytic leukemia protein is required for gain of function by
mutant p53. Cancer Res. 69:4818–4826. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
D'Anna JA, Valdez JG, Habbersett RC and
Crissman HA: Association of G1/S-phase and late S-phase checkpoints
with regulation of cyclin-dependent kinases in Chinese hamster
ovary cells. Radiat Res. 148:260–271. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li N, Li Q, Cao X, et al: The tumor
suppressor p33ING1b upregulates p16INK4a expression and induces
cellular senescence. FEBS Lett. 585:3106–3112. 2011. View Article : Google Scholar : PubMed/NCBI
|