Introduction
Multiple myeloma (MM) is a malignant neoplasia of
plasma cells, which accounts for ~10% of all types of hematological
cancer. According to the latest statistics, the annual incidence of
MM was reported to be 20,450 cases and the mortality rate for MM
was 11,090 cases/year in the USA in 2013 (1). In addition, MM has evolved as one of the
major diseases affecting the aging population in China (2). At present, MM is the second most
prevalent hematological malignancy following non-Hodgkin's lymphoma
(2). Even with the introduction of
immunmodulators, including thalidomide and lenalidomide, and
proteasome inhibitors, including bortezomib (BTZ), over the last
decade, the 5-year survival rate of patients in the USA with MM is
only 42% (1). In addition, almost all
patients with MM undergo relapse and refractory disease,
predominantly due to drug resistance (3). Until now, MM remains an incurable
disorder among hematological malignancies.
Patients with MM are at enhanced risk of
thromboembolism due to disease-specific and treatment-specific risk
factors, particularly when treated with thalidomide and
lenalidomide (4,5). Thus, the prevention of thrombosis has
become important in cases of MM. At present, no significant
differences have been observed among the use of aspirin (ASA),
low-dose warfarin and low-molecular weight heparin in
thromboprophylaxis (6). ASA may be
the optimum candidate to prevent thromboembolism in patients with
MM due to its merits in method of administration, safety and cost
without the requirement for regular coagulation monitoring
(7). ASA has been widely used for
thromboprophylaxis in patients with MM, particularly in cases
treated with thalidomide or lenalidomide combinations.
An increasing number of studies have demonstrated
that ASA possesses antineoplastic actions against a wide range of
solid tumor types, including esophageal, breast, lung and gastric
cancer, and colon cancer in particular (8–12).
Currently, the underlying anticancer mechanisms of ASA have been
ascribed to the inhibition of nuclear factor (NF)-kB, and the
induction of apoptosis by caspase activation, interruption of
extracellular signal-regulated kinase and epidermal growth factor
receptor in various types of cancer (13–17). Our
previous study found that ASA exerted antiproliferative and
pro-apoptotic activities in vivo and in vitro via the
upregulation of B cell lymphoma-2 (Bcl-2)-associated X protein
(Bax), and the suppression of Bcl-2 and vascular endothelial growth
factor (18).
The antimyeloma effect of BTZ, the first proteasome
inhibitor to be approved by the Food and Drug Administration of USA
in 2003, is well-recognized. At present, BTZ has become the
foundation of first- and second-line treatment in MM. The major
mechanisms underlying the action of BTZ in MM include inhibition of
the activation of NF-kB, upregulation of pro-apoptotic Noxa and the
subsequent downregulation of anti-apoptotic Bcl-2 (19). However, the increasing administration
of BTZ is associated with the development of drug resistance
(20). There have been a number of
explanations for the insensitivity to BTZ, including proteasome
subunit β5 mutation, insulin growth factor-1 and the
differentiation status of myeloma cells (21–23). The
activation of AKT and upregulation of survivin induced by BTZ
itself are the primary factors involved in the development of
resistance to BTZ in MM (24).
Coincidentally, survivin, Bcl-2 and AKT have been confirmed to be
the targets of ASA in gastric, cervical and ovarian cancer,
respectively (13,25,26).
In relation to the findings described above, the
present study hypothesized that ASA in combination with BTZ may
produce additive or synergistic effects in the treatment of MM. The
aim of the present study was to investigate the interaction between
ASA and BTZ in the MM1.S and RPMI-8226 myeloma cell lines, and
clarify the underlying mechanisms through detecting the effects of
ASA and BTZ on the Bcl-2, survivin and AKT proteins.
Materials and methods
Drugs and reagents
ASA (Sigma-Aldrich; Merck Millipore, Darmstadt,
Germany) was dissolved in solution containing 10 N sodium hydroxide
and adjusted to pH 7.0. BTZ (Selleck Chemicals, Houston, TX, USA)
was dissolved in dimethyl sulfoxide at a final concentration of 50
µM.
All liquid culture media were acquired from
Invitrogen; Thermo Fisher Scientific, Inc. (Waltham, MA, USA). The
BCA protein assay kit was obtained from Beyotime Institute of
Biotechnology (Nantong, China). Antibodies against GADPH and Bcl-2
were obtained from Goodhere Biotechnology Co., Ltd. (Hangzhou,
China) and Sigma-Aldrich; Merck Millipore, respectively. Antibodies
against Akt, p-Akt (Thr308) and p-Akt (Ser473) were obtained from
CST Biological Reagents Company Limited (Shanghai, China). The
secondary antibody was from EarthOx Company (San Francisco, CA
USA). The EnoGene™ total protein extraction kit was acquired from
EnoGene (Nanjing, China). Phosphosafe™ extraction reagent was
purchased from Merck Millipore. The chemiluminescent detection kit
(Super-Signal West Femto substrate) was from Thermo Fisher
Scientific, Inc.
Cell culture
The MM1.S cell line, harboring the K-Ras mutation,
and RPMI-8226, harboring the N-Ras mutation, were selected, as the
oncogenic mutations of the K- and N-Ras genes were found to exist
in 50% of MM cases and correlated with aggressive disease,
resistance to therapy and poor survival rates (27). The MM1.S human myeloma cell line was
provided by Dr Lu-Gui Qiu (Hematology Hospital, Chinese Academy of
Medical Science, Tianjin, China). The human MM cell line
(RPMI-8226) was purchased from American Type Culture Collection
(Manassas, VA, USA). The cells were cultured at 37°C in a
water-saturated atmosphere of 95% air and 5% CO2 in
RPMI-1640 medium supplemented with 10% heat-inactivated FBS (Thermo
Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin. When indicated, the cells were seeded at a confluence
of 80% (serum-free medium was the vehicle) in 96-well or 6-well
plates and treated with vehicle or ASA.
Cell proliferation assay
The cells were harvested and seeded into 96-well
plates (1×104 cells per well) in a total volume of 200
µl. According to the experimental design, the cells were divided
into four groups, including untreated, ASA-treated, BTZ-treated and
ASA+BTZ co-treated groups. The cells were then incubated for 24, 48
and 72 h at 37°C, respectively. Following treatment of the cells
for indicated durations, Cell Counting Kit-8 (CCK-8) solution
(Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was added
(20 µl per well) and the cells were incubated for 2 h at 37°C. The
plates were then read on an automated microplate spectrophotometer
(DNM-9602; Perlong Medical Equipment Co., Ltd., Nanjing, China) at
450 nm.
Cell apoptosis analysis
In the cell apotosis assay, Annexin-V-fluorescenin
isothiocyanate (FITC; 1:250) and propidium iodide (PI; 1 µg/ml)
were used, according to the protocol of the Annexin V-FITC
apoptosis assay kit (Beyotime Institute of Biotechnology). FITC
specifically binds to the phosphatidyl serine residues on the cell
membrane, whereas PI binds to DNA when the cell membrane becomes
permeable. The cells were stained and analyzed using the FACScan
system (BD Biosciences, Franklin Lakes, NJ, USA). The data were
analyzed using CellQuest (BD Biosceinces) software. For each
analysis, 10,000 events were recorded.
Drug interaction analysis
The ratio (R) was used to analyze the
interaction of the ASA and BTZ combination in the two cell lines,
as previously described (28). The
R ratio was calculated as follows:
R = survival (ASA+BTZ) / (survival ASA ×
survival BTZ)
If R<0.8, the combination was considered
to be synergistic; if 0.8<R<1.2, the combination was
considered additive; if R>1.2, the combination was
considered antagonistic.
Western blot analysis
The treated cells (5–6×106) were washed
in PBS and then lysed in 2X Laemmli buffer. Protein concentrations
were determined using the BCA assay kit.
Following boiling for 5 min, equivalent quantities
of protein (30–40 µg) were separated on 8–12% SDS-polyacrylamide
gels and then transferred onto a nitrocellulose membrane (0.45 µM).
The membranes were blocked with 5% BSA in TBS/Tween20 (0.05% v/v)
for 1 h, followed by incubation at 4°C overnight with primary
antibodies against Bcl-2 (1:500 dilution), survivin (1:870
dilution), Akt (1:1,000 dilution), p-Akt (Thr308; 1:1,000 dilution)
and p-Akt (Ser473; 1:1,000 dilution). GADPH (1:1,000 dilution)
served as a control. The membranes were washed three times with
TBST and then incubated for 1 h at room temperature with
horseradish peroxidase-conjugated anti-rabbit secondary antibodies
(1:10,000 dilution). The protein bands were visualized using
enhanced chemiluminescence on a ChemiDoc™ XRS+ system
with Image Lab™ software version 4.0 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The quantification of protein levels was
performed by Gel-Pro Analyzer (version 4.0) and the integrated
option density was used as the quantity for comparison.
Statistical analysis
All results in bar graphs are expressed as the mean
+ standard deviation obtained from at least three independent
experiments. Statistical differences were evaluated using Student's
t-test (paired) and one-way analysis of variance (ANOVA), as
appropriate, using SPSS 13.0 (SPSS, Inc, Chicago, IL, USA). P≤0.05
was considered to indicate a statistically significant difference.
Differences determined to be significant using ANOVA were further
analyzed using Turkey's pairwise comparison to detect specific
differences between treatments.
Results
Co-treatment of ASA and BTZ reduces
the proliferation of myeloma cells
ASA at a dose of 2.5 mM was selected for use in
experiments in the present study, as 2.5 mM was the concentration
measured in the serum of patients treated with ASA for chronic
inflammatory diseases in a previous study (29). A BTZ concentration of 10 nM was the
plateau concentration following an initial peak at ~300 nM (for 1
h) in the serum of BTZ-treated patients with MM in a previous study
(30). Therefore, 10 nM BTZ was used
as the experimental concentration in the present study. The effects
of ASA, BTZ and ASA in combination with BTZ on the proliferation of
MM1.S and RPMI-8226 cells were evaluated using a CCK-8 assay.
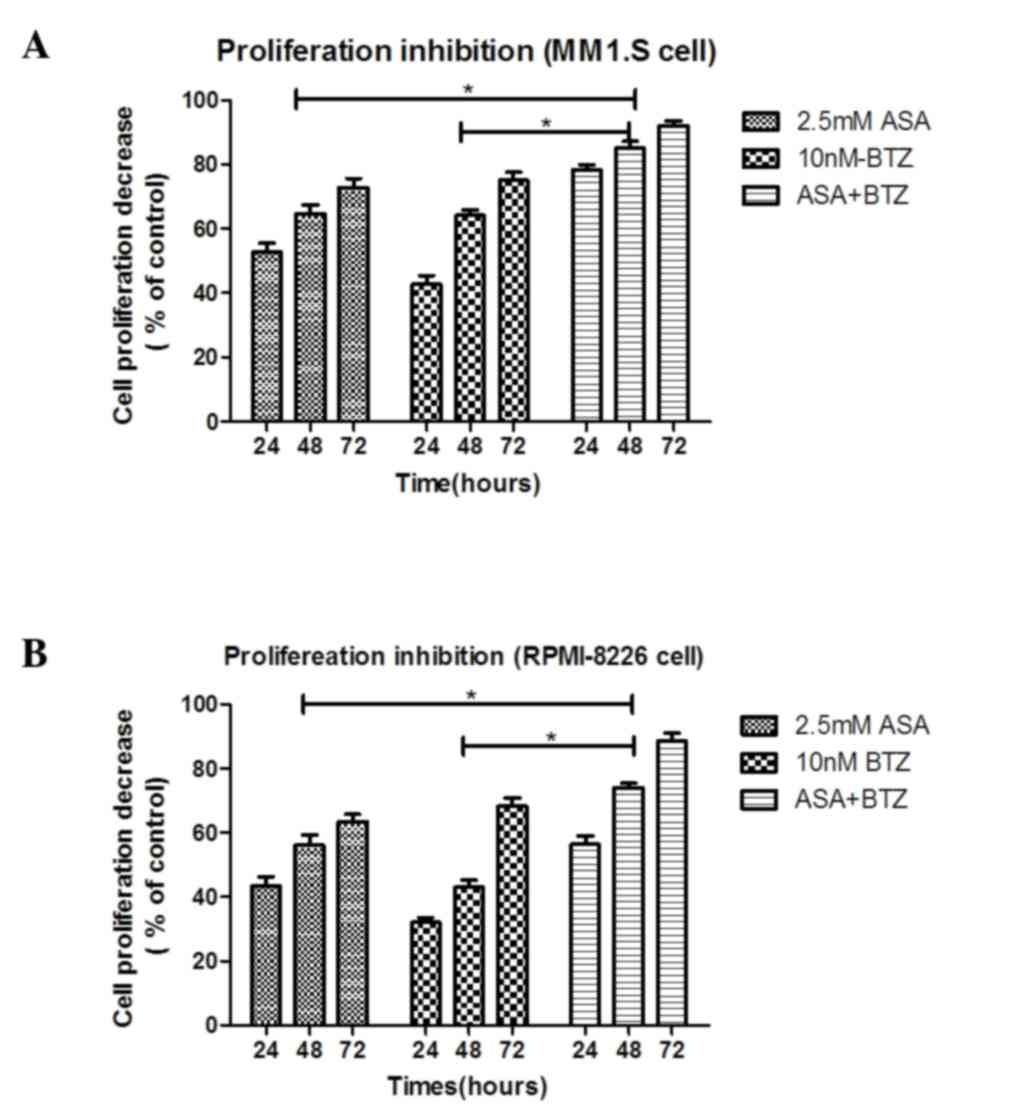
As shown in Fig. 1A,
the MM1.S cells treated with ASA+BTZ for 24, 48 and 72 h led to
significant decreases (P<0.05) in cell proliferation (78.3, 85.1
and 91.9%, respectively), compared with the cells treated with ASA
alone (52.7, 64.6 and 72.8%, respectively) or BTZ alone (43.6, 64.1
and 75.1%, respectively. However, no significant difference was
found between the ASA-treated and BTZ-treated groups.
As shown in Fig. 1B,
treatment with ASA+BTZ significantly (P<0.05) reduced the
viability of the RPMI-8226 cells within 24, 48 and 72 h (56.4, 73.9
and 88.5%, respectively), compared with the cells exposed to ASA
alone (43.4, 56.1 and 63.6%, respectively) or BTZ alone (32.1, 42.9
and 68.4%, respectively). Again, no statistical differences were
found between the ASA-treated and BTZ-treated groups
(P>0.05).
The combination treatment of ASA and BTZ for 24, 48
and 72 h resulted in an additive effect on the inhibitory rate of
MM1.S or RPMI-8226 cells, as R was between 0.8 and 1.2 in
the two cell lines, indicating that ASA enhanced the antimyleoma
activity of BTZ.
Co-exposure of ASA+BTZ augments the
apoptotic rate of myeloma cells
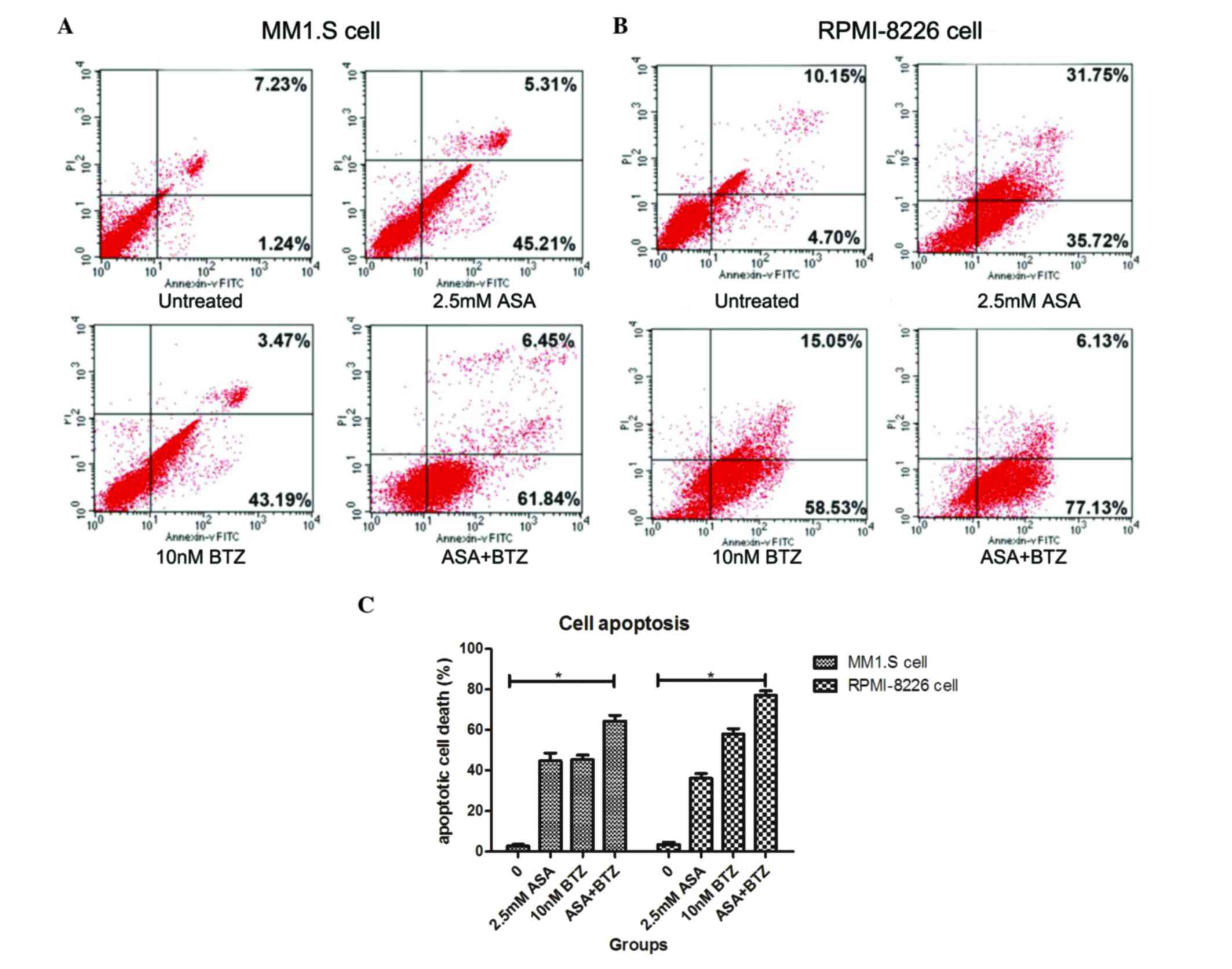
The present study used Annexin V-FITC/PI flow
cytometry to determine whether the additive action of ASA and BTZ
was correlated with the induction of cell apoptosis. The MM1.S and
RPMI-8226 cells were incubated with ASA alone, BTZ alone, and with
the combination of ASA and BTZ at 37°C for 48 h. At the indicated
duration, the cells were harvested for the Annexin V-FITC/PI assay
using flow cytometry.
As shown in Fig. 2,
the apoptotic rates of the untreated cells, and the cells treated
with ASA alone, BTZ alone and with ASA and BTZ were 2.73, 44.8,
45.4 and 64.3%, respectively. Co-exposure of the cells to ASA with
BTZ led to higher rates of apoptosis, compared with the cells
treated with either ASA or BTZ alone (P<0.05). No significant
differences were found between the ASA-treated group and
BTZ-treated group.
In the RPMI-8226 cells, the apoptotic rates were
3.43, 36.1, 57.9 and 77.2% in the untreated, ASA-treated,
BTZ-treated and ASA+BTZ-treated cells, respectively. Similarly,
co-treatment with ASA and BTZ triggered higher rates of cell
apoptosis, compared with either ASA or BTZ alone (P<0.05).
Unlike the MM1.S cells, BTZ treatment induced higher rates of
apoptosis, compared with ASA treatment in the RPMI-8226 cells
(P<0.05). Taken together, these findings revealed that
co-exposure to ASA and BTZ augmented the apoptotic rate of myeloma
cells, compared with ASA or BTZ treatment alone in myeloma
cells.
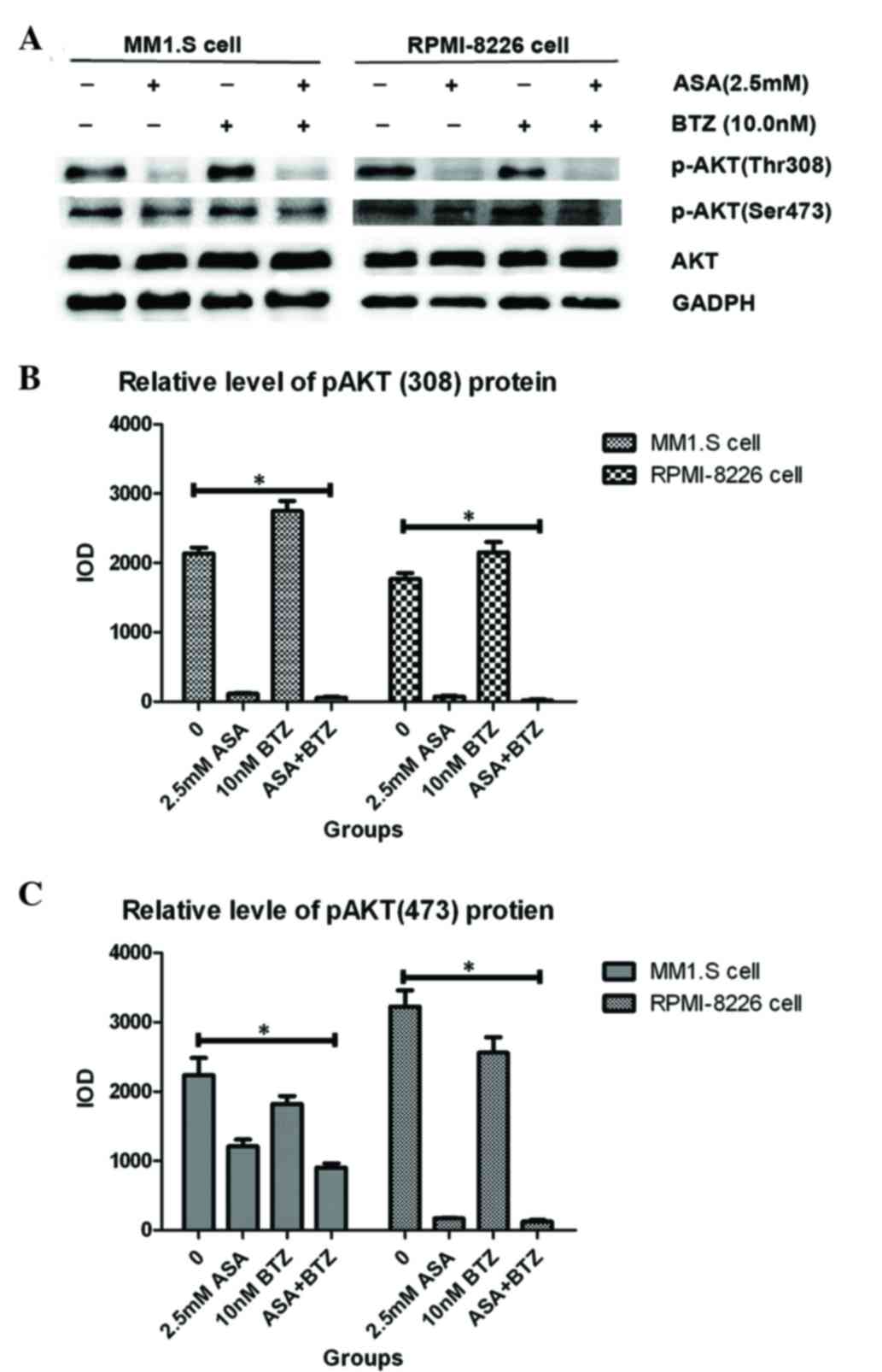
ASA inhibits the phosphorylation of
AKT induced by BTZ in myeloma cells
AKT is important in the carcinogenesis of MM via
controlling cell survival and apoptosis (31). Aberrant AKT activation confers
chemoresistance to dexamethasone (Dex) and BTZ in MM cells
(24). In particular, the
phosphorylation of Thr308 and Ser473 is essential in the activation
of AKT. Therefore, the present study examined the effect of ASA and
BTZ alone or in combination on the phosphorylation of AKT in
myeloma cells over 48 h.
As shown in Fig. 3,
the MM1.S and RPMI-8226 cells expressed high endogenous levels of
p-AKT, indicating the constitutive activation of AKT in the two
cell lines. ASA treatment markedly decreased the levels of p-AKT,
including Thr308 and Ser473, in the MM1.S and RPMI-8226 cells. Of
note, exposure of the two cell lines to ASA almost completely
inhibited the phosphorylation of AKT at Thr308. However, no
significant changes were found in total AKT protein between the two
cell lines. In addition, it was found that BTZ treatment led to the
enhancement of p-AKT, including Thr308 and Ser473. Treatment with
ASA in combination with BTZ markedly reduced the phosphorylation of
AKT, particularly for Thr308, suggesting the potential activity of
ASA in overcoming the resistance to BTZ in myeloma cells.
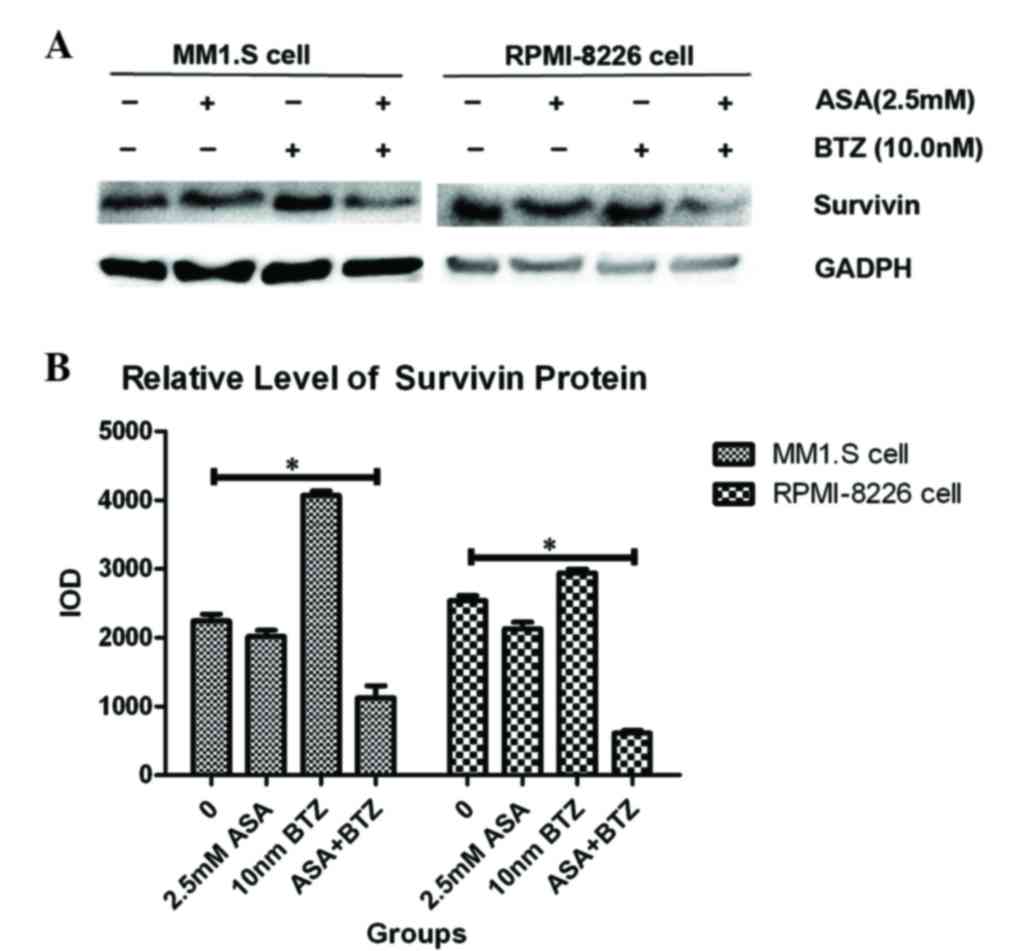
ASA downregulates the expression of
survivin induced by BTZ
As a prominent anti-apoptotic molecule, increased
levels of survivin contribute to uncontrolled cell proliferation
and drug resistance in MM. Therefore; the present study
investigated the effect of the ASA and BTZ combination on the level
of survivin.
As shown in Fig. 4,
exposure of the MM1.S and RPMI-8226 cells to ASA alone for 48 h led
to significant decreases in survivin, compared with the untreated
group. By contrast, BTZ treatment induced the upregulation of
survivin in the MM1.S and RPMI-8226 cells. However, the combined
treatment with ASA and BTZ decreased the protein levels of survivin
in the MM1.S and RPMI-8226 cells, indicating that ASA may inhibit
the upregulation of survivin triggered by BTZ.
ASA in combination with BTZ suppresses
the expression of Bcl-2 in myeloma cells
As a classical anti-apoptotic gene, Bcl-2 regulates
apoptosis and cell death in MM, contributing to the pathogenesis,
progression and chemoresistance of MM. Therefore, the present study
examined the effect of treatment with ASA and/or BTZ on the protein
expression of Bcl-2.
As shown in Fig. 5,
incubation of the myeloma cells with ASA or BTZ for 48 h caused a
marked downregulation in the level of Bcl-2, compared with the
corresponding vehicle. Notably, exposure of myeloma cells to
ASA+BTZ markedly decreased the protein level of Bcl-2, compared
with ASA or BTZ treatment alone. These results supported the causal
association between the suppression of Bcl-2 protein and the
apoptosis induced by ASA and/or BTZ in MM1.S and RPMI-8226 myeloma
cells.
Discussion
At present, BTZ has been incorporated as leading
agent into conditioning, consolidation and maintenance therapy for
patients with MM (19). However, the
prolonged administration of BTZ is associated with the development
of drug resistance (20). The
mechanisms underlying BTZ resistance in MM remain to be elucidated,
however, they have been partly attributed to ‘molecular
side-effects’, including the induction of AKT activation and
upregulation of survivin (24), which
attenuate the cytotoxic action of BZT against MM cells and can lead
to chemoresistance. Therefore, identifying how to enhance the
antimyeloma activity of BTZ, particularly in patients with relapsed
or refractory MM, is required. The antineoplastic role of ASA has
been verified in a wide range of solid tumor types (32–36). Our
previous study reported that ASA exerted antimyeloma activity in
vitro and in vivo accompanying the thromboprophylactic
profile (18). In the present study,
the chemosensitive effect of ASA was examined in myeloma cells
treated with BTZ based on the ‘molecular side-effects’.
The concentrations of 2.5 mM ASA and 10 nM BTZ were
selected as the experimental concentrations in the present study,
as they resemble the serum concentrations in patients with MM with
important clinical implications (29,30).
Initially, the present study found that ASA in combination with BTZ
resulted in enhanced inhibition of cell proliferation, compared
with ASA or BTZ alone in MM1.S and RPMI-8226 cells, and occurred in
a time-dependent manner. Subsequent interaction analysis revealed
an additive effect, indicating that ASA may potentiate the
antimyeloma property of BTZ. In accordance, the co-administration
of ASA and BTZ triggered higher apoptotic rates, compared with
treatment with ASA or BTZ alone in the two cell lines. These
results supported the hypothesis that ASA has a chemosensitive
effect in BTZ-treated myeloma cells.
As a serine-threonine kinase, AKT is a key molecule
of the phosphoinositide 3-kinase/AKT signaling pathway. Once
recruited to the plasma membrane, AKT is activated by
phosphorylation at the Ser473 and Thr308 residues, and then
regulates the function of several cellular proteins involved in
survival/apoptosis, differentiation and proliferation, including
mammalian target of rapamycin, NF-κB and glycogen synthase kinase-3
(37). Several studies have provided
evidence that the aberrant activation of AKT is important in the
pathogenesis of various types of cancer, including MM (38–41). The
ectopic activation of AKT has been documented in ~50% patients with
MM. In addition, p-AKT renders anti-apoptotic activity to MM cells,
indicating the importance of AKT activation in the carcinogenesis
of MM (42). p-AKT is also associated
with the prognosis in MM (38). These
findings provide compelling evidence that AKT may be a potential
therapeutic target in MM treatment. In the present study, treatment
with ASA alone almost completely inhibited the phosphorylation of
AKT (Thr308) and partly suppressed the phosphorylation of AKT
(Ser473) in MM1.S and RPMI-8226 cells, indicating that AKT was the
anticancer target of ASA in myeloma cells. By contrast, exposure of
the myeloma cells to BTZ alone induced the phosphorylation of AKT,
which was in agreement with a report by Hideshima et al
(24). Of note, treatment with ASA
combined with BTZ markedly inhibited the levels of p-AKT (Thr308
and Ser473), particularly for Thr308. These data suggested that ASA
may augment the cytotoxic activity of BTZ against myeloma cells,
and may even overcome the chemoresistance of BTZ in
refractory/relapsed MM.
Survivin is also involved in inhibiting apoptosis,
promoting proliferation and enhancing invasion in malignancies.
Survivin has been reported to be overexpressed ubiquitously in
cells in several types of cancer, including breast cancer (43), lung cancer (44) and MM (45). Furthermore, elevated levels of
survivin have been correlated with poor prognosis, decreased
survival and chemotherapy resistance in MM cells (45,46).
Accordingly, the inhibition of survivin may be an efficient form of
therapy in patients with MM. In the present study, it was
demonstrated that BTZ alone was capable of increasing the level of
survivin in MM1.S and RPMI-8226 cells, which was in accordance with
the results reported in human non-small cell lung cancer cells by
Liu et al (47). By contrast,
the concurrent use of ASA and BTZ substantially inhibited the
expression of survivin in the two cell lines. These findings
provided convincing evidence that ASA may increase the cytotoxicity
of BTZ to myeloma cells, in part, via the inhibition of
survivin.
Cell apoptosis is correlated with a broad spectrum
of cellular factors and pathophysiologic pathways. The Bcl-2 family
is the most prominent in the process of apoptosis. In addition,
Bcl-2, as a classical anti-apoptotic protein, has been confirmed to
be overexpressed in a diverse range of types of cancer in humans
(48–50). The Bcl-2 protein is expressed at high
levels in patients with myeloma and is implicated in the regulation
of chemosensitivity. Dex has been used for the treatment of MM for
>50 years. Dex has also been incorporated into agents in
combination with BTZ, thalidomide or lenalidomide. However,
elevated expression of Bcl-2 confers chemoresistance to Dex in
myeloma cells (51,52). In the present study, ASA exhibited
antineoplastic activity against Dex-sensitive MM1.S cells and
Dex-resistant RPMI-8226 cells. The western blot assay revealed that
treatment with either ASA or BTZ alone downregulated the level of
Bcl-2. Furthermore, ASA in combination with BTZ led to a marked
decrease in the protein level of Bcl-2, compared with either ASA or
BTZ alone, suggesting that ASA in combination with BTZ may be a
promising treatment regimen for patients with MM with elevated
expression of Bcl-2. Thus, the rationale of a combined regimen of
Dex and BTZ in treating MM may be, in part, based on the ability of
BTZ to inhibit Bcl-2. These findings indicated that BTZ may
abrogate chemoresistance to Dex mediated by enhanced Bcl-2, which
was consistent with reports by Ailawadhi et al (53) and Pei et al (54).
Perifosine (PERI), a novel synthetic
alkylphospholipid, targets Akt and exerts potent antineoplastic
activity in a wide variety of neoplasms, particularly MM and
colorectal carcinoma (55). PERI also
induces the significant downregulation of survivin in human myeloma
cells (56). Clinical observations
have found that a treatment regimen of PERI in combination with BTZ
showed marked activity, with an overall response rate (ORR) of 32
and 65%, and median overall survival (mOS) of 22.5 and 30.4 months
in BTZ-refractory and BTZ-relapsed patients, respectively (57). Encouraged by this data, a
placebo-controlled phase III study was designed to evaluate the
effect of adding PERI (50 mg daily) to BTZ and DEX in patients with
MM who had relapsed following a BTZ-based regimen and who had
received between one and four previous treatment regimens. However,
this phase III experiment showed no benefit in PFS or ORR when PERI
was added to BTZ and DEX in patients with resistant, relapsed and
refractory MM. Furthermore, the mOS for PERI was 141.9 weeks and
for the placebo was 83.3 weeks, which showed favorable results for
PERI, but without statistical significance (P=0.356) (58), therefore, the investigation was
discontinued.
Coincidently, in the present study, it was found
that ASA also inhibited the expression of survivin and the
phosphorylation of AKT induced by BTZ in myeloma cells, which
explained the enhanced cytotoxicity of ASA+BTZ against myeloma
cells, compared with either agent alone. Notably, ASA retained its
thromboprophylactic property, which was deficient in the case with
PERI; treatment with PERI (150 mg daily) caused increased
creatinine in 30–60% of patients with MM (59), whereas the continued administration of
ASA may have a protective effect against renal injury (60). Thus, ASA may be a more superior
candidate to augment the antimyeloma activity of BTZ, compared with
PERI.
In conclusion, the present study demonstrated that
the concurrent administration of ASA and BTZ caused higher levels
of proliferative inhibition and apoptotic rates, compared with
either agent alone in myeloma cells in vitro, which
indicated the chemosensitivity towards ASA in MM treated by BTZ.
The underlying mechanisms included the suppression of survivin and
AKT phosphorylation by ASA induced by BTZ, together with the
inhibition of Bcl-2, suggesting the potential of a regimen
comprising ASA+BTZ-containing chemotherapy in patients with
refractory or relapsed MM.
Acknowledgements
This study was funded by the National Nature Science
Foundation of China (grant no. 81460037) and the Medical and
Healthy Research Foundation of Nanjing Military Area of China
(grant no. 14ZD31).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sonneveld P and Broijl A: Treatment of
relapsed and refractory multiple myeloma. Haematologica.
101:9952016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Morgan GJ and Davies FE: Role of
thalidomide in the treatment of patients with multiple myeloma.
Crit Rev Oncol Hematol. 1(88): Supp 1. S14–S22. 2013. View Article : Google Scholar
|
|
5
|
Bagratuni T, Kastritis E, Politou M,
Roussou M, Kostouros E, Gavriatopoulou M, Eleutherakis-Papaiakovou
E, Kanelias N, Terpos E and Dimopoulos MA: Clinical and genetic
factors associated with venous thromboembolism in myeloma patients
treated with lenalidomide-based regimens. Am J Hematol. 88:765–770.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Palumbo A, Cavo M, Bringhen S, Zamagni E,
Romano A, Patriarca F, Rossi D, Gentilini F, Crippa C, Galli M, et
al: Aspirin, warfarin, or enoxaparin thromboprophylaxis in patients
with multiple myeloma treated with thalidomide: A phase III,
open-label, randomized trial. J Clin Oncol. 29:986–993. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Niesvizky R, Martínez-Baños D,
Jalbrzikowski J, Christos P, Furst J, De Sancho M, Mark T, Pearse
R, Mazumdar M, Zafar F, et al: Prophylactic low-dose aspirin is
effective antithrombotic therapy for combination treatments of
thalidomide or lenalidomide in myeloma. Leuk Lymphoma.
48:2330–2337. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Araujo JL, Altorki NK, Sonett JR,
Rodriguez A, Sungur-Stasik K, Spinelli CF, Neugut AI and Abrams JA:
Prediagnosis aspirin use and outcomes in a prospective cohort of
esophageal cancer patients. Therap Adv Gastroenterol. 9:806–814.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Murphy C, Turner N, Wong HL, Sinnathamby
M, Tie J, Lee B, Desai J, Skinner I, Christie M and Hutchinson R:
Examining the impact of regular aspirin use and PIK3CA mutations on
survival in stage 2 colon cancer. Intern Med J. Nov 1–2016.(Epub
ahead of print). PubMed/NCBI
|
|
10
|
Hochmuth F, Jochem M and Schlattmann P:
Meta-analysis of aspirin use and risk of lung cancer shows notable
results. Eur J Cancer Prev. 25:259–268. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bradley MC, Black A, Freedman AN and
Barron TI: Prediagnostic aspirin use and mortality in women with
stage I to III breast cancer: A cohort study in the Prostate, Lung,
Colorectal, and Ovarian Cancer Screening Trial. Cancer.
122:2067–2075. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim YI, Kim SY, Kim JH, Lee JH, Kim YW,
Ryu KW, Park JH and Choi IJ: Long-term low-dose aspirin use reduces
gastric cancer incidence: A nationwide cohort study. Cancer Res
Treat. 48:798–805. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xiang S, Sun Z, He Q, Yan F, Wang Y and
Zhang J: Aspirin inhibits ErbB2 to induce apoptosis in cervical
cancer cells. Med Oncol. 27:379–387. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Park IS, Jo JR, Hong H, Nam KY, Kim JB,
Hwang SH, Choi MS, Ryu NH, Jang HJ, Lee SH, et al: Aspirin induces
apoptosis in YD-8 human oral squamous carcinoma cells through
activation of caspases, down-regulation of Mcl-1, and inactivation
of ERK-1/2 and AKT. Toxicol In Vitro. 24:713–720. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ararat E, Sahin I and Altundag K:
Mechanisms behind the aspirin use and decreased breast cancer
incidence. J BUON. 16:1802011.PubMed/NCBI
|
|
16
|
Im SR and Jang YJ: Aspirin enhances
TRAIL-induced apoptosis via regulation of ERK1/2 activation in
human cervical cancer cells. Biochem Biophys Res Commun. 424:65–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lim WY, Chuah KL, Eng P, Leong SS, Lim E,
Lim TK, Ng A, Poh WT, Tee A, Teh M, et al: Aspirin and non-aspirin
non-steroidal anti-inflammatory drug use and risk of lung cancer.
Lung Cancer. 77:246–251. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding JH, Yuan LY, Huang RB and Chen GA:
Aspirin inhibits proliferation and induces apoptosis of multiple
myeloma cells through regulation of Bcl-2 and Bax and suppression
of VEGF. Eur J Haematol. 93:329–339. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen D, Frezza M, Schmitt S, Kanwar J and
Dou QP: Bortezomib as the first proteasome inhibitor anticancer
drug: Current status and future perspectives. Curr Cancer Drug
Targets. 11:239–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chauhan D, Tian Z, Nicholson B, Kumar KG,
Zhou B, Carrasco R, McDermott JL, Leach CA, Fulcinniti M, Kodrasov
MP, et al: A small molecule inhibitor of ubiquitin-specific
protease-7 induces apoptosis in multiple myeloma cells and
overcomes bortezomib resistance. Cancer Cell. 22:345–358. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oerlemans R, Franke NE, Assaraf YG, Cloos
J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova
K, Lemos C, et al: Molecular basis of bortezomib resistance:
Proteasome subunit beta5 (PSMB5) gene mutation and overexpression
of PSMB5 protein. Blood. 112:2489–2499. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kuhn DJ, Berkova Z, Jones RJ, Woessner R,
Bjorklund CC, Ma W, Davis RE, Lin P, Wang H, Madden TL, et al:
Targeting the insulin-like growth factor-1 receptor to overcome
bortezomib resistance in preclinical models of multiple myeloma.
Blood. 120:3260–3270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gu JL, Li J, Zhou ZH, Liu JR, Huang BH,
Zheng D and Su C: Differentiation induction enhances bortezomib
efficacy and overcomes drug resistance in multiple myeloma. Biochem
Biophys Res Commun. 420:644–650. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hideshima T, Catley L, Yasui H, Ishitsuka
K, Raje N, Mitsiades C, Podar K, Munshi NC, Chauhan D, Richardson
PG and Anderson KC: Perifosine, an oral bioactive novel
alkylphospholipid, inhibits Akt and induces in vitro and in vivo
cytotoxicity in human multiple myeloma cells. Blood. 107:4053–4062.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Uddin S, Ahmed M, Hussain A, Assad L,
Al-Dayel F, Bavi P, Al-Kuraya KS and Munkarah A: Cyclooxygenase-2
inhibition inhibits PI3K/AKT kinase activity in epithelial ovarian
cancer. Int J Cancer. 126:382–394. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang L, Zhu H, Liu D, Liang S, Xu H, Chen
J, Wang X and Xu Z: Aspirin suppresses growth of human gastric
carcinoma cell by inhibiting survivin expression. J Biomed Res.
25:246–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chng WJ, Gonzalez-Paz N, Price-Troska T,
Jacobus S, Rajkumar SV, Oken MM, Kyle RA, Henderson KJ, Van Wier S,
Greipp P, et al: Clinical and biological significance of RAS
mutations in multiple myeloma. Leukemia. 22:2280–2284. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fischel JL, Formento P and Milano G:
Epidermal growth factor receptor double targeting by a tyrosine
kinase inhibitor (Iressa) and a monoclonal antibody (Cetuximab).
Impact on cell growth and molecular factors. Br J Cancer.
92:1063–1068. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Book Review: Goodman and Gilman's the
Pharmacological Basis of Therapeutics. Digital Edition; 11th. 40.
pp. 12182006
|
|
30
|
Terpos E, Roussou M and Dimopoulos MA:
Bortezomib in multiple myeloma. Expert Opin Drug Metab Toxicol.
4:639–654. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Harvey RD and Lonial S: PI3 kinase/AKT
pathway as a therapeutic target in multiple myeloma. Future Oncol.
3:639–647. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Choe KS and Liauw SL: Effects of aspirin
on cancer initiation and progression. Expert Rev Anticancer Ther.
13:115–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wilson JC, Murray LJ, Hughes CM, Black A
and Anderson LA: Non-steroidal anti-inflammatory drug and aspirin
use and the risk of head and neck cancer. Br J Cancer.
108:1178–1181. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rothwell PM: Alternate-day, low-dose
aspirin and cancer risk. Ann Intern Med. 159:148–150. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jonsson F, Yin L, Lundholm C, Smedby KE,
Czene K and Pawitan Y: Low-dose aspirin use and cancer
characteristics: A population-based cohort study. Br J Cancer.
109:1921–1925. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Algra AM and Nortier JW: Aspirin and
cancer: Evidence of a prophylactic effect. Ned Tijdschr Geneeskd.
157:A51892013.(In Dutch). PubMed/NCBI
|
|
37
|
Coutte L, Dreyer C, Sablin MP, Faivre S
and Raymond E: PI3K-AKT-mTOR pathway and cancer. Bull Cancer.
99:173–180. 2012.(In French). PubMed/NCBI
|
|
38
|
Hsu J, Shi Y, Krajewski S, Renner S,
Fisher M, Reed JC, Franke TF and Lichtenstein A: The AKT kinase is
activated in multiple myeloma tumor cells. Blood. 98:2853–2855.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hasson SP, Rubinek T, Ryvo L and Wolf I:
Endocrine resistance in breast cancer: Focus on the
phosphatidylinositol 3-Kinase/Akt/mammalian target of rapamycin
signaling pathway. Breast Care (Basel). 8:248–255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bi LK, Zhou N, Liu C, Lu FD, Lin TX, Xuan
XJ, Jiang C, Han JL, Huang H, Zhang CX, et al: Kidney cancer cells
secrete IL-8 to activate Akt and promote migration of mesenchymal
stem cells. Urol Oncol. 32:607–612. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ito S, Igishi T, Takata M, Ueda Y,
Matsumoto S, Kodani M, Takeda K, Izumi H, Sakamoto T, Yamaguchi K,
et al: Synergistic cell growth inhibition by the combination of
amrubicin and Akt-suppressing agents in K-ras mutation-harboring
lung adenocarcinoma cells: Implication of EGFR tyrosine kinase
inhibitors. Int J Oncol. 44:685–692. 2014.PubMed/NCBI
|
|
42
|
Steinbrunn T, Stühmer T, Gattenlöhner S,
Rosenwald A, Mottok A, Unzicker C, Einsele H, Chatterjee M and
Bargou RC: Mutated RAS and constitutively activated Akt delineate
distinct oncogenic pathways, which independently contribute to
multiple myeloma cell survival. Blood. 117:1998–2004. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xu C, Yamamoto-Ibusuki M, Yamamoto Y,
Yamamoto S, Fujiwara S, Murakami K, Okumura Y, Yamaguchi L, Fujiki
Y and Iwase H: High survivin mRNA expression is a predictor of poor
prognosis in breast cancer: A comparative study at the mRNA and
protein level. Breast Cancer. 21:482–490. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang M, Liu BG, Yang ZY, Hong X and Chen
GY: Significance of survivin expression: Prognostic value and
survival in stage III non-small cell lung cancer. Exp Ther Med.
3:983–988. 2012.PubMed/NCBI
|
|
45
|
Tsubaki M, Satou T, Itoh T, Imano M, Komai
M, Nishinobo M, Yamashita M, Yanae M, Yamazoe Y and Nishida S:
Overexpression of MDR1 and survivin, and decreased Bim expression
mediate multidrug-resistance in multiple myeloma cells. Leuk Res.
36:1315–1322. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tsubaki M, Komai M, Itoh T, Imano M,
Sakamoto K, Shimaoka H, Takeda T, Ogawa N, Mashimo K, Fujiwara D,
et al: By inhibiting Src, verapamil and dasatinib overcome
multidrug resistance via increased expression of Bim and decreased
expressions of MDR1 and survivin in human multidrug-resistant
myeloma cells. Leuk Res. 38:121–130. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu X, Yue P, Chen S, Hu L, Lonial S,
Khuri FR and Sun SY: The proteasome inhibitor PS-341 (bortezomib)
up-regulates DR5 expression leading to induction of apoptosis and
enhancement of TRAIL-induced apoptosis despite up-regulation of
c-FLIP and survivin expression in human NSCLC cells. Cancer Res.
67:4981–4988. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Geng M, Wang L and Li P: Correlation
between chemosensitivity to anticancer drugs and Bcl-2 expression
in gastric cancer. Int J Clin Exp Pathol. 6:2554–2559.
2013.PubMed/NCBI
|
|
49
|
Du P, Cao H, Wu HR, Zhu BS, Wang HW, Gu
CW, Xing CG and Chen W: Blocking Bcl-2 leads to autophagy
activation and cell death of the HEPG2 liver cancer cell line.
Asian Pac J Cancer Prev. 14:5849–5854. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang D, Chen MB, Wang LQ, Yang L, Liu CY
and Lu PH: Bcl-2 expression predicts sensitivity to chemotherapy in
breast cancer: A systematic review and meta-analysis. J Exp Clin
Cancer Res. 32:1052013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Iyer R, Ding L, Batchu RB, Naugler S,
Shammas MA and Munshi NC: Antisense p53 transduction leads to
overexpression of bcl-2 and dexamethasone resistance in multiple
myeloma. Leuk Res. 27:73–78. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gazitt Y, Fey V, Thomas C and Alvarez R:
Bcl-2 overexpression is associated with resistance to
dexamethasone, but not melphalan, in multiple myeloma cells. Int J
Oncol. 13:397–405. 1998.PubMed/NCBI
|
|
53
|
Ailawadhi S, Miecznikowski J, Gaile DP,
Wang D, Sher T, Mulligan G, Bryant B, Wilding GE, Mashtare T, Stein
L, et al: Bortezomib mitigates adverse prognosis conferred by Bcl-2
overexpression in patients with relapsed/refractory multiple
myeloma. Leuk Lymphoma. 53:1174–1182. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Pei XY, Dai Y and Grant S: The proteasome
inhibitor bortezomib promotes mitochondrial injury and apoptosis
induced by the small molecule Bcl-2 inhibitor HA14-1 in multiple
myeloma cells. Leukemia. 17:2036–2045. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Vink SR, Schellens JH, van Blitterswijk WJ
and Verheij M: Tumor and normal tissue pharmacokinetics of
perifosine, an oral anti-cancer alkylphospholipid. Invest New
Drugs. 23:279–286. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hideshima T, Catley L, Raje N, Chauhan D,
Podar K, Mitsiades C, Tai YT, Vallet S, Kiziltepe T, Ocio E, et al:
Inhibition of Akt induces significant downregulation of survivin
and cytotoxicity in human multiple myeloma cells. Br J Haematol.
138:783–791. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Richardson PG, Wolf J, Jakubowiak A,
Zonder J, Lonial S, Irwin D, Densmore J, Krishnan A, Raje N, Bar M,
et al: Perifosine plus bortezomib and dexamethasone in patients
with relapsed/refractory multiple myeloma previously treated with
bortezomib: Results of a multicenter phase I/II trial. J Clin
Oncol. 29:4243–4249. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Richardson PG, Nagler A, Dina BY and
Ashraf Badros: Randomized placebo-controlled phase III study of
perifosine combined with bortezomib and dexamethasone in relapsed,
refractory multiple myeloma patients previously treated with
bortezomib. ASH Annual Meeting abstract. 31892013.
|
|
59
|
Jakubowiak AJ, Richardson PG, Zimmerman T,
Alsina M, Kaufman JL, Kandarpa M, Kraftson S, Ross CW, Harvey C,
Hideshima T, et al: Perifosine plus lenalidomide and dexamethasone
in relapsed and relapsed/refractory multiple myeloma: A phase I
multiple myeloma research consortium study. Br J Haematol.
158:472–480. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gerrah R, Ehrlich S, Tshori S and Sahar G:
Beneficial effect of aspirin on renal function in patients with
renal insufficiency postcardiac surgery. J Cardiovasc Surg
(Torino). 45:545–550. 2004.PubMed/NCBI
|