Introduction
The proto-oncogene MET encodes the receptor
tyrosine kinase MET. Its primary function is to mediate
morphogenetic embryonic development and tissue repair in
vertebrates. The natural ligand for this receptor is the hepatocyte
growth factor; the binding of this ligand to MET induces tyrosine
phosphorylation of the receptor and activation of downstream
signaling pathways mediated by phosphoinositide 3-kinase and AKT,
by signal transducer and activator of transcription 3, or RAS and
mitogen-activated protein kinase (MAPK) (1,2).
Garcinia hanburyi (G. hanburyi) is a
traditional herbal medicine, which is used for anti-inflammation
and hemostasis in South Asia. Gambogic acid (GA) is the main active
component extracted from G. hanburyi. Gamboge resin has
previously been used as a coloring material and in Traditional
Chinese Medicine for the treatment of human diseases (3). A previous study demonstrated that GA
exerts antitumor effects in vitro and in vivo
(4). GA has been demonstrated to
inhibit proliferation, induce apoptosis, reverse multidrug
resistance and possess anti-angiogenic properties (5).
GA has been approved by the Chinese Food and Drug
Administration for the treatment of different types of cancer in
clinical trials (6,7). Therefore, identification of the specific
molecular targets responsible for the observed GA-mediated
antitumor effects may be of clinical significance. A number of
potential molecular targets of GA have been reported, which may
contribute to its cytotoxic and antitumor activities, including
binding to the transferrin receptor, suppressing nuclear factor-κB
(NF-κB) signaling transduction (8)
and inhibiting the KDR signaling pathway (9). GA was also found to induce apoptosis in
the non-small cell lung cancer (NSCLC) cell lines SPC-A1 and
SK-MES-1 via Caspase 2, Caspase 9, Caspase 10, Bax and involved
signaling pathways (10). Lung cancer
is the leading cause of cancer mortalities worldwide, accounting
for 18.2% of all cancers. The ratio of mortality to incidence is
0.86, and NSCLC represents ~80% of all lung cancers (11).
Although GA has been demonstrated to exert an
antitumor effect on NSCLC, there are few reports regarding the
mechanisms underlying this activity at present. The current study
aimed to elucidate the potential mechanisms involved.
Materials and methods
Reagents
GA was purchased from Sigma-Aldrich (St. Louis, MO,
USA). The MET selective inhibitor, PHA665752, was purchased from
Selleck Chemicals (Houston, TX, USA). All drugs used in the present
study were dissolved in sterile dimethylsulfoxide (DMSO;
Sigma-Aldrich); a 10 mM working solution was prepared and stored in
aliquots at −22°C. Rabbit polyclonal IgG antibodies against human
phosphorylated (p-) MET (#sc-101736), p-AKT (#sc-101629),
p-extracellular-signal-regulated kinase (ERK; #sc-101760), MET
(#sc-10), AKT (#sc-8312) and ERK (#sc-292838) were purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Rabbit anti-Ki-67
monoclonal IgG antibodies for immunohistochemistry (IHC) were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA;
#9129). All the chemicals used in the present study were of
analytical reagent grade.
Cell culture
The human NSCLC cell line, NCI-H1993, which harbors
a MET gene amplification (12), was obtained from the American Type
Culture Collection (Manassas, VA, USA). The cells were cultured in
DMEM supplemented with 10% fetal bovine serum, 105 U/l
penicillin and 100 mg/l streptomycin (GE Healthcare Life Sciences,
Logan, UT, USA) at 37°C in an atmosphere containing 5%
CO2.
Animals
BALB/c female nude mice, obtained from Vital River
Laboratory Animal Technology Co., Ltd. (Beijing, China), were used
when they were 7–9 weeks old. The health of all animals was
monitored daily by gross observation, and the experimental animals
were housed in the laminar airflow cabinet. All the animals were
allowed to acclimatize and recover from any stress associated with
shipping for at least three days prior to experimental
manipulation. Autoclaved water and irradiated food (Vital River
Laboratory Animal Technology Co., Ltd.) were provided ad
libitum, and the animals were maintained in a 12 h light and
dark cycle. Cages, bedding and water bottles were autoclaved prior
to use and were changed twice weekly. All the animal experiments
were performed in accordance with protocols approved by the
Experimental Animal Center of the Second Military Medical
University Animal Care and Use Committee (Zhuozhou, China).
NCI-H1993 xenograft model
establishment
NCI-H1993 cells were harvested, pelleted by
centrifugation at 600 × g for 10 min, and resuspended in sterile
serum-free medium supplemented with 50% Matrigel (BD Biosciences,
Franklin Lakes, NJ, USA). The cells (5×106 in 100 µl)
were then subcutaneously implanted into the hind-flank region of
each mouse and allowed to grow to a volume of 150–200
mm3 prior to the administration of GA.
Efficacy study
Nude mice bearing NCI-H1993 tumors (150–200
mm3) received vehicle [10% DMSO, 15% ethanol and 75%
phosphate-buffered saline (PBS) (Sigma-Aldrich)] or 10, 20 or 30
mg/kg GA (10% DMSO, 15% ethanol and 75% PBS via intraperitoneal
(i.p.) injection; or 10 mg/kg PHA665752 (L-lactate and 10%
polyethylene glycol; Selleck Chemicals, Houston, TX, USA) via tail
intravenous (i.v.) injection for 21 consecutive days. On day 21 of
the efficacy study, at 2 h following the final treatment with GA,
the mice were humanely sacrificed by CO2 overexposure,
and the tumors were resected. The tumor volume (TV) was determined
by measurement with electronic vernier calipers, and the TV was
calculated using the formula: TV=length × width2/2. TV
was expressed on the indicated days as the median TV ± standard
deviation for the indicated groups of mice.
Western blotting analysis
The protein expression levels of p-MET, p-AKT,
p-ERK, MET, AKT and ERK were analyzed by western blot analysis. On
day 21 of the efficacy study, at 2 h following the final injection
of GA, the mice were humanely sacrificed. The tumors were harvested
in lysis buffer (Cell Signaling Technology, Inc.) and homogenized
using Misonix Sonicator 4000 (Misonix Inc., Farmingdale, NY, USA);
protein lysates were generated and protein concentrations were
determined using a bicinchoninic acid assay (Pierce Biotechnology,
Inc., Rockford, IL, USA). Equal amounts of protein (50 µg) were
then separated by SDS-PAGE on 10% gels, blotted on polyvinylidene
difluoride membranes (Sigma-Aldrich) and probed with p-MET, p-AKT,
p-ERK, MET, AKT and ERK rabbit polyclonal primary antibodies
(dilution, 1:1,000; incubation, overnight at 4°C) and subsequently
with goat anti-rabbit horseradish peroxidase-conjugated secondary
antibody (#sc-2040; dilution, 1:1,000; incubation, 1 h at room
temperature), and detected with an enhanced chemiluminescence kit
(Sigma-Aldrich).
IHC
The tumor specimens were fixed in 10% buffered
formalin for 24 h prior to being transferred to 70% ethanol. The
tumor samples were subsequently paraffin-embedded, and 4-mm
sections were cut and baked onto microscope slides (formalin,
paraffin and slides from Sigma-Aldrich). The slides were incubated
with the primary Ki-67 antibody (dilution, 1:1,000; incubation,
overnight at 4°C), then secondary antibodies (dilution, 1:1,000;
incubation, 1 h at room temperature), and visualized using a
colorimetric method (EnVision+ System-HRP DAB kit; Dako North
America, Inc., Carpinteria, CA, USA). All of the immunostained
sections were counterstained using hematoxylin. An automated
Ventana Discovery XT Staining Module (Ventana Medical Systems,
Inc., Tucson, AZ, USA) was used to conduct histological staining.
The stained sections were analyzed using an Olympus BX46 microscope
(Olympus Corporation, Tokyo, Japan), and quantitative analysis of
section staining was performed using the Automated Cellular Imaging
system (GE Healthcare Life Sciences, Chalfont, UK). The number of
Ki-67-positive nuclei was counted regardless of the immunointensity
in 4 random fields at ×100 magnification (60% center field).
Caspase activity assay
As a measure of the level of apoptosis, caspase-3,
−8 and −9 activity were measured using caspase colorimetric
protease kits (Abnova Corporation, Walnut, CA, USA). Fresh tumors
in each group were resected following the final treatment with 10,
20 or 30 mg/kg GA for 2 h on day 21 of the efficacy study, and the
tumor lysis containing 200 µg protein was incubated with 5 µl 4 mM
pNA-conjugated caspase substrates (4-amino acid sequences;
DEVD-pNA, IETD-pNA and LEHD-pNA) at 37°C for 2 h. The quantity of
pNA released was measured at 405 nm using a FLx800™ Multi-Detection
microplate reader (BioTek Instruments, Inc., Winooski, VT,
USA).
Statistical analysis
All the results and data were confirmed in at least
three separate experiments. The data are expressed as the mean ±
standard deviation, and were analyzed by Student's t-test
using SPSS software, version 13.0 (SPSS Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
GA inhibits tumor growth of NCI-H1993
xenografts
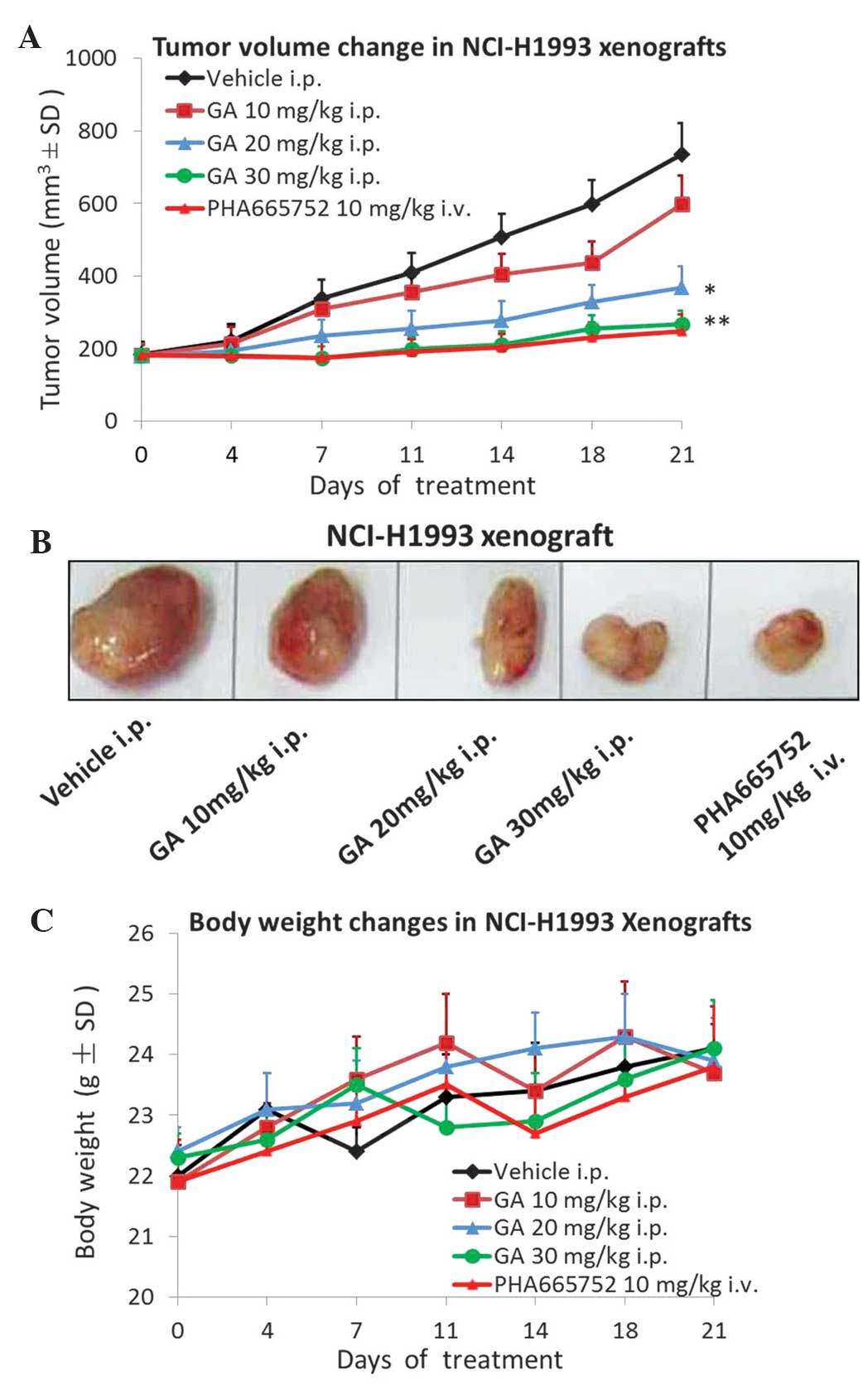
In order to investigate the tumor growth inhibition
effect of GA on NCI-H1993 xenografts in vivo, NCI-H1993
tumor-bearing mice received i.p. injection with 10, 20 or 30 mg/kg
GA once a day for 21 days. As demonstrated in Fig. 1A and B, treatment with 10 mg/kg GA
only slightly inhibited tumor growth, however, 20 mg/kg GA markedly
inhibited tumor growth (P=0.021) and 30 mg/kg GA treatment almost
completely inhibited tumor growth (P=0.008) compared with the
vehicle control group. Throughout the duration of the efficacy
study, no body weight loss was observed in any of the groups
(Fig. 1C). The MET selective
inhibitor PHA665752 was used as a positive control.
GA suppresses the protein expression
of p-MET, p-AKT and p-ERK in NCI-H1993 xenografts
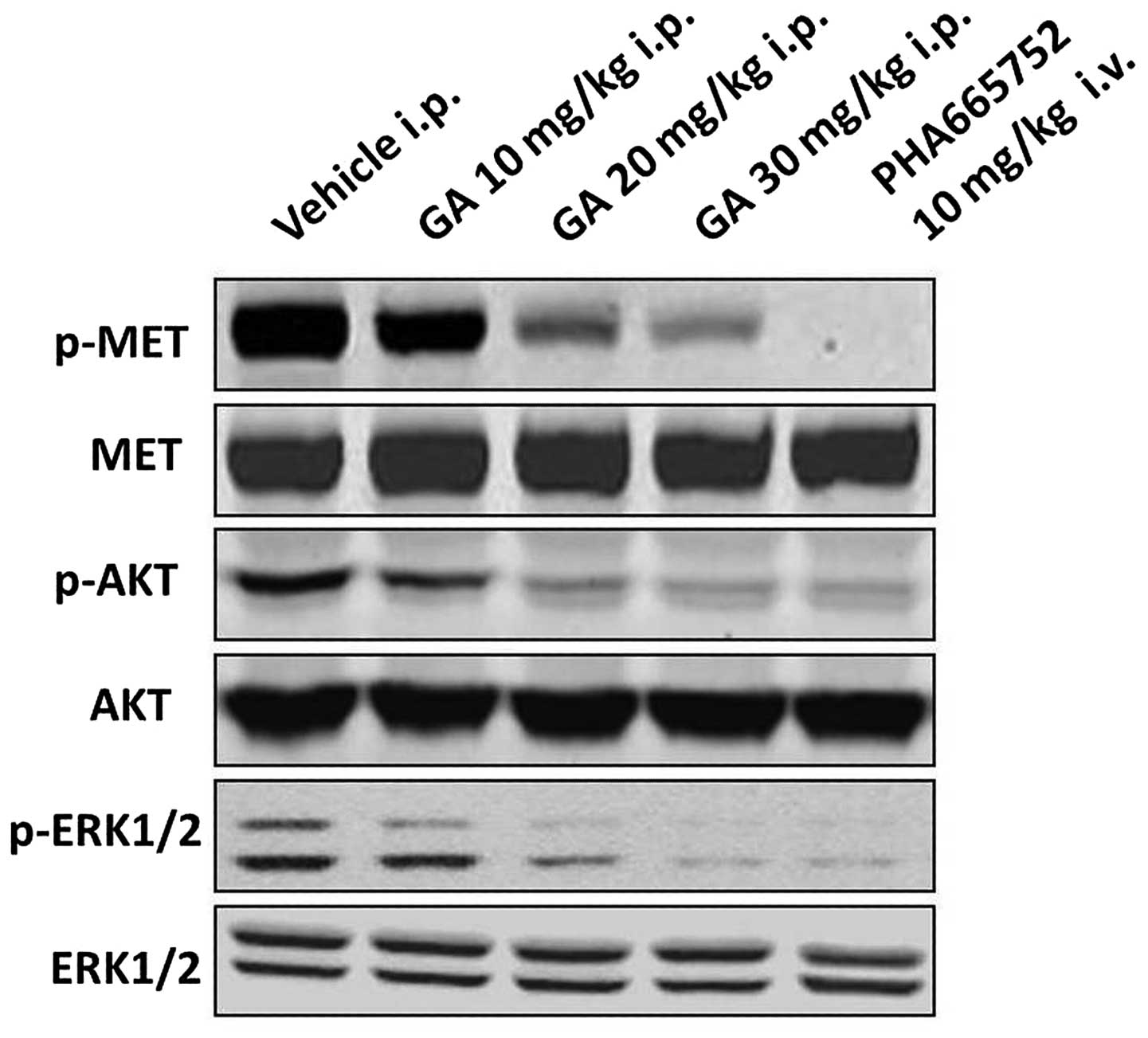
To ascertain whether the expression of p-MET in
NCI-H1993 tumors was affected by GA treatment, the tumor tissues
were analyzed by western blotting. The results demonstrated that
the protein expression levels of p-MET in the NCI-H1993 tumor
tissues were downregulated by GA in a concentration-dependent
manner compared with those of the vehicle group. In addition, the
protein expression levels of the downstream signaling molecules
p-AKT and p-ERK were also markedly downregulated in a
concentration-dependent manner, compared with those of the vehicle
group (Fig. 2). The MET selective
inhibitor PHA665752 was used as a positive control.
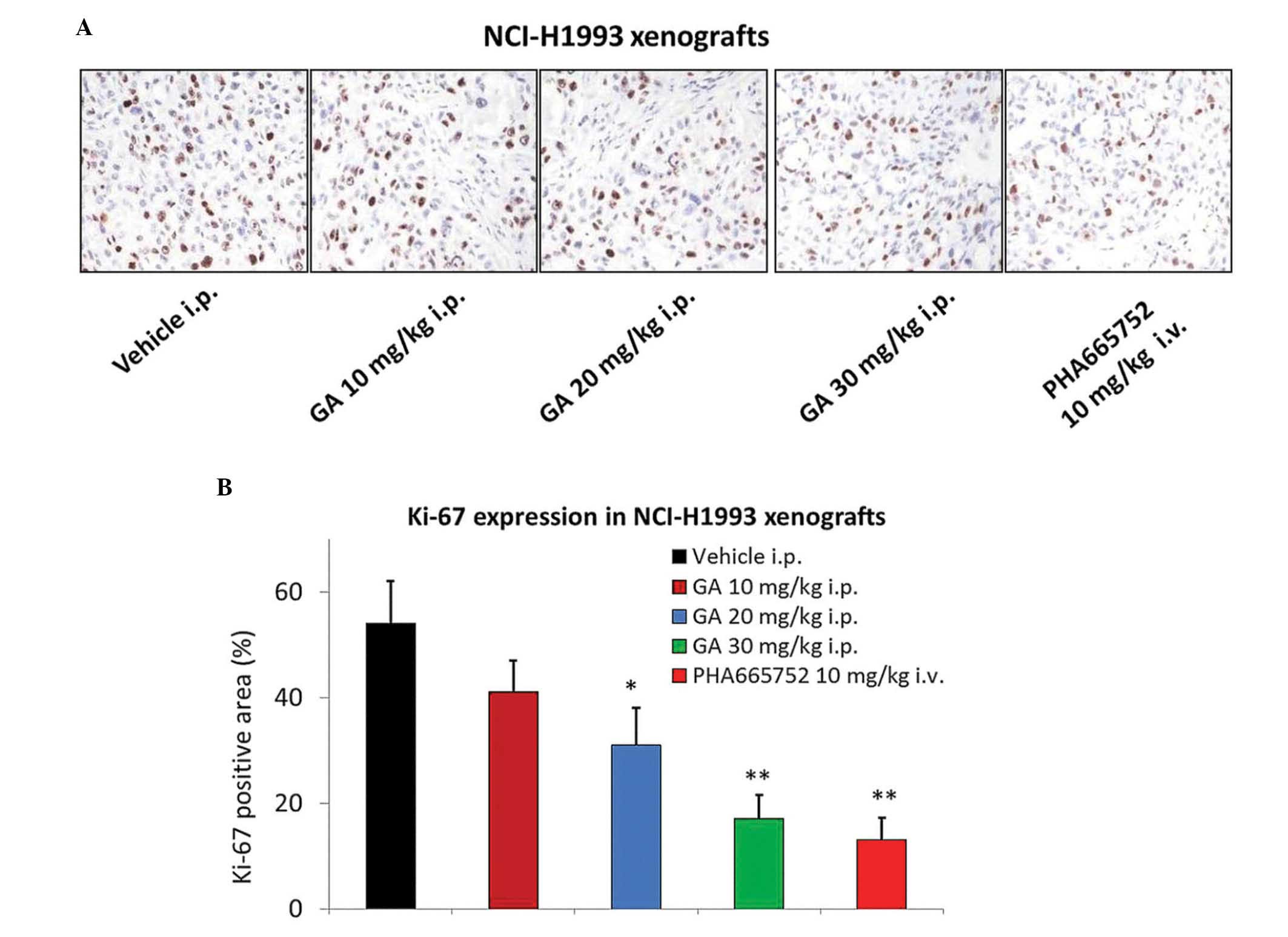
GA treatment significantly inhibits
the expression of Ki-67 in the NCI-H1993 xenograft model
GA was also assessed for its effect on the tumor
mitotic index (Ki-67) using IHC. A significant 2–3-fold reduction
in Ki-67 levels was observed 2 h following the administration of 20
or 30 mg/kg GA in the NCI-H1993 tumor tissues (P=0.046 and 0.009,
respectively; Fig. 3A and B), which
was consistent with the results of the efficacy study. The MET
selective inhibitor PHA665752 also significantly reduced the
expression of Ki-67 in tumor tissues (P=0.007).
GA does not alter caspase-3, −8 or −9
activities in NCI-H1993 xenografts
In order to investigate whether GA induces apoptosis
in the NCI-H1993 xenograft model, the activities of caspase-3, −8
and −9 were measured using a colorimetric assay. The results
demonstrated that GA had no effect on caspase-3, −8 and −9
activities in the NCI-H1993 xenograft model (Fig. 4).
Discussion
Previous studies have demonstrated that GA exerts
significant anti-proliferative and pro-apoptotic effects on a
variety of human cancer cell lines in vitro and in
vivo (13–15). In addition, GA has been approved for
use in clinical trials in China, although its anticancer mechanisms
are not yet fully understood (16).
To the best of our knowledge, the present study is the first to
indicate that GA inhibits the tumor growth of NSCLC cells harboring
a MET amplification in a dose-dependent manner. Wang et
al (11) reported that GA
synergistically potentiates cisplatin-induced apoptosis in NSCLC
through suppressing NF-κB and MAPK/HO-1 signaling. Zhu et al
(10) also reported that mechanisms
of GA-induced apoptosis exist in NSCLC cells and are associated
with transferrin receptors. However, in the present study, the
antitumor mechanisms of GA on NCI-H1993 xenograft were demonstrated
to be associated with MET downregulation. These results indicate
that GA may be investigated further as a potential anticancer
candidate for clinical applications.
A previous study demonstrated that MET and its
receptor are overexpressed by ~70% and ~40% in human lung cancer
tissues, respectively; such values are increased compared with
those in breast (16%) and ovarian cancer (31%), but reduced
compared with renal (72%) and colorectal cancers (78%) (17). However, p-MET expression is observed
to be at the highest levels in lung cancer (73%), followed by
ovarian (33%), breast (23%) and renal (18%) cancer (18). Therefore, MET may be a promising
target for the treatment of lung cancer. In the present study, the
expression of p-MET in NCI-H1993 tumors was inhibited by GA
treatment in a dose-dependent manner. In addition, the downstream
signaling molecules p-AKT and p-ERK were also downregulated by GA,
thereby resulting in tumor growth inhibition. However, AKT and ERK
are downstream signaling molecules not only for MET, but also for
other members of the receptor tyrosine kinase (RTK) family. It is
not clear whether p-MET is the only RTK family member that is
inhibited by GA treatment and therefore further investigation is
required.
In a number of previous reports, GA was found to
induce apoptosis in tumor cells (19,20).
However, in the present study, no effect of GA on caspase-3, 8 and
9 activities was identified in the NCI-H1993 xenograft model.
Another previous study also reported that GA significantly
inhibited U87 tumor growth without inducing apoptosis (21).
In conclusion, the present study demonstrated that
GA exerted significant antitumor effects on NCI-H1993 xenografts by
downregulating the p-MET protein expression level. The expression
of the downstream molecules p-AKT and p-ERK were also inhibited by
GA. Ki-67 analysis in tumor tissues demonstrated that the antitumor
effect of GA was primarily associated with its anti-proliferation
activity. These results may aid in explaining the molecular
mechanisms underlying the antiumor effects of GA on NSCLC.
References
|
1
|
Maestrini E, Tamagnone L, Longati P, et
al: A family of transmembrane proteins with homology to the
MET-hepatocyte growth factor receptor. Proc Natl Acad Sci USA.
93:674–678. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sattler M and Salgia R: c-Met and
hepatocyte growth factor: Potential as novel targets in cancer
therapy. Curr Oncol Rep. 9:102–108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guo Q, Qi Q, You Q, Gu H, Zhao L and Wu Z:
Toxicological studies of gambogic acid and its potential targets in
experimental animals. Basic Clin Pharmacol Toxicol. 99:178–184.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu ZQ, Guo QL, You QD, et al: Gambogic
acid inhibits proliferation of human lung carcinoma SPC-A1 cells
in vivo and in vitro and represses telomerase
activity and telomerase reverse transcriptase mRNA expression in
the cells. Biol Pharm Bull. 27:1769–1774. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guo QL, Lin SS, You QD, et al: Inhibition
of human telomerase reverse transcriptase gene expression by
gambogic acid in human hepatoma SMMC-7721 cells. Life Sci.
78:1238–1245. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang X, Lu N, Yang Q, et al: Studies on
chemical modification and biology of a natural product, gambogic
acid (III): Determination of the essential pharmacophore for
biological activity. Eur J Med Chem. 46:1280–1290. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang J, Zhao L, Hu Y, et al: Studies on
chemical structure modification and biology of a natural product,
Gambogic acid (I): Synthesis and biological evaluation of oxidized
analogues of gambogic acid. Eur J Med Chem. 44:2611–2620. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pandey MK, Sung B, Ahn KS, et al: Gambogic
acid, a novel ligand for transferrin receptor, potentiates TNF
induced apoptosis through modulation of the nuclear factor-kappaB
signaling pathway. Blood. 110:3517–3525. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yi T, Yi Z, Cho SG, et al: Gambogic acid
inhibits angiogenesis and prostate tumor growth by suppressing
vascular endothelial growth factor receptor 2 signaling. Cancer
Res. 68:1843–1850. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhu X, Zhang H, Lin Y, et al: Mechanisms
of gambogic acid-induced apoptosis in non-small cell lung cancer
cells in relation to transferrin receptors. J Chemother.
21:666–672. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang LH, Li Y, Yang SN, et al: Gambogic
acid synergistically potentiates cisplatin-induced apoptosis in
non-small-cell lung cancer through suppressing NF-κB and MAPK/HO-1
signaling. Br J Cancer. 110:341–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sunaga N, Shames DS, Girard L, et al:
Knockdown of oncogenic KRAS in non-small cell lung cancers
suppresses tumor growth and sensitizes tumor cells to targeted
therapy. Mol Cancer Ther. 10:336–346. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gu HY, Guo QL, You QD, et al: Gambogic
acid inducing apoptosis in human hepatoma SMMC-7721 cells with p53
and Bax up-regulated. Chin J Nat Med. 3:169–171. 2005.
|
|
14
|
Liu W, Guo QL, You QD, et al: Anticancer
effect and apoptosis induction of gambogic acid in human gastric
cancer line BGC-823. World J Gastroenterol. 11:3655–3659. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao L, Guo QL, You QD, et al: Gambogic
acid induces apoptosis and regulates expressions of Bax and Bcl-2
protein in human gastric carcinoma MGC-803 cells. Biol Pharm Bull.
27:998–1003. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou Z and Wang J: Phase I human
tolerability trial of gambogic acid. Chin J New Drugs. 16:79–82.
2007.
|
|
17
|
Paez JG, Jänne PA, Lee JC, et al: EGFR
mutations in lung cancer: Correlation with clinical response to
gefitinib therapy. Science. 304:1497–1500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maulik G, Kijima T, Ma PC, et al:
Modulation of the c-Met/hepatocyte growth factor pathway in small
cell lung cancer. Clin Cancer Res. 8:620–627. 2002.PubMed/NCBI
|
|
19
|
Zhao W, Zhou SF, Zhang ZP, et al: Gambogic
acid inhibits the growth of osteosarcoma cells in vitro by
inducing apoptosis and cell cycle arrest. Oncol Rep. 25:1289–1295.
2011.PubMed/NCBI
|
|
20
|
Yang Y, Yang L, You QD, et al:
Differential apoptotic induction of gambogic acid, a novel
anticancer natural product, on hepatoma cells and normal
hepatocytes. Cancer Lett. 256:259–266. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
He XY, Liu XJ, Chen X, et al: Gambogic
acid induces EGFR degradation and Akt/mTORC1 inhibition through
AMPK dependent-LRIG1 upregulation in cultured U87 glioma cells.
Biochem Biophys Res Commun. 435:397–402. 2013. View Article : Google Scholar : PubMed/NCBI
|