Introduction
Cronkhite-Canada syndrome (CCS), also known as
polyposis pigmentation-alopecia-onycholrophia syndrome, is a
syndrome distinguished by gastrointestinal (GI) polyposis and
ectodermal changes (1). It has been
demonstrated that the incidence of CCS is 1 in a million (2). CCS affects more men compared with women,
with a ratio of 3:2, and commonly occurs in the fifth decade of
life, with a mean age of onset between 50–60 years (3). Cronkhite and Canada first described CCS
in 1955, and CCS is a rare disease of unknown etiology (4). Following its identification, >500
cases have been described in the literature (5). Although the incidence of CCS is low, it
is associated with a high mortality; 5-year mortality may be as
high as 55% (6). At present, the
treatments for CSS include corticosteroids, nonsteroidal
anti-inflammatory drugs, proton pump inhibitors, H2-receptor
antagonists, hyperalimentation, cromolyn sodium, antibiotics,
anabolic steroids, surgery, 5-aminosalicylate acid, antitumor
necrosis factor α agents and the eradication of Helicobacter
pylori, or a combination of these therapies (7).
In the present case study, a 58-years-old male with
CCS diagnosed at the Department of Gastroenterology, The Third
Xiangya Hospital of Central South University (Hunan, China) is
reported. The patient experienced a history of diarrhea and
hematochezia for 4 months, with abdominal pain for 1 month and
additional nail and toenail loss for half a month. The clinical,
endoscopic and histological data confirmed the diagnosis. The
patient was treated with proton pump inhibitors, gastric mucosal
protective agents, endoscopic electrocision of colon polyps,
glutamine capsule, nutrition support and Bifid triple viable
capsules. The patient eventually recovered.
Case report
A 58-year-old male visited The Third Xiangya
Hospital of Central South University (Hunan, Changsha, China) on
June 6, 2014, with the primary complaint of diarrhea and
hematochezia for 4 months, abdominal pain for 1 month and nail and
toenail loss for half a month. Informed written consent was
obtained from the patient for publication of the present study. The
patient additionally felt tiredness and had experienced a weight
loss of 5 kg in half a month. There were no abnormalities,
including GI polyposis or colorectal cancer, in the family history
of the individual. However, the patient had a history of alcoholic
cirrhosis for >10 years, and 13 years prior to visiting The
Third Xiangya Hospital of Central South University he had suffered
from gastrorrhagia. The patient had been drinking 400 ml rice wine
and smoking 20 cigarettes/day for 40 years.
Physical examination revealed that the patient was
suffering from malnutrition, he was emaciated and had an anemic
appearance, with a dermatological triad of hyperpigmentation in his
oral mucosa (Fig. 1) and a brown
pigmentation in his palms and feet (Fig.
2). Atrophy of fingernail and toenails were later observed, in
addition to eventual toenail and nail loss.
Laboratory tests revealed that the patient's white
blood cell count was 7.5×109/l (normal range,
4–10×109/l), platelet count was 216×109/l
(normal range, 100–300×109/l), hemoglobin was 61 g/l,
fecal occult blood test (+), C-reactive protein and erythrocyte
sedimentation rate were normal, serum albumin was 28.5 g/l (normal
range, 40–60 g/l) and serum total protein was 46.1 g/l (normal
range, 60–80 g/l).
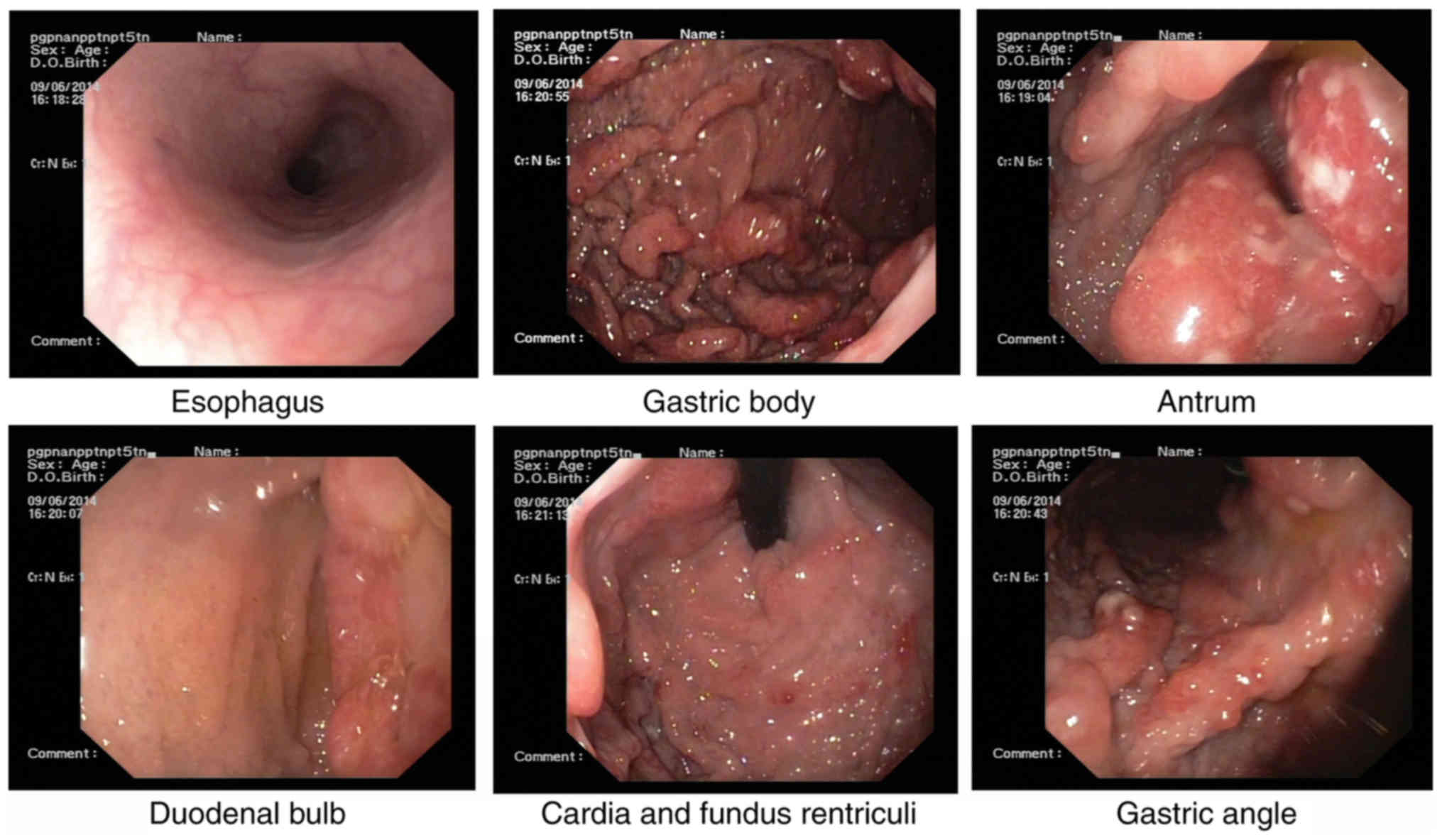
Esophagogastroduodenoscopy, performed for the
further evaluation of the GI tract, revealed multiple nodules and
granular polyps in the stomach and duodenum (Fig. 3).
In the present study, biopsy specimens were fixed in
4% paraformaldehyde at room temperature, dehydrated (75% ethanol
for 45 min, 85% ethanol for 45 min, 95% ethanol for 45 min, 100%
ethanol for 45 min, immersed in 100% isobutanol overnight and
lastly transferred to 100% butanol for 3 h) and embedded in
paraffin. Paraffin-embedded sections measuring 4 µm-thick were then
stained with 0.5% hematoxylin for 2 min and 1% eosin for 1 min at
room temperature (H&E). Finally, the sections were observed
under a light microscope (magnification, ×100; Olympus BX51;
Olympus Corporation, Tokyo, Japan,).
Biopsy specimens from the gastric antrum mucosa
displayed mucosal chronic inflammation, edematous stroma with
inflammatory cell infiltration, small blood vessel proliferation
and regional glandular epithelial hyperplasia (Fig. 4).
As these findings could not confirm the diagnosis, a
further colonoscopy was performed for differential diagnosis, which
revealed numerous, dense polyps throughout the terminal ileum,
colon and rectum (Figs. 5 and
6). Biopsy specimens from the colon
displayed colorectal villus-tubiform adenoma, glandular epithelial
hyperplasia and mild-to-moderate atypical hyperplasia (Fig. 7). GI radiography revealed that the GI
multiple filling defect may be a result of the multiple polyps
(Fig. 8).
The patient presented with diffuse GI polyposis
associated with ectodermal changes including hyperpigmentation and
onychatrophy, and these findings resulted in the diagnosis of CCS.
We intended to treat the patient with corticosteroids, but he
refused due to the potential side-effects. Therefore, the patient
was treated with proton pump inhibitors to inhibit gastric acid
secretion (40% pantoprazole sodium, 100 ml, was administered
intravenously, once a day), gastric mucosal protective agents to
protect gastric mucosa (Hydrotalcite Chewable Tablets, 1 g, orally,
three times a day), endoscopic electrocision of colon polyps,
glutamine to protect the intestinal intima (3% alanine glutamine,
50 ml, ivgtt, once a day) and nutritional support (7% amino acid
compound infusion18AA-II, 250 ml ivgtt, once a day). We also used
bifid triple viable capsules to modulate intestinal flora (Bifid
triple viable capsules, 420 mg, orally, three times a day). The
patient eventually recovered and was discharged from the hospital
within 1 month.
Following 1 month of treatment, gastroscopy revealed
gastric duodenal mucosal swelling and mucous with nodular and
polypoid hyperplasia (Fig. 9). Biopsy
specimens from the gastric antrum mucosal displayed mucosal chronic
inflammation, regional glandular epithelial hyperplasia and a
growth tendency of inflammatory hyperplastic polyps with
inflammatory cell infiltration (Fig.
10). Colonoscopy revealed terminal ileum, colon and rectal
hyperplastic polyps, with colon polyps emerging subsequent to
endoscopic mucosal resection and titanium clip clipping operation
(Figs. 11 and 12). Biopsy specimens from the colon
displayed a tubular adenoma 40 cm from the anus and ascending
colon. Additionally, part of the glandular epithelium had
light-mild atypical hyperplasia, a stroma of acidophilic ball
infiltration (in the terminal ileum and appendix) and mucosal
chronic inflammation (in the appendix) within the interstitial
eosinophilic inflammation (Fig. 13).
In order to exclude lymphoma, ultrasonic gastroscopy was advised,
and the patient was further advised to take corticosteroids, but he
refused and asked for discharging from the hospital following 1
month of treatment.
Discussion
Cronkhite and Canada first described CCS in 1955
(4). CCS is a rare, acquired,
nonhereditary syndrome with diffuse GI polyposis associated with
ectodermal changes, including hyperpigmentation, alopecia and
onychatrophy (8,9). Additionally, it has been reported that
the CCS has been associated with poor prognosis and
life-threatening malignant complications (7). The etiology of CCS remains unknown, and
genetic abnormalities (10), mental
stress (11), immune dysregulation
(12,13), low turnover cell differentiation
(14) and fatigue are regarded as
triggering factors for CCS (2).
CCS may occur in all ethnic groups, and the
estimated incidence of CCS is extremely rare, approximately one
case in a million individuals (2). At
present, >500 cases of CCS have been reported globally, and
patients from Europe and Asia are most frequently affected
(2). Of all reported cases of CCS,
75% have been from Japan (15).
Furthermore, it has been reported that CCS is
sporadic and there is no strong evidence to suggest a hereditary
predisposition (2). CCS affects more
men compared with women, with a ratio of 3:2, and commonly occurs
in the fifth decade of life. A total of 80% of patients are >50
years of age at the time of presentation (6). According to a previous study, it has
been revealed that the mean age of onset of CCS is 63.5 (range,
31–86) years (7).
CCS is also known as polyposis
pigmentation-alopecia-onycholrophia syndrome, and GI polyposis and
ectodermal changes are its two main features (16). For patients with CCS, diarrhea is the
most common initial symptom, which may develop to substantially
watery diarrhea, followed with symptoms of malabsorption, including
weakness, anemia, weight loss, edema and dysgeusia (17). Subsequent endoscopic and radiological
evaluation may reveal sessile polyps throughout the GI tract
(18).
Histopathological reviews of biopsies obtained from
these polyps revealed that these polyps are similar to that of
juvenile, adenomatous polyps or inflammatory type polyps, however
they were additionally marked by striking stromal and lamina
propria edematous changes, eosinophilic inflammation, were
cystically dilated and had distorted glands with inflammatory
infiltration (7).
Ectodermal changes include alopecia, nail dystrophy
and hyperpigmentation. These changes often occur later during the
disease progression, usually several weeks or months subsequent to
the GI symptoms (8). Consistent with
this, in the present case study, the patient initially experienced
diarrhea and hematochezia, followed by abdominal pain, nail and
toenail loss, onychatrophy and hyperpigmentation.
Complications of CCS include GI bleeding with
anemia, intussusception, hypoproteinemia, rectal prolapse,
malabsorption, electrolyte turbulences, enteropathy and
hypovitaminosis (19). In addition,
CCS was reported to be associated with various rare complications
including recurrent severe acute pancreatitis (20), GI tract cancer, portal thrombosis, a
high titer of antinuclear antibodies and membranous
glomerulonephritis (21). Among them,
the risk of GI tract tumor types substantially increases. It has
been reported that between 1980 and 2011, there were 383 patients
diagnosed with CCS in Japan, and of these patients, 10.4% (40
patients) of them were also diagnosed with gastric cancer and 69
lesions (51 patients) were also diagnosed with colon cancer
(15). Due to this, endoscopic
surveillance is strongly recommended.
Differential diagnosis of CCS includes Menetrier
disease, familial adenomatous polyposis, juvenile polyposis, Cowden
syndrome, Peutz-Jeghers syndrome, inflammatory bowel disease,
Whipple disease and small intestinal lymphoma (22–24). In
the present case, the patient was initially diagnosed with familial
polyposis, but eventually was diagnosed with CCS due to the
dermatological triad of hyperpigmentation in oral mucosa (Fig. 1), brown pigmentation in palms and feet
(Fig. 2) and toenail and nail
loss.
Due to the rarity of CCS, evidence-based therapies
have yet to be developed, however, to the best of our knowledge,
there are no systematic investigations of medical or surgical
interventions. The treatments and strategy of CSS currently include
corticosteroids, nonsteroidal anti-inflammatory drugs, proton pump
inhibitors, H2-receptor antagonists, hyperalimentation, cromolyn
sodium, antibiotics, anabolic steroids, surgery, 5-aminosalicylate
acid, antitumor necrosis factor α agents and the eradication of
Helicobacter pylori and combinations of these therapies
(7). Steroids are considered to be
the mainstay of medical treatment, however until now, there have
been no guidelines for the recommended dose and duration of their
use.
The prognosis of CCS is poor, with a 5-year
mortality rate of 55% and the majority of mortality being
associated with malnutrition, hypoalbuminemia, repetitive
infection, sepsis, heart failure and GI bleeding (8). The natural history of CCS appears to be
substantially improved owing to the sufficient dose and duration of
corticosteroid therapy accompanied by nutritional support and
periodic endoscopic surveillance (7).
Altogether, when a patient presents with the
symptoms of CCS, early diagnosis and treatment of CCS is necessary,
in addition to receiving endoscopic follow-up or polypectomy when
necessary. In the present case, the results from a telephone
follow-up implied that the patient is in a good condition; he feels
well and does not experience any symptoms, therefore has refused to
return to the hospital for a follow-up.
CCS is a rare but serious disease with an increased
mortality rate if clinical intervention is received late (25). Delays in diagnosis are common,
primarily due to the non-familiarity of physicians with this rare
entity, resulting in a poor outcome (26). This patient did not present with the
cardinal manifestations of CCS which resulted in a delayed
diagnosis. Therefore, in order to avoid the misdiagnosis of CCS
without typical features in future, physicians are recommended to
analyze the histopathology of the polyps and to search for the
presence of characteristic dermatological changes: The changes in
the shape and colour of toenail and nail abnormalities.
Acknowledgements
The present case study was supported by the National
Natural Science Foundation of China (grant nos. 81272736, 81670504
and 81472287), the National Key Clinical Specialty of National
Health and Family Planning Commission and The Third Xiangya
Hospital ‘Xiangya Doctors Heritage Plan’.
References
|
1
|
Sweetser S, Alexander GL and Boardman LA:
A case of Cronkhite-Canada syndrome presenting with adenomatous and
inflammatory colon polyps. Nat Rev Gastroenterol Hepatol.
7:460–464. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Goto A: Cronkhite-Canada syndrome:
Epidemiological study of 110 cases reported in Japan. Nihon Geka
Hokan. 64:3–14. 1995.PubMed/NCBI
|
|
3
|
Ward EM and Wolfsen HC: Review article:
The non-inherited gastrointestinal polyposis syndromes. Aliment
Pharmacol Ther. 16:333–342. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gardner EJ, Burt RW and Freston JW:
Gastrointestinal polyposis: Syndromes and genetic mechanisms. West
J Med. 132:488–499. 1980.PubMed/NCBI
|
|
5
|
Rubio CA and Björk J: Cronkhite-Canada
syndrome-A case report. Anticancer Res. 36:4215–4217.
2016.PubMed/NCBI
|
|
6
|
Daniel ES, Ludwig SL, Lewin KJ, Ruprecht
RM, Rajacich GM and Schwabe AD: The Cronkhite-Canada syndrome. An
analysis of clinical and pathologic features and therapy in 55
patients. Medicine. 61:293–309. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Watanabe C, Komoto S, Tomita K, Hokari R,
Tanaka M, Hirata I, Hibi T, Kaunitz JD and Miura S: Endoscopic and
clinical evaluation of treatment and prognosis of Cronkhite-Canada
syndrome: A Japanese nationwide survey. J Gastroenterol.
51:327–336. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yun SH, Cho JW, Kim JW, Kim JK, Park MS,
Lee NE, Lee JU and Lee YJ: Cronkhite-Canada syndrome associated
with serrated adenoma and malignant polyp: A case report and a
literature review of 13 cronkhite-Canada syndrome cases in Korea.
Clin Endosc. 463:301–305. 2013. View Article : Google Scholar
|
|
9
|
Wen XH, Wang L, Wang YX and Qian JM:
Cronkhite-Canada syndrome: Report of six cases and review of
literature. World J Gastroenterol. 20:7518–7522. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Patil V, Patil LS, Jakareddy R, Verma A
and Gupta AB: Cronkhite-Canada syndrome: A report of two familial
cases. Indian J Gastroenterol. 32:119–122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murata I, Yoshikawa I, Endo M, Tai M,
Toyoda C, Abe S, Hirano Y and Otsuki M: Cronkhite-Canada syndrome:
Report of two cases. J Gastroenterol. 35:706–711. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sweetser S, Ahlquist DA, Osborn NK,
Sanderson SO, Smyrk TC, Chari ST and Boardman LA: Clinicopathologic
features and treatment outcomes in Cronkhite-Canada syndrome:
Support for autoimmunity. Dig Dis Sci. 57:496–502. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Riegert-Johnson DL, Osborn N, Smyrk T and
Boardman LA: Cronkhite-Canada syndrome hamartomatous polyps are
infiltrated with IgG4 plasma cells. Digestion. 75:96–97. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Freeman K, Anthony PP, Miller DS and Warin
AP: Cronkhite Canada syndrome: A new hypothesis. Gut. 26:531–536.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matsui S, Kibi M, Anami E, Anami T,
Inagaki Y, Kanouda A, Yoshinaga H, Watanabe A, Sugahara A, Mukai H,
et al: A case of Cronkhite-Canada syndrome with multiple colon
adenomas and early colon cancers. Nihon Shokakibyo Gakkai Zasshi.
108:778–786. 2011.(In Japanese). PubMed/NCBI
|
|
16
|
Wang J, Zhao L, Ma N, Che J, Li H and Cao
B: Cronkhite-Canada syndrome associated with colon cancer
metastatic to liver: A case report. Medicine (Baltimore).
968:e74662017. View Article : Google Scholar
|
|
17
|
Chakrabarti S: Cronkhite-Canada syndrome
(CCS)-A rare case report. J Clin Diagn Res. 9:OD08–OD09.
2015.PubMed/NCBI
|
|
18
|
Goto A, Mimoto H, Shibuya C and Matsunami
E: Cronkhite-Canada syndrome: An analysis of clinical features and
follow-up studies of 80 cases reported in Japan. Nihon Geka Hokan.
57:506–526. 1988.PubMed/NCBI
|
|
19
|
Seshadri D, Karagiorgos N and Hyser MJ: A
case of cronkhite-Canada syndrome and a review of gastrointestinal
polyposis syndromes. Gastroenterol Hepatol (NY). 8:197–201.
2012.
|
|
20
|
Yasuda T, Ueda T, Matsumoto I, Shirasaka
D, Nakajima T, Sawa H, Shinzeki M, Kim Y, Fujino Y and Kuroda Y:
Cronkhite-Canada syndrome presenting as recurrent severe acute
pancreatitis. Gastrointest Endosc. 67:570–572. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takeuchi Y, Yoshikawa M, Tsukamoto N,
Shiroi A, Hoshida Y, Enomoto Y, Kimura T, Yamamoto K, Shiiki H,
Kikuchi E and Fukui H: Cronkhite-Canada syndrome with colon cancer,
portal thrombosis, high titer of antinuclear antibodies, and
membranous glomerulonephritis. J Gastroenterol. 38:791–795. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sweetser S and Boardman LA:
Cronkhite-Canada syndrome: An acquired condition of
gastrointestinal polyposis and dermatologic abnormalities.
Gastroenterol Hepatol (NY). 8:201–203. 2012.
|
|
23
|
Samet JD, Horton KM, Fishman EK and
Iacobuzio-Donahue CA: Cronkhite-Canada syndrome: Gastric
involvement diagnosed by MDCT. Case Rep Med. 2009:1487952009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kopáčová M, Urban O, Cyrany J, Laco J,
Bureš J, Rejchrt S, Bártová J and Tachecí I: Cronkhite-Canada
syndrome: Review of the literature. Gastroenterol Res Pract.
2013:8568732013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Iqbal U, Chaudhary A, Karim MA, Anwar H
and Merrell N: Cronkhite-Canada syndrome: A rare cause of chronic
diarrhea. Gastroenterology Res. 10:196–198. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fan RY, Wang XW, Xue LJ, An R and Sheng
JQ: Cronkhite-Canada syndrome polyps infiltrated with IgG4-positive
plasma cells. World J Clin Cases. 4:248–252. 2016. View Article : Google Scholar : PubMed/NCBI
|