Introduction
Oral cancer is the eighth most common type of cancer
among men worldwide, with an age-standardized rate of 6.3 per
100,000 (1,2), and is the fourth leading cause of
cancer-associated mortality among men in Taiwan (3). In addition, oral cancer has been ranked
highest with respect to the rates of incidence and mortality among
men aged 25–44 years in Taiwan since 2005 (3). Chemotherapy has been used as the primary
treatment for patients exhibiting recurrent or metastatic cancers,
including oral cancer (4). Of the
various chemotherapy drugs available, cisplatin and 5-fluorouracil
(5-FU) are the two most frequently utilized for the treatment of
oral cancer (5). However, the
efficacy of cancer chemotherapy is often limited by the development
of drug resistance by the cancer cells in clinical practice
(6).
It has been demonstrated that multidrug resistance
results largely from the expression of adenosine
triphosphate-binding cassette (ABC) transporters with broad drug
specificity (7–9). Multidrug resistance-associated protein 2
(MRP2), a member of the ABC transporter superfamily, has been
observed in various tumors to confer resistance to a number of
therapeutic drugs, including cisplatin (10–12). The
levels of MRP2 messenger (m)RNA in certain human cancer cells are
inversely correlated with their sensitivity to cisplatin (13,14). Thus,
increased expression of the gene encoding MRP2 in cancer cells may
reduce the intracellular accumulation of cisplatin (15). By contrast, a reduction in MRP2
expression using the RNA interference (RNAi) approach was
demonstrated to reverse MRP2-dependent cisplatin resistance in
human ovarian carcinoma (16) and
nasopharyngeal carcinoma cells (17).
In addition, it has been confirmed that the mRNA levels of
MRP2 are markedly correlated with the half-maximal
inhibitory concentration (IC50) value of 5-FU in
esophageal carcinoma cells (18).
Therefore, knockdown of MRP2 may influence the sensitivity
of cancer cells to 5-FU.
Epidermal growth factor receptor (EGFR), which is
one of most well-studied receptor tyrosine kinases, promotes cancer
cell growth and is associated with drug resistance (19). Thus, it has emerged as a significant
target for the development of anticancer therapy (20). Although cancer patients may initially
benefit from EGFR-targeted therapies, drug resistance frequently
develops due to crosstalk or inappropriate activation of downstream
signaling pathways of EGFR (21).
Instead of using EGFR-targeted monoclonal antibodies or
small-molecule inhibitors to inhibit the activity of EGFR, the
expression of its gene, EGFR, in cancer cells may be
silenced using RNAi techniques (22).
Therefore, lentivirus vector-mediated RNAi was used in the present
study to silence the expression of EGFR and MRP2 in
oral cancer. The aim of the present study was to enhance the
chemosensitivity of oral cancer cells to the chemotherapeutic drugs
cisplatin and 5-FU, and to reverse the drug resistance exhibited by
these cells.
Materials and methods
Cell cultures
The oral squamous cell carcinoma (OSCC) cell lines
OC2 and OCSL (23,24), derived from two Taiwanese male
patients who demonstrated habits of alcohol drinking, betel quid
chewing and cigarette smoking, were maintained in RPMI 1640 medium
(catalog no., 11875-085; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
catalog no., 10437-028; Invitrogen; Thermo Fisher Scientific,
Inc.). The OSCC cell lines SCC25 [obtained from American Type
Culture Collection (ATCC), Manassas, VA, USA] and HSC3 (obtained
from Health Science Research Resources Bank, Osaka, Japan) were
maintained in Dulbecco's modified Eagle's medium (DMEM)/Ham's
Nutrient Mixture F-12 (1:1 mixture; catalog no., 11330-032, Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% FBS. SCC25
cells were additionally supplied with 400 ng/ml hydrocortisone
(Sigma-Aldrich, St. Louis, MO, USA). The transformed human
embryonic kidney cell line 293T (obtained from ATCC) was maintained
in DMEM with 10% FBS.
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
Total RNA was extracted from cells using REzol™ C
& T reagent (catalog no., KP200CT; Protech Technology
Enterprise Co., Ltd., Taipei, Taiwan), according to the
manufacturer's protocol. Complementary DNA (cDNA) was synthesized
from aliquots of RNA using SuperScript® III First-Strand
Synthesis System (catalog no., 18080-051; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. DNase I (catalog
no., 18068-015; Thermo Fisher Scientific, Inc.) included in this
system was used to treat the RNA sample to eliminate DNA. RT-PCR
was performed using Taq DNA Polymerase Master Mix Red (catalog no.,
A180301; Ampliqon, Odense M, Denmark) and MyCycler™ Thermal Cycler
(catalog no., 170-9703; Bio-Rad Laboratories, Inc., Hercules, CA,
USA). EGFR and MRP2 cDNA was amplified using the
following primers (Protech Technology Enterprise Co., Ltd.): EGFR
forward, 5′-CCAAACAATTAGCCTGGACA-3′ and reverse,
5′-CGCGACCCTTAGGTATTCTG-3′; and MRP2 forward,
5′-TGCAGCCTCCATAACCATGAG-3′ and reverse,
5′-GATGCCTGCCATTGGACCTA-3′. Glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) cDNA was additionally amplified as
an internal control with primers forward,
5′-GCCATCAATGACCCCTTAATT-3′ and reverse,
5′-TTGACGGTGCCATGGAATTT-3′. The PCR programs were set to perform
the following steps: For EGFR, 95°C for 5 min, followed by
30 cycles of successive incubation at 95°C for 30 sec, 54°C for 30
sec and 72°C for 30 sec; for MRP2, 95°C for 5 min, followed
by 38 cycles of successive incubation at 95°C for 30 sec, 63°C for
30 sec and 72°C for 30 sec; for GAPDH, 95°C for 5 min,
followed by 26 cycles of successive incubation at 95°C for 30 sec,
54°C for 30 sec and 72°C for 30 sec. The PCR products and DNA
ladder (catalog no., 02001-500; OmicsBio, Taipei, Taiwan) were
resolved on a 2% agarose gel (catalog no., 0710-250G; Amresco,
Solon, OH, USA) and visualized by ethidium bromide (catalog no.,
X328; Protech Technology Enterprise Co., Ltd.) staining. Expression
of EGFR and MRP2 was quantified using ImageJ software
v1.49 (http://imagej.nih.gov/ij/download.html; accessed
October 21, 2014) and normalized against that of GAPDH.
Lentivirus vector production and
transfection
Lentivirus vectors were produced by transient
transfection of pCMVdeltaR8.91, pMD.G and pLKO.1-puro or
pLKO-TRC008 vectors, carrying a short-hairpin RNA (shRNA), into
293T cells using calcium phosphate precipitation [2.5 M
CaCl2, catalog no., 1332-01 (J.T. Baker, Phillipsburg,
NJ, USA); 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
(HEPES), catalog no., 15630-080 (Gibco; Thermo Fisher Scientific,
Inc.); 2X HEPES-buffered saline, catalog no., 7021 (Sigma-Aldrich);
10 mM KCl, catalog no., 3040-01 (J.T. Baker); 1.4 mM
Na2HPO4, catalog no., 0404-500G (Amresco);
240 mM NaCl, catalog no., 0241-2.5KG (Amresco); 10 mM chloroquine,
catalog no., c6628 (Sigma-Aldrich)], as described previously
(25). pLKO plasmids were obtained
from the National RNAi Core Facility at the Academia Sinica in
Taipei, Taiwan. The shRNA sequences specific for EGFR and
MRP2 were 5′-GCTGGATGATAGACGCAGATA-3′ and
5′-GCCGGTGGTCAGATTATCATT-3′, respectively. OSCC cells infected with
shRNA-expressing lentivirus vectors (shEGFR or shMRP2) were
selected by incubation with 2 µg/ml puromycin (Sigma-Aldrich) or 20
µg/ml blasticidin (Sigma-Aldrich) for 2 days. Cells infected with
sh-red fluorescent protein (RFP) were used as a control.
Western blot analysis
Cells were lysed by the treatment of RIPA Lysis and
Extraction Buffer (catalog no., 89900; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. The cell lysates
were resuspended in sodium dodecyl sulfate (SDS) gel-loading buffer
[62.5 mM Tris-HCl (pH 6.8); 2% SDS; 10% glycerol; 100 mM
dithiothreitol; 0.01% bromophenol blue) and boiled for 5 min.
Protein samples were run on SDS-10% polyacrylamide gels with
protein marker (catalog no., PT-PS03; Protech Technology Enterprise
Co., Ltd.) at 120 volts for 90 min and transferred onto
polyvinylidene difluoride membranes (catalog no., 162-0177, Bio-Rad
Laboratories, Inc.) at 90 volts for 80 min. The membranes were
blocked for 2 h at room temperature in TBST [100 mM Tris-HCl (pH
7.5); 150 mM NaCl; 0.1% Tween 20] with 5% non-fat dry milk (catalog
no., 1706404, Bio-Rad Laboratories, Inc.), and probed with rabbit
polyclonal anti-EGFR antibody (dilution, 1:1,000; catalog no.,
ab2430; Abcam, Cambridge, MA, USA) or mouse monoclonal anti-β-actin
antibody (dilution, 1:5,000; catalog no., A5316; Sigma-Aldrich).
The specific protein signals were detected by using an enhanced
chemiluminescence kit (catalog no., WBKLS0500; Merck Millipore,
Darmstadt, Germany).
Cell proliferation analysis by
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay
To determine the proliferation rate of cells
transfected with shEGFR, the cells were seeded onto replicate
96-well plates (catalog no., 30096; SPL Life Sciences, Co., Ltd.,
Gyeonggi, Korea) at a density of 400 cells/well. Following
overnight culture, cell proliferation was determined daily by MTS
assay, using the CellTiter 96® AQueous One Solution Cell
Proliferation Assay kit (Promega Corporation, Madison, WI,
USA).
Colony formation assay
A total of 400 cells were seeded onto 6-well plates
(catalog no., 30006; SPL Life Sciences, Co., Ltd.) and cultured for
1–2 weeks or until colonies of cells were visible with the naked
eye. Cell colonies were stained with 0.4% crystal violet
(Sigma-Aldrich) in 50% methanol (catalog no., 322415;
Sigma-Aldrich) for 10 min, followed by washing with
phosphate-buffered saline (PBS), air-drying at room temperature for
2 days and counting with the naked eye. Experiments were performed
in triplicate.
Determination of the IC50 values of
drugs
To determine the IC50 values of 5-FU
(catalog no., F6627; Sigma-Aldrich) and cisplatin (catalog no.,
P4394; Sigma-Aldrich), cells were seeded onto replicate 96-well
plates at a density of 2,000 cells/well. Following overnight
culture, the cells were incubated with 5-FU or cisplatin at various
concentrations (0.00, 0.25, 0.50, 1, 2, 4, 6, 8, 10, 12, 14 and 16
µg/ml) for 3 days, and the remaining viable cells were determined
by MTS assay.
Apoptosis analysis
OC2 and OC2 cisplatin-resistant (CisR) cells were
exposed to increasing concentrations of cisplatin, ranging from
1–16 µg/ml for 24 h. The cells were subsequently harvested with
trypsin (catalog no., 25200-072; Gibco; Thermo Fisher Scientific,
Inc.), washed with PBS and resuspended in binding buffer (catalog
no., AVF250, Strong Biotech Corp., Taipei, Taiwan) containing the
optimal concentration of calcium required for Annexin V to bind to
phosphotidylserine on the cell surface. Annexin V-fluorescein
isothiocyanate (Strong Biotech Corp.) and propidium iodide (PI;
Sigma-Aldrich) were added to the cell suspension and allowed to
incubate in the dark for 15 min at room temperature. The cells were
subsequently analyzed by flow cytometry using a FACSCalibur™ with
BD CellQuest™ Pro Software version 6.0 (BD Biosciences, San Jose,
CA, USA), and the percentages of early (Annexin V+,
PI−) and late apoptotic cells (Annexin V+,
PI+) were calculated according to the manufacturer's
instructions.
Development of the OC2CisR cell
line
OC2 cells were seeded onto 6-cm culture plates
(catalog no., 20101, SPL Life Sciences, Co., Ltd.) at 50%
confluency. Following overnight culture, the cells were incubated
with cisplatin at a concentration of 0.10 µg/ml, until the cultures
approached confluency. The cells were subcultured onto another 6-cm
plate at 50% confluency and incubated with cisplatin at a
concentration of 0.25 µg/ml. Following the repetition of the
procedure with the increased concentrations of 0.50 and 0.75 µg/ml,
the cells were able to survive in a medium containing 0.75 µg/ml
cisplatin and the cells were designated as the OC2CisR cell
line.
In vivo experiments
OSCC cells were implanted into the right dorsal
flank of athymic female BALB/c nude mice (randomly divided into
groups, n=6/group; National Laboratory Animal Center, Taipei,
Taiwan). When the tumor volumes reached ~100 mm3, the
mice were administered intraperitoneal injections of 5-FU (15
mg/kg/day) for a total of 6 treatments, or cisplatin at a weekly
dose of 3 mg/kg for a total of 3 treatments. The tumor volumes were
measured daily, and the maximum allowable size of the tumors was 15
mm in diameter. The housing conditions of the mice were as follows:
12-h light/12-h dark cycle; temperatures of 18–23°C with 40–60%
humidity; and water and food accessible at all times. All
experiments were performed under license from the Institutional
Animal Care and Use Committee of the National Chung Cheng
University.
Statistical analysis
Student's t-tests were performed for
statistical analysis of relative gene expression, cell
proliferation, cell apoptosis, colony-formation, IC50
value and relative tumor volume. All data are presented as the mean
± standard error (SE). For the comparison of tumor growth rates,
generalized linear models with generalized estimated equations were
used to account for the correlation between the repeated
measurements. A significant interaction between time and group in
the generalized linear models meant that the growth rates of the
groups were changing over time in various ways. A P-value of
<0.05 was considered to indicate a statistically significant
difference. Analyses were performed using SAS version 9.3 (SAS
Institute Inc., Cary, NC, USA).
Results
Expression levels of EGFR and MRP2 in
OSCC cell lines
RT-PCR was employed to examine the expression of
EGFR and MRP2 in the OSCC cell lines OC2, OCSL, SCC25
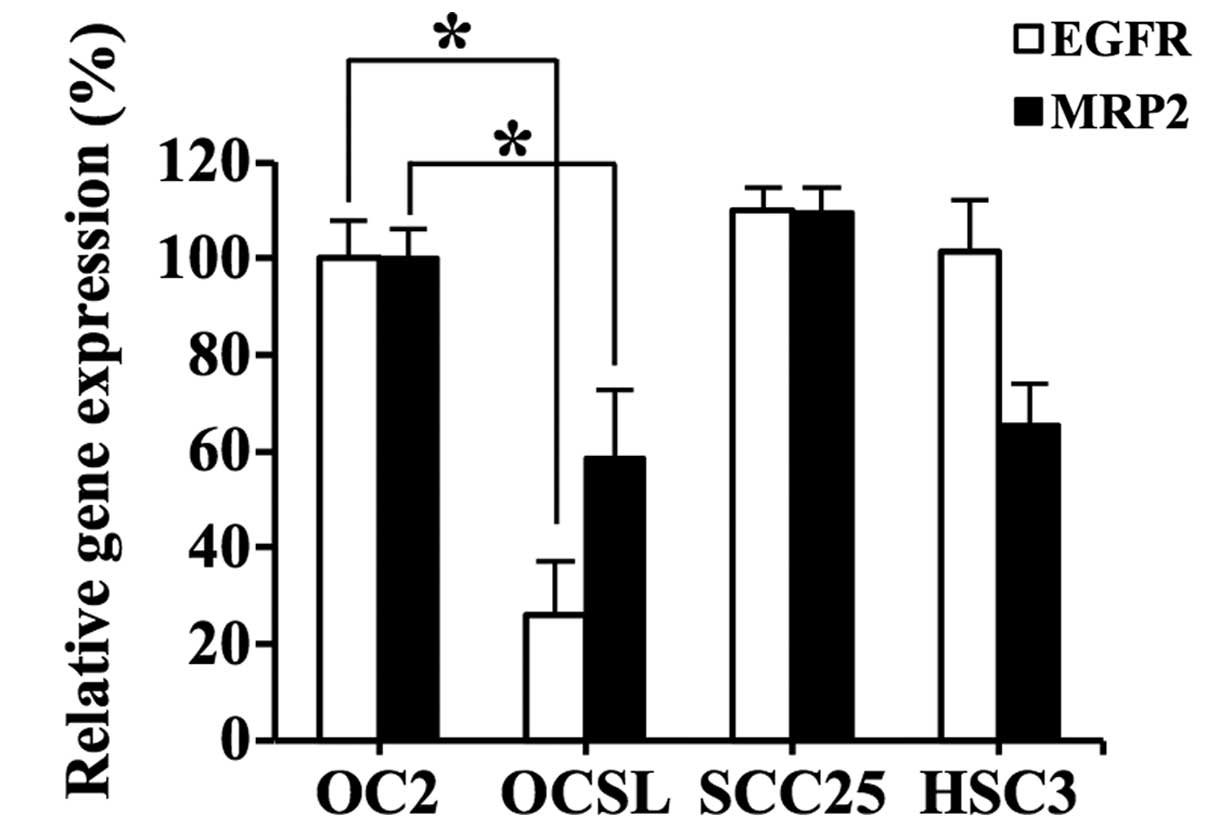
and HSC3. Fig. 1 shows the
differential expression of these two genes among the four cell
types. With respect to the two OSCC cell types derived from
Taiwanese male patients who habitually drank alcohol, chewed
betel-quid and smoked cigarettes, namely, OC2 and OCSL, the former
exhibited increased expression of EGFR and MRP2
compared with the the latter (P<0.05). Thus, OC2 was selected as
the target cell line for evaluation of RNAi-mediated downregulation
of EGFR and MRP2.
Downregulation of EGFR expression
inhibits the growth of OC2 cells
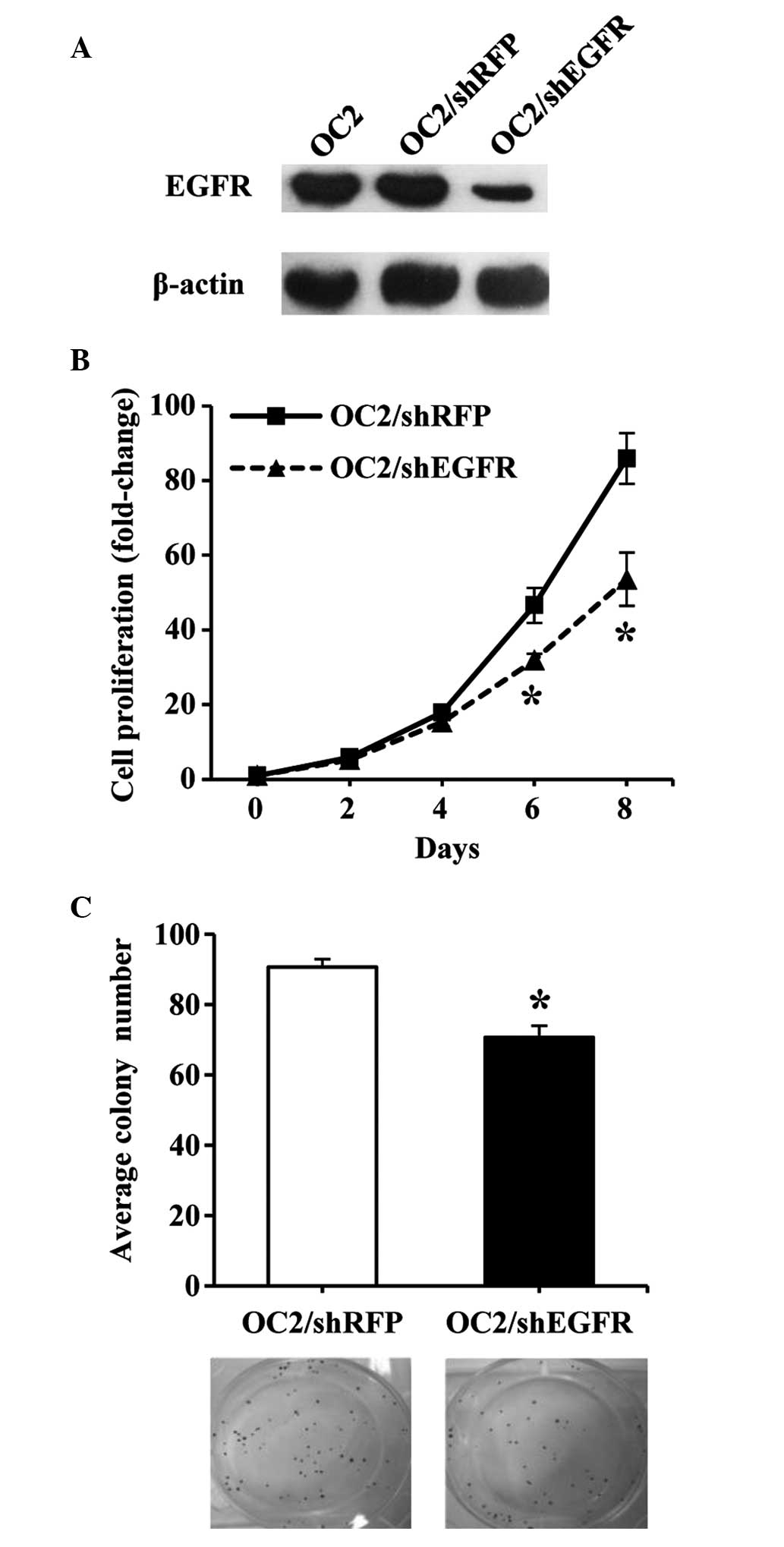
Western blot analysis was utilized to determine the
expression of EGFR protein in OC2 cells transfected with shEGFR,
thus confirming the specificity of RNAi-mediated silencing of
EGFR. It was observed that shEGFR transfection effectively
reduced the expression of EGFR protein in OC2 cells (Fig. 2A). In addition, MTS and colony
formation assays were employed to investigate the proliferation
rate and colony-forming ability, respectively, of the transfected
OC2 cells. A significant difference was observed between the growth
of shEGFR- and shRFP-transfected OC2 cells (P<0.01; Fig. 2B and C), indicating that
downregulation of EGFR expression inhibited the growth of
the OC2 cells.
Downregulation of EGFR and MRP2
enhances the sensitivity of OC2 cells to 5-FU
OC2 cells transfected with shMRP2 alone or in
combination with shEGFR were established to determine whether
MRP2 downregulation enhanced the chemosensitivity of OC2
cells to 5-FU. Fig. 3A shows the
reduced expression of EGFR and MRP2 in
OC2/shEGFR/shMRP2 cells compared with the control group OC2/shRFP
(P<0.01). Transfection of OC2 cells with shEGFR was effective at
sensitizing the cells to 5-FU according to the IC50
value, which was determined using the MTS assay (Fig. 3B). In addition, downregulation of
MRP2 increased the sensitivity of the cells to 5-FU.
However, knockdown of MPR2 and EGFR did not further
decrease the IC50 value of 5-FU, compared with the cells
transfected with shEGFR alone.
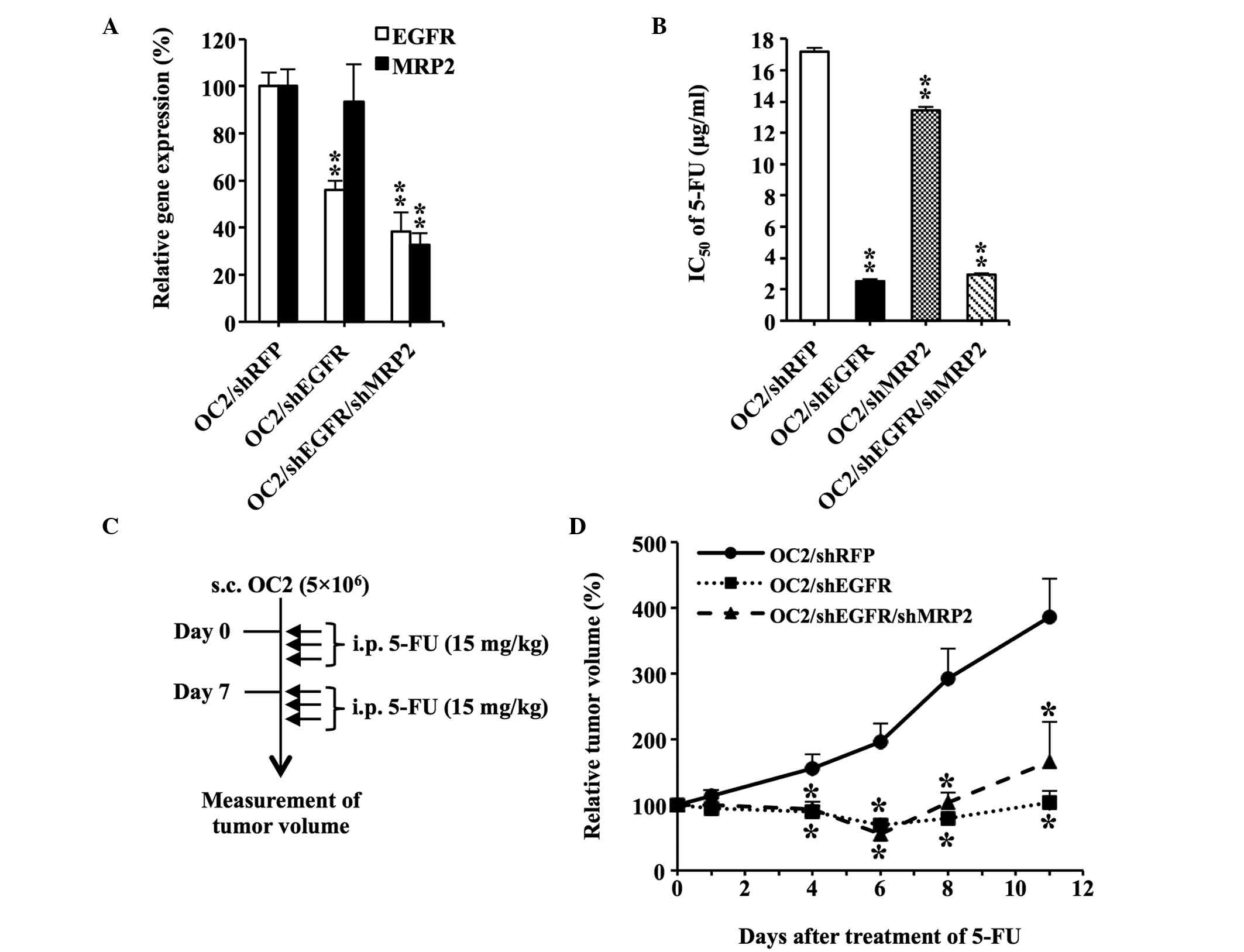 | Figure 3.Downregulation of EGFR and
MRP2 enhances the sensitivity of OC2 cells to 5-FU. (A)
Reduced expression of EGFR and MRP2 in
OC2/shEGFR/shMRP2 cells. The messenger RNA levels of EGFR
and MRP2 in the cells were quantified using reverse
transcription-polymerase chain reaction analysis. The expression
levels of EGFR and MRP2 in OC2/shRFP were assigned a
value of 100%. (B) Determination of the IC50 values of
5-FU in shRNA-transfected OC2 cells. The IC50 value of
5-FU in OC2/shRFP was used as the control. (C) Diagram illustrating
the procedure of the in vivo experiments. A total of
5×106 shRNA-transfected OC2 cells were subcutaneously
implanted into the right dorsal flank of nude mice (n=6/group).
When the tumor volumes reached ~100 mm3 (set as day 0),
the mice were administered intraperitoneal injections of 5-FU (15
mg/kg) for a total of 6 treatments. The tumor volumes were measured
daily. Solid arrow indicates 5-FU treatments on days 0, 1, 2, 7, 8
and 9. (D) In vivo therapeutic effect achieved by the
downregulation of EGFR and MRP2. The tumor volumes on day 0
were assigned a value of 100%. The tumor volumes of OC2/shRFP group
were used as the controls. Data are presented as the mean ±
standard error. *P<0.05. **P<0.01. EGFR, epidermal growth
factor receptor; MRP2, multidrug resistance-associated protein 2;
5-FU, 5-fluorouracil; sh, small hairpin; RFP, red fluorescent
protein; IC50, half-maximal inhibitory concentration;
s.c., subcutaneously; i.p., intraperitoneal. |
Mice bearing subcutaneous OC2 tumors were treated
with 5-FU to determine whether the RNAi-induced downregulation of
EGFR and MRP2 expression confers a therapeutic
benefit in vivo (Fig. 3C).
Downregulation of EGFR resulted in a significant suppression
of tumor growth following 5-FU administration, compared with the
control group (OC2/shRFP; Fig. 3D).
However, downregulation of MRP2 and EGFR did not
further suppress tumor growth, indicating that MRP2 does not have a
significant role in the chemosensitivity of
EGFR-downregulated OC2 cells to 5-FU.
Development of the OC2CisR cell
line
Previous studies have demonstrated that MRP2 confers
resistance to cisplatin in certain types of cancer (9–11). A
cisplatin-resistant OC2 cell line (designated OC2CisR) was
generated for use as the target cell for subsequent experiments, in
order to determine the role of MRP2 in OSCC. These cells were
generated by treating OC2 cells with cisplatin at progressively
increasing concentrations (0.10, 0.25, 0.50 and 0.75 µg/ml) until
they were able to survive in a medium containing 0.75 µg/ml
cisplatin. The expression of EGFR and MRP2 in OC2 and
OC2CisR cells was determined using RT-PCR. MRP2 expression
was significantly increased in OC2CisR cells, compared with OC2
cells (P<0.05), whereas EGFR expression did not differ
significantly between the two cell lines (Fig. 4A). Cell viability analysis confirmed
that OC2CisR cells exhibited an increased resistance to cisplatin,
compared with OC2 cells (P<0.05; Fig.
4B). A cell apoptosis assay additionally confirmed that the
OC2CisR cells were more resistant to the killing effect of
cisplatin, compared with the OC2 cell line (Fig. 4C).
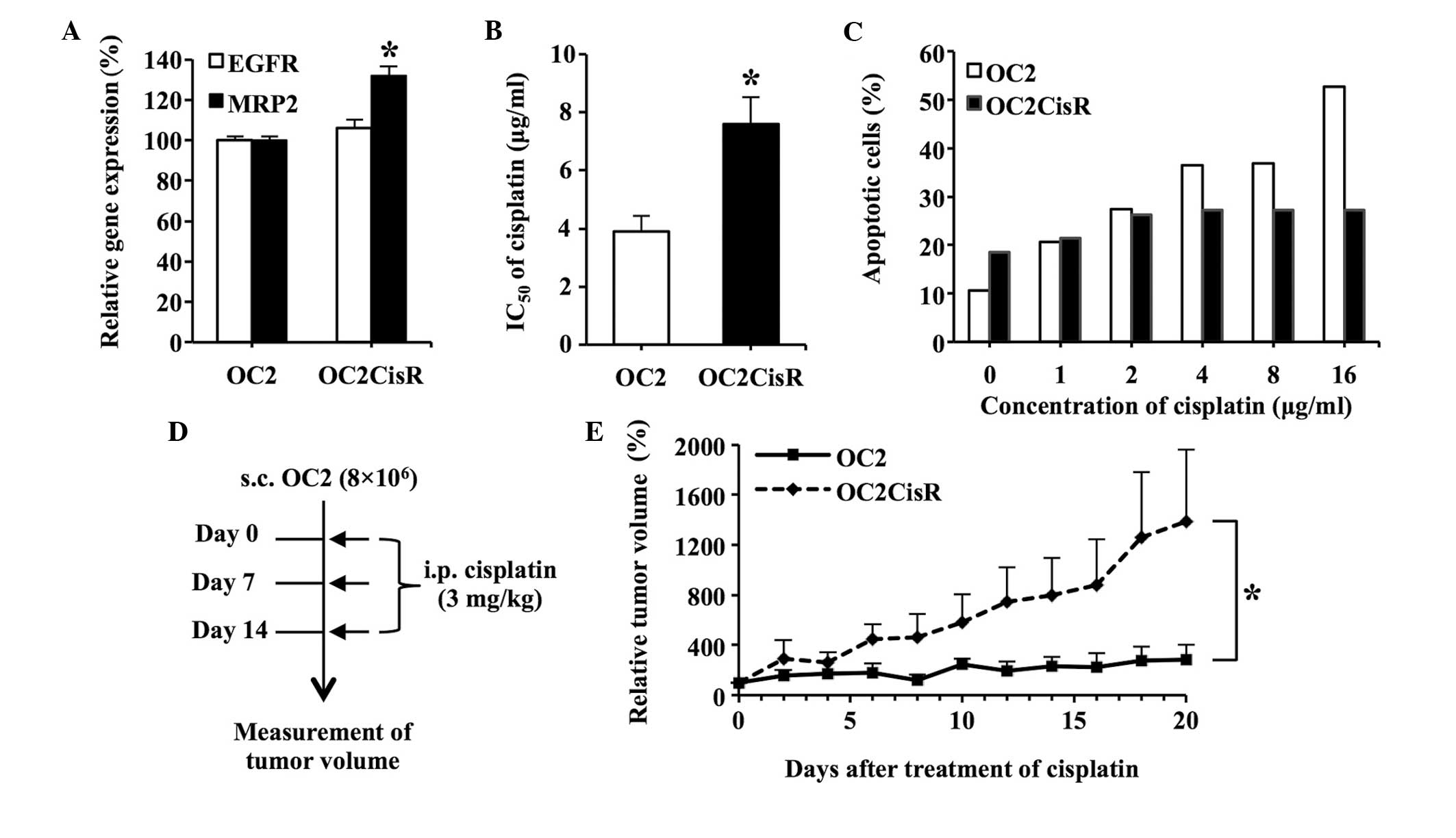 | Figure 4.Development of the OC2CisR cell line.
(A) Expression of EGFR and MRP2 in OC2CisR cells. The
messenger RNA levels of EGFR and MRP2 in OC2CisR
cells were quantified using reverse transcription-polymerase chain
reaction analysis. The expression levels of EGFR and
MRP2 in OC2 cells were assigned a value of 100% as controls.
(B) Determination of the IC50 values of cisplatin in
cells. The IC50 value of cisplatin in OC2 was used as
the control. (C) Cell apoptosis assay. OC2 and OC2CisR cells were
exposed to increasing concentrations of cisplatin ranging from 1 to
16 µg/ml for 24 h, and stained with Annexin V-fluorescein
isothiocyanate and propidium iodide, followed by flow cytometric
analysis. The percentage of total apoptotic cells is shown. (D)
Diagram illustrating the procedure of the in vivo
experiments. A total of 8×106 cells were subcutaneously
implanted into the right dorsal flank of nude mice (n=6/group).
When the tumor volumes reached ~100 mm3 (set as day 0),
the mice were administered intraperitoneal. injections of cisplatin
(3 mg/kg) for a total of 3 treatments. The tumor volumes were
measured daily. Solid arrow indicates cisplatin treatment on days
0, 7 and 14. (E) Comparison of tumor growth rates in the mice
following cisplatin administration. The tumor volumes on day 0 were
assigned a value of 100%. The interaction of time and group in the
generalized linear model with generalized estimated equation was
52.1% (P=0.024), which indicated that the linear growth rate was
52.1% higher per day for the OC2CisR group compared with the OC2
group. Data are presented as the mean ± standard error. *P<0.05.
CisR, cisplatin-resistant; EGFR, epidermal growth factor receptor;
MRP2, multidrug resistance-associated protein 2; IC50,
half-maximal inhibitory concentration; s.c., subcutaneously; i.p.,
intraperitoneal. |
Furthermore, mice bearing OC2 and OC2CisR
subcutaneous tumors were treated with cisplatin to evaluate whether
OC2CisR cells were more resistant to cisplatin, compared with OC2
cells in vivo (Fig. 4D). It
was observed that the growth rate of OC2CisR subcutaneous tumors
was significantly higher than that of OC2 subcutaneous tumors in
the mice following cisplatin administration (Fig. 4E). The tumor growth rate was 52.1%
higher per day in the OC2CisR group, compared with the OC2 group
(SE=23.0%; P=0.024). These observations confirmed that OC2CisR
cells are more resistant to cisplatin, compared with OC2 cells
in vitro and in vivo, suggesting that elevated
expression of MRP2 in OC2CisR cells contributed to their
resistance to cisplatin.
Downregulation of EGFR and MRP2
increases the inhibitory effects of cisplatin on the growth of
OC2CisR tumors
OC2CisR cells were transfected with shMRP2 and
shEGFR to establish whether MRP2 had a significant role in
cisplatin resistance in these cells, and to observe whether
downregulation of MRP2 was able to enhance the therapeutic
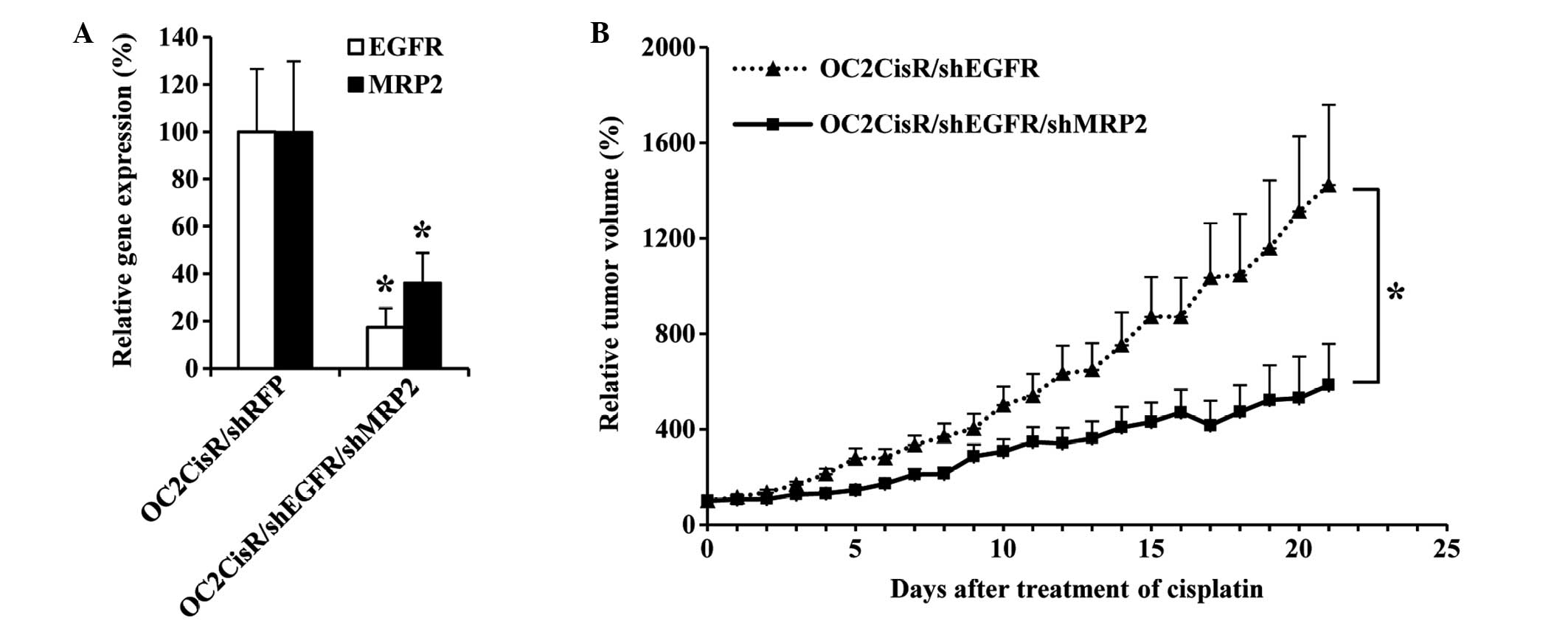
effects of cisplatin in EGFR-downregulated cells. Fig. 5A demonstrates the RNAi-mediated
downregulation of EGFR and MRP2 expression in
OC2CisR/shEGFR/shMRP2 cells in vitro, compared with the
control group OC2CisR/shRFP (P<0.05). In vivo, the growth
rate of OC2CisR/shEGFR/shMRP2 subcutaneous tumors was significantly
lower, compared with that of OC2CisR/shEGFR subcutaneous tumors in
the mice following cisplatin administration (Fig. 5B). The tumor growth rate was 37.4%
lower per day in the OC2CisR/shEGFR/shMRP2 group, compared with the
OC2CisR/shEGFR group (SE=17.8%; P=0.035). These results confirmed
that MRP2 overexpression confers resistance to cisplatin in
OC2CisR cells, and that downregulation of MRP2 is able to
significantly enhance the therapeutic effects of cisplatin in
EGFR-downregulated cells.
Discussion
Overexpression of EGFR is frequently observed
in numerous types of cancer, including OSCC (26,27).
EGFR-targeted therapies, including gefitinib (Iressa®;
AstraZeneca, London, UK) (26,28–30)
and cetuximab (Erbitux®; Eli Lilly and Company,
Indianapolis, IN, USA) (31,32), have been studied extensively as
potential methods of blocking the cancer-promoting functions of
EGFR in OSCC. However, although it has been reported that EGFR
inhibitors are initially beneficial for the treatment of cancer,
resistance to the inhibitors eventually develops (21,33).
Therefore, an alternative strategy was investigated in the present
study, in which rather than targeting the EGFR protein, the
expression of its gene, EGFR, in OSCC was downregulated
using RNAi techniques. The results of the present study demonstrate
that reduction of EGFR expression inhibits the growth and
enhances the cytotoxicity of 5-FU in OSCC cells.
Although MRP2 downregulation also increased
the sensitivity of OC2 cells to 5-FU, it did not contribute to the
suppression of EGFR-downregulated OC2 tumors following 5-FU
administration. Therefore, the ability of MRP2
downregulation to achieve further suppression of
EGFR-downregulated OC2 tumors in vivo during
cisplatin administration was investigated. Following establishment
of the cisplatin-resistant cell line OC2CisR, it was observed that
the expression of MRP2 was positively correlated with the
level of cisplatin resistance in the cells, whereby the OC2CisR
cells became highly resistant to the killing effect of cisplatin,
compared with the parental cell line OC2. These results imply that
MRP2 may have a significant role in modulating the response of
OC2CisR cells to cisplatin. Furthermore, downregulation of
MRP2 significantly enhanced the therapeutic effect of
cisplatin in EGFR-downregulated cells.
In conclusion, MRP2-targeted therapy may be a
promising strategy for overcoming the resistance to cisplatin in
OSCC. Inhibitors of ABC transporters, including MRP2, have been
extensively investigated in previous studies (34). However, there has been little impact
of these studies on clinical outcomes (35,36).
Therefore, an RNAi-mediated targeting strategy was applied in the
present study, with a view to reversing multidrug resistance in
tumors. Due to its specificity and effectiveness, RNAi-mediated
downregulation of MRP2 may be applicable as a therapeutic
approach toward reversing MRP2-dependent cisplatin resistance in
OSCC.
Acknowledgements
The present study was supported by the Ditmanson
Medical Foundation Chia-Yi Christian Hospital (Chia-Yi, Taiwan;
grant no. R100-3) and the National Science Council of Taiwan
(Taipei, Taiwan; grant no. NSC99-2320-B-194-004-MY3).
References
|
1
|
Petersen PE: Strengthening the prevention
of oral cancer: The WHO perspective. Community Dent Oral Epidemiol.
33:397–399. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
de Camargo Cancela M, Voti L, Guerra-Yi M,
Chapuis F, Mazuir M and Curado MP: Oral cavity cancer in developed
and in developing countries: Population-based incidence. Head Neck.
32:357–367. 2010.PubMed/NCBI
|
|
3
|
2009 Statistics of Causes of Death.
Ministry of Health and Welfare. Taipei, Taiwan: 2010.
|
|
4
|
Chabner BA and Roberts TG Jr: Timeline:
Chemotherapy and the war on cancer. Nat Rev Cancer. 5:65–72. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Andreadis C, Vahtsevanos K, Sidiras T,
Thomaidis I, Antoniadis K and Mouratidou D: 5-Fluorouracil and
cisplatin in the treatment of advanced oral cancer. Oral Oncol.
39:380–385. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ramos P and Bentires-Alj M:
Mechanism-based cancer therapy: Resistance to therapy, therapy for
resistance. Oncogene. 34:3617–3626. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gottesman MM, Fojo T and Bates SE:
Multidrug resistance in cancer: Role of ATP-dependent transporters.
Nat Rev Cancer. 2:48–58. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dean M, Rzhetsky A and Allikmets R: The
human ATP-binding cassette (ABC) transporter superfamily. Genome
Res. 11:1156–1166. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stavrovskaya AA and Stromskaya TP:
Transport proteins of the ABC family and multidrug resistance of
tumor cells. Biochemistry (Mosc). 73:592–604. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Surowiak P, Materna V, Kaplenko I,
Spaczynski M, Dolinska-Krajewska B, Gebarowska E, Dietel M, Zabel M
and Lage H: ABCC2 (MRP2, cMOAT) can be localized in the nuclear
membrane of ovarian carcinomas and correlates with resistance to
cisplatin and clinical outcome. Clin Cancer Res. 12:7149–7158.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hall MD, Okabe M, Shen DW, Liang XJ and
Gottesman MM: The role of cellular accumulation in determining
sensitivity to platinum-based chemotherapy. Annu Rev Pharmacol
Toxicol. 48:495–535. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yamasaki M, Makino T, Masuzawa T, Kurokawa
Y, Miyata H, Takiguchi S, Nakajima K, Fujiwara Y, Matsuura N, Mori
M and Doki Y: Role of multidrug resistance protein 2 (MRP2) in
chemoresistance and clinical outcome in oesophageal squamous cell
carcinoma. Br J Cancer. 104:707–713. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kool M, de Haas M, Scheffer GL, Scheper
RJ, van Eijk MJ, Juijn JA, Baas F and Borst P: Analysis of
expression of cMOAT (MRP2), MRP3, MRP4, and MRP5, homologues of the
multidrug resistance-associated protein gene (MRP1), in human
cancer cell lines. Cancer Res. 57:3537–3547. 1997.PubMed/NCBI
|
|
14
|
Guminski AD, Balleine RL, Chiew YE,
Webster LR, Tapner M, Farrell GC, Harnett PR and Defazio A: MRP2
(ABCC2) and cisplatin sensitivity in hepatocytes and human ovarian
carcinoma. Gynecol Oncol. 100:239–246. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Taniguchi K, Wada M, Kohno K, Nakamura T,
Kawabe T, Kawakami M, Kagotani K, Okumura K, Akiyama S and Kuwano
M: A human canalicular multispecific organic anion transporter
(cMOAT) gene is overexpressed in cisplatin-resistant human cancer
cell lines with decreased drug accumulation. Cancer Res.
56:4124–4129. 1996.PubMed/NCBI
|
|
16
|
Materna V, Stege A, Surowiak P, Priebsch A
and Lage H: RNA interference-triggered reversal of ABCC2-dependent
cisplatin resistance in human cancer cells. Biochem Biophys Res
Commun. 348:153–157. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xie SM, Fang WY, Liu Z, Wang SX, Li X, Liu
TF, Xie WB and Yao KT: Lentivirus-mediated RNAi silencing targeting
ABCC2 increasing the sensitivity of a human nasopharyngeal
carcinoma cell line against cisplatin. J Transl Med. 6:552008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Minegaki T, Takara K, Hamaguchi R,
Tsujimoto M and Nishiguchi K: Factors affecting the sensitivity of
human-derived esophageal carcinoma cell lines to 5-fluorouracil and
cisplatin. Oncol Lett. 5:427–434. 2013.PubMed/NCBI
|
|
19
|
Agulnik M: New approaches to EGFR
inhibition for locally advanced or metastatic squamous cell
carcinoma of the head and neck (SCCHN). Med Oncol. 29:2481–2491.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mendelsohn J and Baselga J: Epidermal
growth factor receptor targeting in cancer. Semin Oncol.
33:369–385. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chong CR and Jänne PA: The quest to
overcome resistance to EGFR-targeted therapies in cancer. Nat Med.
19:1389–1400. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang M, Zhang X, Bai CX, Song XR, Chen J,
Gao L, Hu J, Hong QY, West MJ and Wei MQ: Silencing the epidermal
growth factor receptor gene with RNAi may be developed as a
potential therapy for non small cell lung cancer. Genet Vaccines
Ther. 3:52005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wong DY, Chang KW, Chen CF and Chang RC:
Characterization of two new cell lines derived from oral cavity
human squamous cell carcinomas - OC1 and OC2. J Oral Maxillofac
Surg. 48:385–390. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lai YH, He RY, Chou JL, Chan MW, Li YF and
Tai CK: Promoter hypermethylation and silencing of tissue factor
pathway inhibitor-2 in oral squamous cell carcinoma. J Transl Med.
12:2372014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shichinohe T, Bochner BH, Mizutani K,
Nishida M, Hegerich-Gilliam S, Naldini L and Kasahara N:
Development of lentiviral vectors for antiangiogenic gene delivery.
Cancer Gene Ther. 8:879–889. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shintani S, Li C, Mihara M, Nakashiro K
and Hamakawa H: Gefitinib (‘Iressa’), an epidermal growth factor
receptor tyrosine kinase inhibitor, mediates the inhibition of
lymph node metastasis in oral cancer cells. Cancer Lett.
201:149–155. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takaoka S, Iwase M, Uchida M, Yoshiba S,
Kondo G, Watanabe H, Ohashi M, Nagumo M and Shintani S: Effect of
combining epidermal growth factor receptor inhibitors and cisplatin
on proliferation and apoptosis of oral squamous cell carcinoma
cells. Int J Oncol. 30:1469–1476. 2007.PubMed/NCBI
|
|
28
|
Shintani S, Li C, Mihara M, Terakado N,
Yano J, Nakashiro K and Hamakawa H: Enhancement of tumor
radioresponse by combined treatment with gefitinib (Iressa,
ZD1839), an epidermal growth factor receptor tyrosine kinase
inhibitor, is accompanied by inhibition of DNA damage repair and
cell growth in oral cancer. Int J Cancer. 107:1030–1037. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shintani S, Li C, Mihara M, Yano J,
Terakado N, Nakashiro K and Hamakawa H: Gefitinib (‘Iressa’,
ZD1839), an epidermal growth factor receptor tyrosine kinase
inhibitor, up-regulates p27KIP1 and induces G1 arrest in oral
squamous cell carcinoma cell lines. Oral Oncol. 40:43–51. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee EJ, Whang JH, Jeon NK and Kim J: The
epidermal growth factor receptor tyrosine kinase inhibitor ZD1839
(Iressa) suppresses proliferation and invasion of human oral
squamous carcinoma cells via p53 independent and MMP, uPAR
dependent mechanism. Ann N Y Acad Sci. 1095:113–128. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dai W, Li Y, Zhou Q, Xu Z, Sun C, Tan X
and Lu L: Cetuximab inhibits oral squamous cell carcinoma invasion
and metastasis via degradation of epidermal growth factor receptor.
J Oral Pathol Med. 43:250–257. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qian M, Qian D, Jing H, Li Y, Ma C and
Zhou Y: Combined cetuximab and celecoxib treatment exhibits a
synergistic anticancer effect on human oral squamous cell carcinoma
in vitro and in vivo. Oncol Rep. 32:1681–1688.
2014.PubMed/NCBI
|
|
33
|
Ohnishi Y, Minamino Y, Kakudo K and Nozaki
M: Resistance of oral squamous cell carcinoma cells to cetuximab is
associated with EGFR insensitivity and enhanced stem cell-like
potency. Oncol Rep. 32:780–786. 2014.PubMed/NCBI
|
|
34
|
Zhou SF, Wang LL, Di YM, Xue CC, Duan W,
Li CG and Li Y: Substrates and inhibitors of human multidrug
resistance associated proteins and the implications in drug
development. Curr Med Chem. 15:1981–2039. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Falasca M and Linton KJ: Investigational
ABC transporter inhibitors. Expert Opin Investig Drugs. 21:657–666.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu M, Ocana A and Tannock IF: Reversal of
ATP-binding cassette drug transporter activity to modulate
chemoresistance: Why has it failed to provide clinical benefit?
Cancer Metastasis Rev. 32:211–227. 2013. View Article : Google Scholar : PubMed/NCBI
|