Introduction
Triple-negative breast cancer (TNBC) accounts for
10–15% of all breast cancer cases (1). These tumors can currently only be
treated by conventional chemotherapy as they lack expression of
estrogen receptor α (ERα) and progesterone receptors and cannot be
treated with HER2 antibody trastuzumab or other HER2 directed
therapies. To date, no successful targeted therapy is available for
TNBC patients (2). Consequently,
the mortality rate of TNBC patients is still twice as high as for
patients having ERα-positive tumors (3). A high proportion (30–52%) of TNBCs
overexpress epidermal growth factor receptor (EGFR) and this high
expression of EGFR is associated with poor prognosis (4). A number of tyrosine kinase inhibitors
(TKIs) preferentially acting on EGFR have been developed in the
last decades. Gefitinib (Iressa®) and erlotinib are
compounds possessing a high affinity to the hydrophobic ATP-binding
pocket of the tyrosine kinase domain of EGFR (5). Gefitinib (ZD1839 or
Iressa®) was found to inhibit proliferation of
EGFR-overexpressing cancer cell lines by more than 50%, including
the TNBC cell line MDA-MB-231 (6).
In a phase II multicenter trial with advanced breast
cancer patients, treatment with 500 mg gefitinib per day achieved
stable disease for 6 months in 38.7% of patients.
Immunohistochemical staining of biopsies of treated patients
demonstrated a complete inhibition of EGFR phosphorylation
(7). Particularly, breast tumors
that developed resistance to tamoxifen benefited from gefitinib
treatment showing stable disease for 24 weeks in 54% of patients.
In the same study, only 11.5% of ER-negative patients showed stable
disease under gefitinib treatment (8).
GPER, also known as GPR30, is a G protein-coupled
receptor responsible for nongenomic actions of 17β-estradiol
(9,10). Binding of 17β-estradiol to GPER
leads to a dissociation of the heterotrimeric G-protein complex.
The βγ-subunit activates the tyrosine kinase Src (11). Subsequently, matrix metalloproteases
release EGF from the extracellular matrix that initiates
autophosphorylation of the EGFR finally leading to the activation
of the ras-MAP-kinase pathway (9).
In addition, it has been shown that stimulation of
GPER by the synthetic agonist G1 suppressed EMT in the TNBC cell
line MDA-MB-231 by downregulation of NF-κB signaling (12). High GPER expression in the breast is
one reason for acquired tamoxifen resistance of breast tumors as
tamoxifen has been shown to be an agonist of GPER (13).
In TNBC, GPER is also frequently expressed very
strongly and high GPER expression in this subgroup of breast cancer
was found to correlate with increased recurrence (14). Recently, we provided evidence, that
inhibition of the transcription of GPR30 by siRNA is able to
prevent 17β-estradiol-dependent growth stimulation of TNBC
(15). As siRNA is not applicable
in patients, other approaches to lower GPER are necessary. Vivacqua
et al reported that in breast cancer cells high expression
of GPER correlates with overexpression of the EGFR (16). On the other hand, EGFR is
overexpressed in approximately 50% of TNBCs and high EGFR
expression is a predictor of a poor prognosis of these breast
cancer patients (17).
In the present study, we therefore analyzed the
impact of gefitinib on GPER expression and on
17β-estradiol-dependent growth stimulation in TNBC cells.
Materials and methods
Reagents
Gefitinib (Iressa®) was purchased from
Selleck Chemicals (Houston, TX, USA). 17β-estradiol (E2), insulin
and transferrin were purchased from Sigma-Aldrich (Deisendorf,
Germany).
Cell lines
TNBC cell lines HCC70 and HCC1806 both of the
basal-like subtype were purchased from the American Type Culture
Collection (ATCC; Manassas, VA, USA) and maintained in Dulbecco's
modified Eagle's medium (DMEM) (Biochrom GmbH, Berlin, Germany)
supplemented with 2 mM glutamine, 6 ng/ml insulin, 10 ng/ml
transferrin, penicillin (50 U/ml), streptomycin (50 µg/ml) from
Gibco (Paisley, UK) and 10% fetal bovine serum (Biochrom GmbH).
Proliferation assays
Proliferation assays were performed as previously
described (18). Charcoal depleted
serum (CD-FCS) was prepared according to the procedure described by
Stanley et al (19). Cell
number was determined by a colorimetric method using Alamar Blue
(Biosource, Solingen, Germany).
Proliferation assays were performed at least three
times in quadruplicates with different passages. Means and standard
deviations of the optical density (OD) of the replicates were
calculated.
Treatment of cells
Four million cells of either cell line (HCC70 and
HCC1806) were treated for 48 h with 200 nM gefitinib. Twenty-four
hours before harvest, the cells were starved from serum. Finally,
the cells were stimulated for 15 min with 10−8 M
17β-estradiol. Cells were detached with 1 mM EDTA/PBS and the
pellets were lysed in 50 µl CelLytic M containing protease and
phosphatase inhibitors (Sigma-Aldrich).
Western blotting
Lysates of cells were cleared and protein was
determined using the method of Bradford. Twenty micrograms of each
sample were separated in a 7.5% polyacrylamide gel, blotted on a
PVDF membrane and detected with rabbit anti-human primary
antibodies: anti-GPR30 (sc-48524; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), anti-phospho-Src (2113) and anti-Src (2109) from
Cell Signaling Technology, Inc. (Danvers, MA, USA),
anti-phospho-Tyr1173 EGFR (324864) from Calbiochem
(Darmstadt, Germany), anti-EGFR antibody (2235) supplied by
Epitomics (Hamburg, Germany) and anti-actin by Sigma-Aldrich. Blots
were washed four times in TBST and further incubated with a
1:20,000 dilution of horseradish peroxidase conjugated goat
anti-rabbit antibody (ECL; GE Healthcare, Freiburg, Germany). After
washing again for four times, the blots were incubated with
chemoluminescence reagent Femto (Thermo Fisher Scientific,
Rockford, IL, USA) for 5 min and emitted light was detected on a
LI-COR chemoluminescence detector (LI-COR Biosciences, Lincoln, NE,
USA). Densitometric evaluation of the protein bands was performed
with Image Studio Digits program from LI-COR and expression values
of the proteins were normalized to actin.
RT-PCR
RNA of the variously treated TNBC cells was purified
using the RNeasy kit (Qiagen, Hilden, Germany). Reverse
transcription-polymerase chain reaction of c-fos, cyclin D1 and
aromatase was performed as previously described (15).
Agarose gels of PCR products were stained in
ethidium bromide (2 µg/ml) for 30 min and photographed on a
transiluminator using a CDS camera (Biometra, Göttingen, Germany).
The band intensities of the PCR products were evaluated by the
Digital Science 1D software (Eastman Kodak, Rochester, NY, USA).
Values of the RT-PCR products were normalized to the ribosomal
protein L7.
Statistical analysis
The data were tested for significant differences by
one-way analysis of variance followed by Student-Newman-Keuls test
for comparison of individual groups, after a Bartlett's test had
shown that variances were homogenous.
Results
Expression of EGFR and GPER in TNBC
cell lines
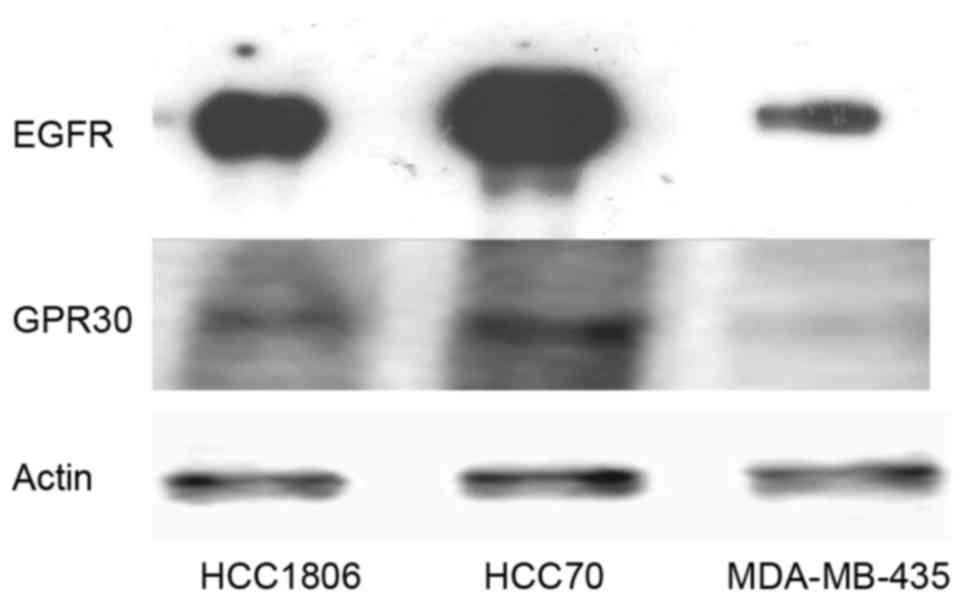
In order to verify the assumption that expression of
GPER parallels the expression of EGFR, 20 µg protein of the TNBC
cell lines HCC1806, HCC70 and MDA-MB-435 were analyzed on western
blotting for expression of EGFR and GPER (Fig. 1). MDA-MB-435 cells, not amplified
for the EGFR gene, contained the lowest amount of EGFR protein.
HCC1806 cells expressed 15±3.2-fold the amount of EGFR expressed in
the MDA-MB-435 cells. The signal of EGFR was strongest in the HCC70
cells, expressing 48±5.8-fold more EGFR than that noted in the
MDA-MB-435 cells.
The analysis of GPER expression in TNBC cell lines
confirmed the observation of Vivacqua et al that GPER
expression correlates with the amount of EGFR pointing to a direct
regulation of GPER by EGF (16). In
the cell lines tested, GPER expression was lowest in the MDA-MB-435
cells similar to EGFR and GPER expression was higher in cell lines
expressing more EGFR. In the HCC1806 cells, GPER expression was
3.8±0.9-fold the amount detected in the MDA-MB-435 cells and HCC70
cells expressed 8.4±1.4-fold as much GPER as cells of the cell line
MDA-MB-435.
Reduction in GPER expression following
treatment with gefitinib
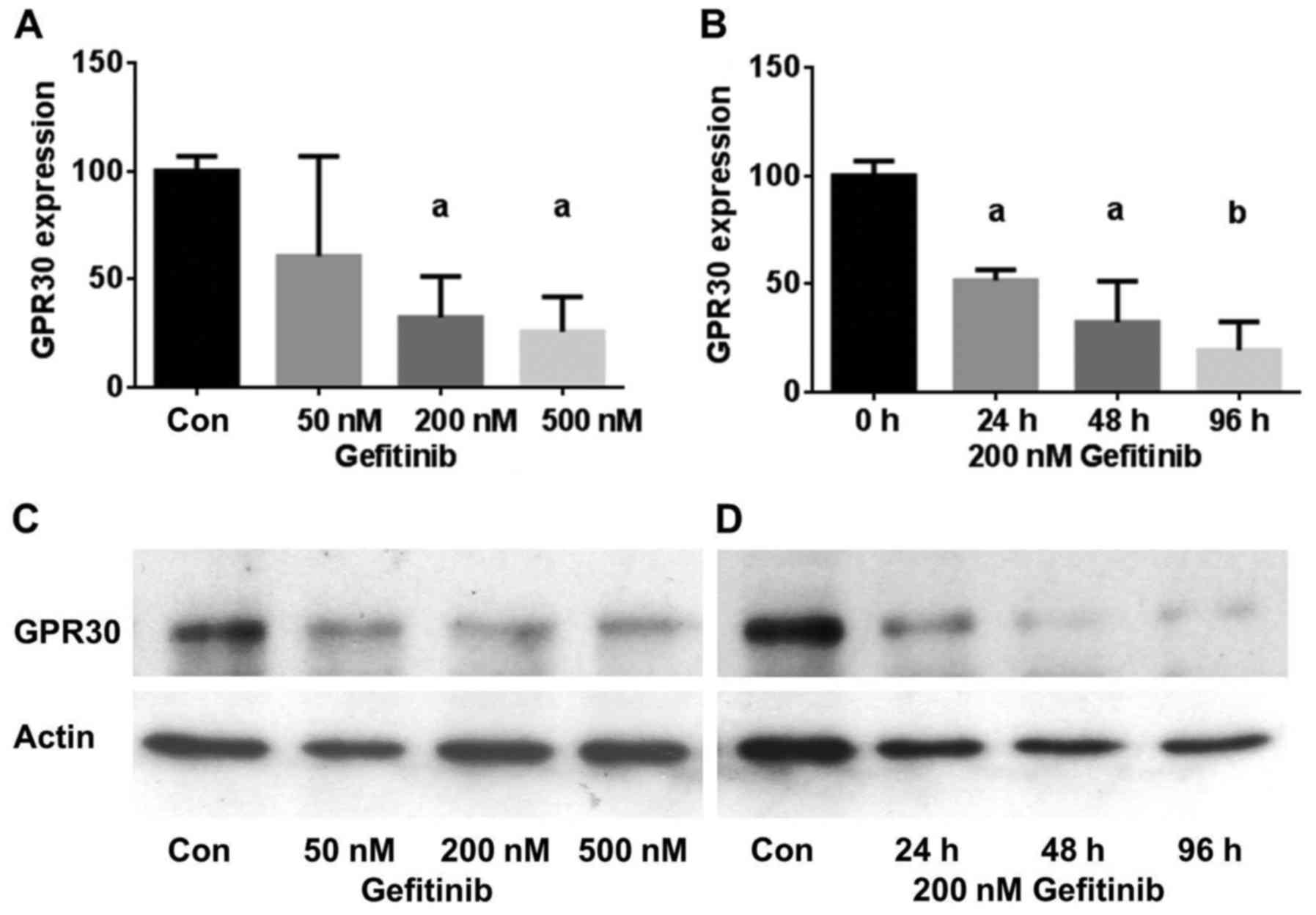
To test the hypothesis that inhibition of EGFR
reduces expression of GPER, the TNBC cell lines were treated with
increasing concentrations of gefitinib for up to 4 days and GPER
expression was analyzed by western blotting of the lysates of the
treated cells (Fig. 2). A 2-day
treatment of HCC1806 cells with 50 nM gefitinib reduced GPER
expression to 60.5±46% of the control (NS). Using 200 nM gefitinib,
GPER expression was reduced to 32±18% (p<0.05) and 500 nM
gefitinib lowered GPER expression in the HCC1806 cells further to
26±16% (p<0.05) (Fig. 2A). The
inhibition of GPER expression was also found to be more effective
with increasing time of exposure to gefitinib. In the HCC1806 cells
treated with 200 nM gefitinib for 24 h, GPER expression decreased
to 52±5% (p<0.05) and after 96 h the GPER level reached 39±5%
(p<0.01) of the control (Fig.
2B). Fig. 2C shows a
representative western blot of the concentration-dependent
reduction in GPER expression by gefitinib in the HCC1806 cells.
Similar results were obtained with the TNBC cell line HCC70. In
Fig. 2D, a representative western
blot of the time-dependent decrease in GPER expression after
treatment with 200 nM gefitinib for 24–96 h is presented.
Treatment of HCC70 cells with 200 nM gefitinib led
to an almost maximal reduction in GPER expression after 24 h to
25±7% (p<0.01) of the control and GPER expression further
decreased only slightly after a 96-h treatment with 200 nM
gefitinib (15±11%) (p<0.01) (Table
I).
 | Table I.Quantitative evaluation of GPER
expression after treatment of TNBC cells with various
concentrations of gefitinib for 24 to 96 h. |
Table I.
Quantitative evaluation of GPER
expression after treatment of TNBC cells with various
concentrations of gefitinib for 24 to 96 h.
|
| Treatment of HCC1806
cells |
|---|
|
|
|
|---|
| Concentrations | 50 nM | 200 nM | 500 nM |
|---|
| Duration |
|
|
|
| 24 h | 64±16%
(p<0.01) | 52±5%
(p<0.001) | 32±6%
(p<0.001) |
| 48 h | 60±46% (NS) | 32±18%
(p<0.05) | 26±16%
(p<0.05) |
| 96 h | 49±32%
(p<0.05) | 39±5%
(p<0.01) | 26±18%
(p<0.01) |
|
|
| Treatment of
HCC1806 cells |
|
|
|
| Concentrations | 50 nM | 200 nM | 500 nM |
|
| Duration |
|
|
|
| 24 h | 28±8%
(p<0.001) | 25±7%
(p<0.001) | 25±10%
(p<0.001) |
| 48 h | 31±18%
(p<0.01) | 29±6%
(p<0.01) | 21±8%
(p<0.01) |
| 96 h | 35±19%
(p<0.01) | 15±11%
(p<0.01) | 28±19%
(p<0.01) |
Reduction of GPER expression by
gefitinib prevents growth stimulation of TNBC by 17β-estradiol
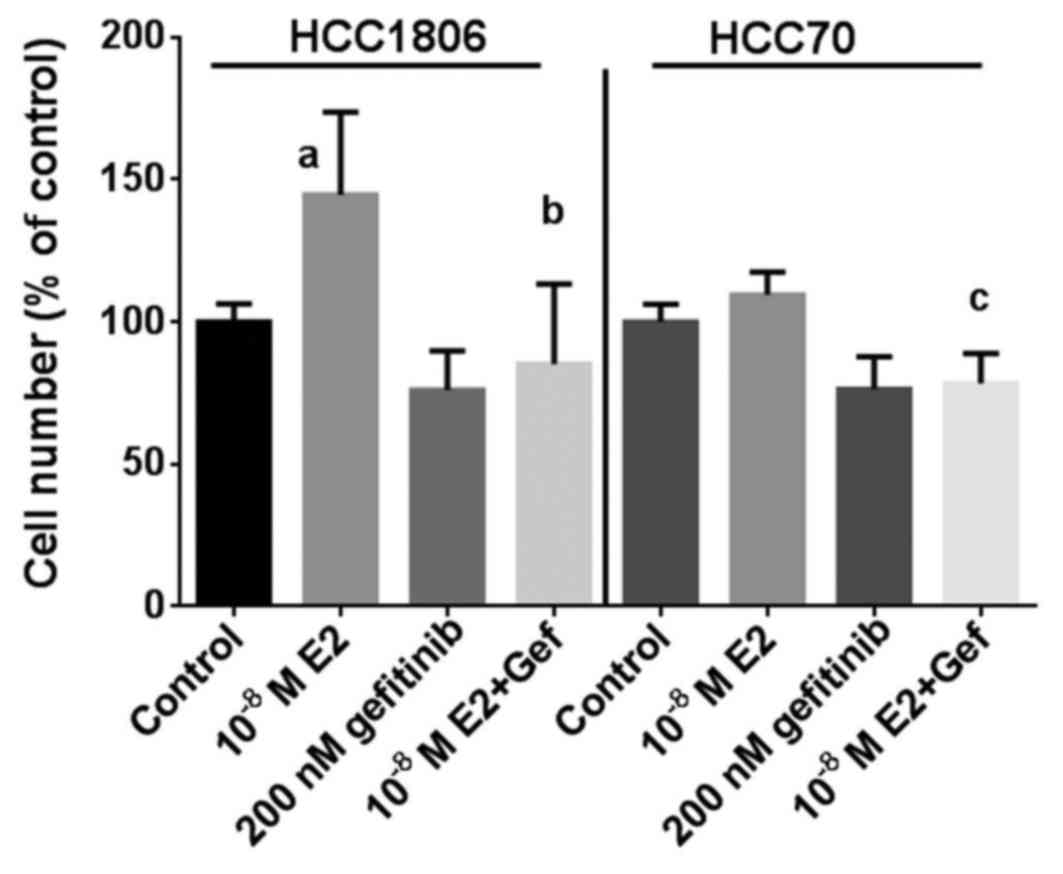
As additional proof that the inhibitory effect of
gefitinib on the growth of TNBC depends in part on a reduction in
GPER expression, the effect of 17β-estradiol on the proliferation
of the TNBC cell lines was analyzed at a concentration of
10−8 M in the absence or in the presence of 200 nM
gefitinib (Fig. 3). Within 7 days
of culture, the percentage of HCC1806 cells increased to 145±29%
(p<0.01) in the presence of 17β-estradiol compared to the
controls. Treatment of HCC1806 cells with 200 nM gefitinib alone
reduced the percentage of cells to 76±14% of the control. When
HCC1806 cells treated with gefitinib were stimulated by
17β-estradiol, the percentage of cells only slightly increase to
85±28% and remained below the control level (Fig. 3, left). The induction of growth of
HCC70 cells by 10−8 M 17β-estradiol was less pronounced
with an increase of 110±8% of the control; following treatment with
200 nM gefitinib the percentage of HCC70 cells was 76±11% of the
control. In the HCC70 cells co-treated with gefitinib stimulation
with 17β-estradiol failed to increase the cell percentage after 7
days of culture (78±11%) (Fig. 3,
right).
Gefitinib inhibits GPER-dependent
phosphorylation of c-src and subsequent activation of EGFR
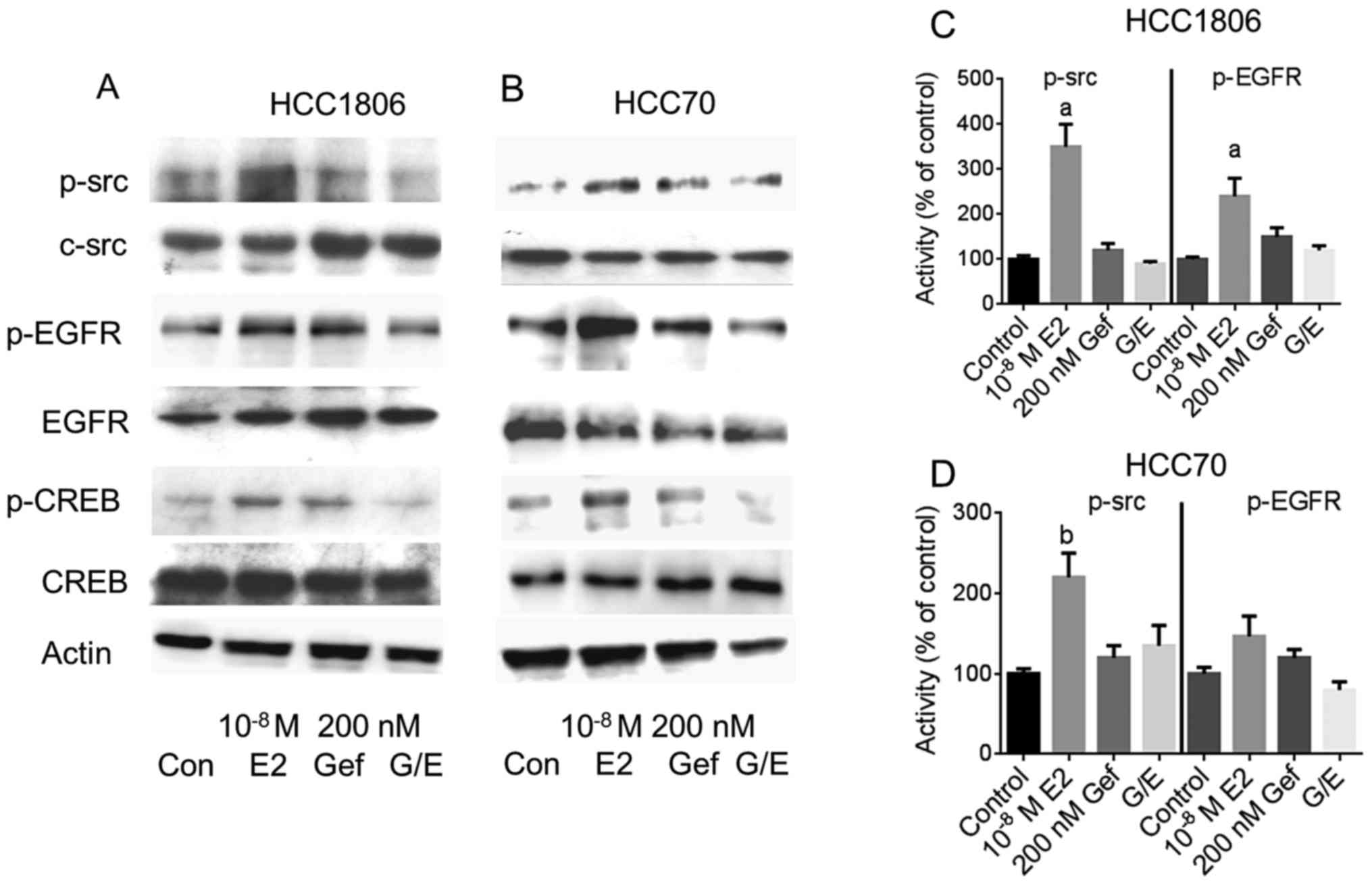
Next, the impact of the reduction in GPER expression
by gefitinib was analyzed on the indirect activation of c-src and
EGFR by 10−8 M 17β-estradiol. The two TNBC cell lines,
either pretreated with 200 nM gefitinib for 4 days or not, were
stimulated with 17β-estradiol and phosphorylation of c-src and EGFR
was analyzed by western blotting (Fig.
4A and B).
Even in the serum-starved HCC1806 and HCC70 cells a
basal phosphorylation of c-src at Tyr416 was detectable
(lane 1). In both cell lines, phosphorylation of c-src was strongly
increased after stimulation with 10−8 M 17β-estradiol
(Fig. 4A and B, lane 2).
Densitometric analysis of the p-src bands revealed that the amount
of p-src was increased in the HCC1806 cells to 350±50% of the
control (p<0.01) (Fig. 4C) and
in HCC70 cells to 220±30% (p<0.05) (Fig. 4D). Pretreatment of the cells for 96
h with 200 nM gefitinib did not change c-src phosphorylation
(Fig. 4A and B, lane 3). In both
TNBC cell lines pretreatment with 200 nM gefitinib prevented
activation of c-src by 10−8 M 17β-estradiol. Whereas in
the HCC1806 cells, phosphorylation of c-src by estradiol was
diminished by gefitinib from 350 to 90% of the control (Fig. 4C). In the HCC70 cells, activation of
c-src was reduced from 220 to 135% of the control, when pretreated
for 4 days with 200 nM gefitinib (Fig.
4D).
In the signal transduction of GPER, phosphorylated
src activates matrix metalloproteases that release heparin-bound
EGF from the extracellular matrix. After binding of EGF to its
cognate receptor, EGFR is autophosphorylated at several sites of
the cytosolic domain. The increased phosphorylation of the EGFR at
Tyr1173 after stimulation of the HCC1806 and HCC70 cells
with 10−8 M 17β-estradiol is shown in Fig. 4A and B, lane 2. In HCC1806 cells,
Tyr1173 phosphorylation of EGFR increased to 240±40%
(p<0.01) of the non-stimulated control (Fig. 4C) and in HCC70 cells to 147±25%
(Fig. 4D).
In both cell lines induction of Tyr1173
phosphorylation by 17β-estradiol was almost completely prevented in
the cells pretreated with 200 nM gefitinib: pEGFR in HCC1806,
120±10% of the control; in HCC70 cells, 80±10% of the control
(Fig. 4C and D).
Pretreatment of TNBC with gefitinib
reduces activation of CRE by 17β-estradiol
In addition to the Gβγ-dependent activation of the
GPER signaling pathway described above, stimulation of GPER by
17β-estradiol also results in the release of Gα from the
heterotrimeric G-protein complex that activates adenylyl cyclase.
As a consequence, activity of protein kinase A (PKA) is increased
by cAMP leading to the phosphorylation of the cAMP-responsive
element binding (CREB) protein.
Phosphorylation of CREB was analyzed by western
blotting from cell lysates from the TNBC cell lines (Fig. 4, panels 5 and 6). Stimulation of
HCC1806 cells with 10−8 M 17β-estradiol increased
phosphorylation of CREB to 144±8% (lane 2) compared to
non-stimulated control cells (lane 1). In HCC70 cells, CREB
phosphorylation increased to 169±60% of the control due to
stimulation with 17β-estradiol. When EGFR was inhibited by 200 nM
gefitinib prior to stimulation of HCC1806 and HCC70 cells with
10−8 M 17β-estradiol, activation of CREB was completely
prevented: pCREB in HCC1806 cells, 95±21%; HCC70 cells, 82±17%
(Fig. 4, lane 4).
Previously, we showed by electrophoretic mobility
shift that after stimulation of TNBC cell lines HCC1806 and HCC70
with 17β-estradiol, phospho-CREB binds to the promoter of cyclin D1
(15).
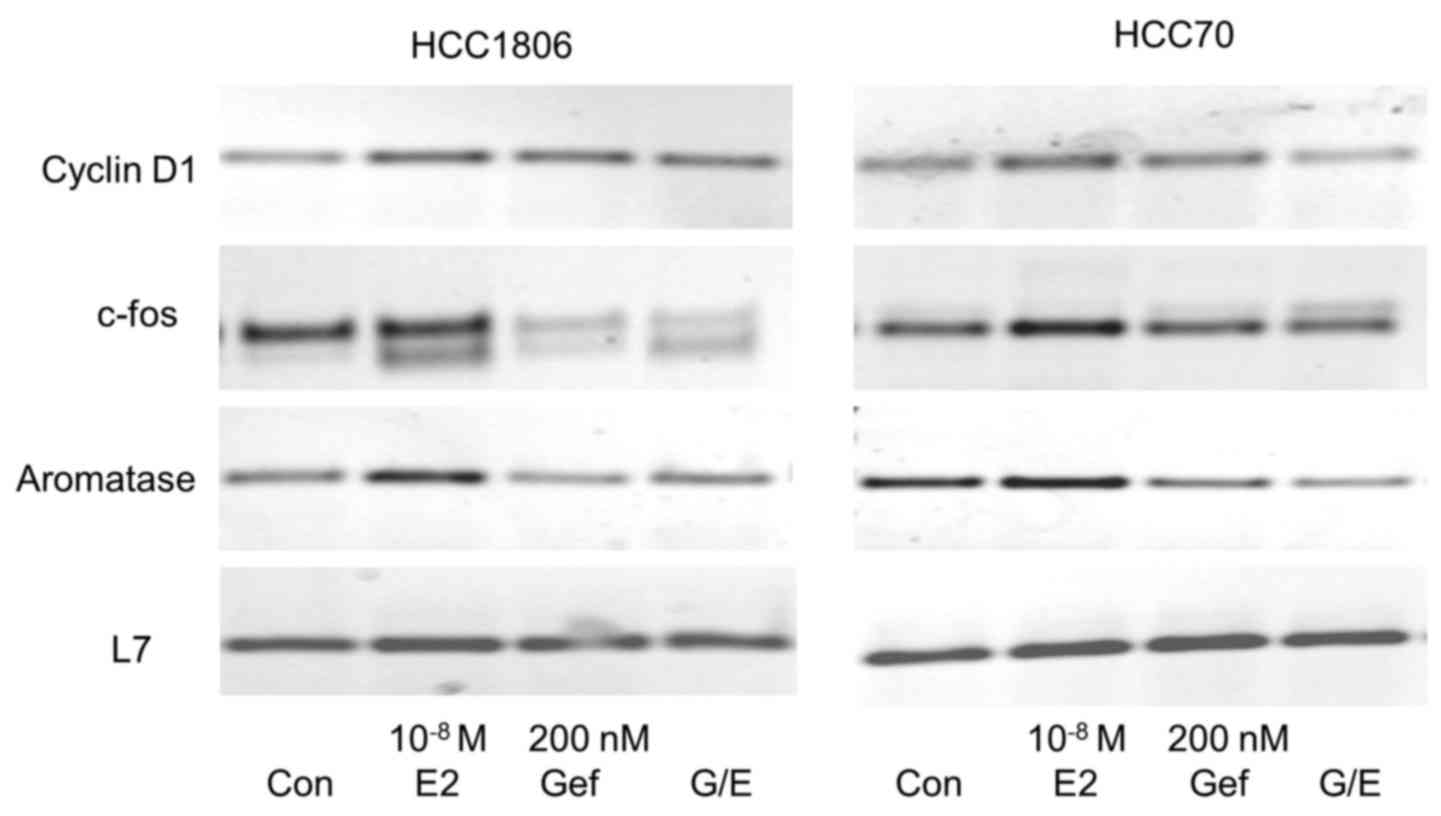
Induction of cyclin D1 expression by
17β-estradiol is inhibited by gefitinib
Cyclin D1 is an important regulator of the
transition from G1 to S phase of the cell cycle necessary for the
induction of proliferation. Expression of cyclin D1 was analyzed
after stimulation of both TNBC cell lines with 10−8 M
17β-estradiol for 30 min (Fig. 5,
first panel).
In HCC1806 cells we observed a distinct expression
of cyclin D1 mRNA in the serum-starved control cells. This high
basal cyclin D1 expression increased to 125±16% after a 30-min
stimulation with 10−8 M 17β-estradiol (lane 2). In
HCC1806 cells pretreated for 96 h with 200 nM gefitinib, cyclin D1
expression was lower than in the control cells (lane 3). In the
gefintinib-treated HCC1806 cells stimulation with 17β-estradiol was
not able to increase cyclin D1 expression (96±12%).
In the non-stimulated HCC70 cells, cyclin D1 was
more strongly expressed than in the HCC1806 cells. A 30-min
stimulation of these cells with 10−8 M 17β-estradiol
elevated the cyclin D1 mRNA content to 129±13% of the control
(Fig. 5, lane 2). Pretreatment of
HCC70 cells with 200 nM gefitinib completely prevented induction of
cyclin D1 expression by 17β-estradiol (82±12%) (Fig. 5, lane 4).
Gefitinib prevents induction of c-fos
expression by 17β-estradiol
Following activation of EGFR, the growth promoting
signal is forwarded via MAP kinase Erk to the nucleus where
subsequent expression of c-fos is induced. Serum-starved cells of
the two different TNBC cell lines (HCC1806 and HCC70) were
stimulated for 30 min with 10−8 M 17β-estradiol. mRNA of
these cells was analyzed for c-fos expression by RT-PCR and
compared to the expression in non-stimulated cells. Stimulation of
HCC1806 cells with 17β-estradiol increased c-fos expression to
155±35% (p<0.05) of the control (Fig. 5, second panel, lane 2). In the HCC70
cells, induction of c-fos by estradiol was less pronounced reaching
only 140±10% compared to the serum-starved control. In HCC1806
cells, wherein expression of GPER was reduced after treatment with
200 nM gefitinib for 96 h, c-fos expression was 45% lower than that
in the control cells (Fig. 5, lane
3) and induction of c-fos expression by 17β-estradiol was
completely prevented (48±9%) (Fig.
5, lane 4). In HCC70 cells pretreated with gefitinib a slight
increase in c-fos expression was still observed but only by the
factor 1.1±0.2.
Aromatase expression in TNBC
cells
Aromatase (CYP19A1) is an enzyme of the cytochrome
450 family catalyzing the formation of the non-saturated A-ring of
estradiol. The promoter of the aromatase gene contains a
cAMP-responsive element (20). For
this reason we expected that aromatase is regulated by GPER.
Stimulation of TNBC cell line HCC1806 cells with 17β-estradiol led
to an increase in mRNA for aromatase to 128±25% (Fig. 5, third panel, lane 2). The reduction
in GPER expression by pretreatment of HCC1806 cells with 200 nM
gefitinib for 96 h prevented the induction of aromatase expression
by 17β-estradiol completely (96±5%) (Fig. 5, lane 4). In HCC70 cells
17β-estradiol increased aromatase expression to 137±10% but in
cells pretreated with gefitinib mRNA of aromatase was less abundant
than that in the control cells (65±18%).
Discussion
Patients with TNBC have very poor prognosis due to
the lack of expression of ERα, progesterone receptor and
HER2-amplification in their tumors. EGFR is overexpressed in about
50% of TNBC and high EGFR expression predicts the poor prognosis of
these breast cancer patients (17).
Although TNBC cells per definition do not express
the nuclear estrogen receptor ERα, we observed a growth stimulation
of 45% in the TNBC cell line HCC1806 by 17β-estradiol (Fig. 3). This enhanced growth is caused by
GPER, the estrogen receptor responsible for the non-genomic effects
of 17β-estradiol. Previously, we showed that knockdown of GPER
using specific siRNA prevented this growth stimulation by
17β-estradiol in TNBC cell lines (15). In search of more physiological ways
to downregulate GPER expression, we found in the literature the
indication that GPER expression parallels the expression of EGFR
(16). In TNBC, strong expression
of GPER is also prevalent in addition to overexpression of EGFR and
strong GPER expression in this cohort was associated with young age
and poor outcome (14).
There are small molecular compounds, such as
gefitinib or erlotinib, binding to the kinase domain of the EGFR
and successfully inhibiting phosphorylation of the cytosolic domain
of EGFR and downregulating EGFR signaling (5).
Here, we proved the interrelation between GPER
expression and EGFR activity, as inhibition of EGFR using gefitinib
dramatically reduced GPER expression for example by up to 85% in
the HCC70 cells after 96 h (Table
I).
In fact, this downregulation of GPER expression by
gefitinib reduced the induction of cell growth of HCC1806 and HCC70
cells by 10−8 M 17β-estradiol below the level of
non-stimulated cells (85±28 and 76±11%, respectively).
Clinical trials treating an unselected population of
breast cancer patients with gefitinib as a single agent were not
successful. Nonetheless, immunohistochemical analysis of biopsies
of treated tumors revealed complete inhibition of EGFR
phosphorylation (7). In a further
study, the treatment of 58 heavily pretreated patients with
metastatic breast cancer using 500 mg gefitinib per day resulted in
partial tumor response in one patient (21).
In one clinical trial particularly targeting EGFR in
TNBC patients with gefitinib, stable disease for 24 weeks was
achieved at least in two of 25 patients (22). There are probably alternative
mechanisms of pathway activation circumventing EGFR activation
(23).
In a phase II trial of 181 women with ER-negative
breast cancer, pathologic complete response (pCR) to gefitinib was
achieved in 17% of TNBC patients, whereas in only 2% of non-TNBC
patients pCR was observed after gefitinib treatment for 12 weeks
(24).
In the present study, we investigated a new strategy
to target GPER in TNBC. We used 200 nM gefitinib
(Iressa®) as a selective reversible inhibitor of EGFR
tyrosine kinase to downregulate GPER expression. Clinical trials
for breast cancer have been performed with a daily dosage of 500 mg
gefitinib resulting in an estimated serum concentration of
approximately 100 µM (7).
In order to confirm the specificity of the
downregulation of GPER expression by gefitinib, we analyzed the
downstream signaling of GPER after downregulation of its expression
by gefitinib. As described by Filardo et al the tyrosine
kinase Src is activated by the βγ-subunit of heterotrimeric
G-proteins after binding of 17β-estradiol to GPER and EGF released
from the extracellular matrix by matrix metalloproteases induces
autophosphorylation of the EGFR (9).
As shown in Fig. 4,
downregulation of GPER expression completely prevented activation
of the non-receptor tyrosine kinase c-src and of EGFR activation by
17β-estradiol. Attempts to pharmacologically inhibit GPER in TNBC
cell lines using estriol or the GPER-specific inhibitor G15 also
proved promising but the concentrations of these inhibitors
necessary to achieve a sufficient reduction of
17β-estradiol-induced cell proliferation were not achievable in
vivo (25). This fact rules out
an application of these compounds in clinical trials.
In this study, we showed that the proportion of
growth of TNBC cells that was induced by 17β-estradiol was
successfully inhibited by gefitinib via reduction of GPER
expression. The use of gefitinib may be a therapeutic option
particularly in TNBCs expressing high amounts of GPER. We expect
from our results that in TNBC patients selected for high expression
of GPER, gefitinib will prove to be more effective than in an
unselected population of breast cancer patients.
Acknowledgements
We thank Sonja Blume for the excellent technical
assistance. This study was supported by grant GR 1895/10-1 of the
German Research Foundation.
References
|
1
|
Petrelli F, Cabiddu M, Ghilardi M and
Barni S: Current data of targeted therapies for the treatment of
triple-negative advanced breast cancer: Empiricism or
evidence-based? Expert Opin Investig Drugs. 18:1467–1477. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Podo F, Buydens LM, Degani H, Hilhorst R,
Klipp E, Gribbestad IS, Van Huffel S, van Laarhoven HW, Luts J,
Monleon D, et al: FEMME Consortium: Triple-negative breast cancer:
Present challenges and new perspectives. Mol Oncol. 4:209–229.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carey LA, Dees EC, Sawyer L, Gatti L,
Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML and Perou
CM: The triple negative paradox: Primary tumor chemosensitivity of
breast cancer subtypes. Clin Cancer Res. 13:2329–2334. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reis-Filho JS and Tutt AN: Triple negative
tumours: A critical review. Histopathology. 52:108–118. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wakeling AE, Guy SP, Woodburn JR, Ashton
SE, Curry BJ, Barker AJ and Gibson KH: ZD1839 (Iressa): An orally
active inhibitor of epidermal growth factor signaling with
potential for cancer therapy. Cancer Res. 62:5749–5754.
2002.PubMed/NCBI
|
|
6
|
Anderson NG, Ahmad T, Chan K, Dobson R and
Bundred NJ: ZD1839 (Iressa), a novel epidermal growth factor
receptor (EGFR) tyrosine kinase inhibitor, potently inhibits the
growth of EGFR-positive cancer cell lines with or without erbB2
overexpression. Int J Cancer. 94:774–782. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baselga J, Albanell J, Ruiz A, Lluch A,
Gascón P, Guillém V, González S, Sauleda S, Marimón I, Tabernero
JM, et al: Phase II and tumor pharmacodynamic study of gefitinib in
patients with advanced breast cancer. J Clin Oncol. 23:5323–5333.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gutteridge E, Agrawal A, Nicholson R,
Cheung K Leung, Robertson J and Gee J: The effects of gefitinib in
tamoxifen-resistant and hormone-insensitive breast cancer: A phase
II study. Int J Cancer. 126:1806–1816. 2010.PubMed/NCBI
|
|
9
|
Filardo EJ, Quinn JA, Frackelton AR Jr and
Bland KI: Estrogen action via the G protein-coupled receptor,
GPR30: Stimulation of adenylyl cyclase and cAMP-mediated
attenuation of the epidermal growth factor receptor-to-MAPK
signaling axis. Mol Endocrinol. 16:70–84. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Haynes MP, Li L, Sinha D, Russell KS,
Hisamoto K, Baron R, Collinge M, Sessa WC and Bender JR: Src kinase
mediates phosphatidylinositol 3-kinase/Akt-dependent rapid
endothelial nitric-oxide synthase activation by estrogen. J Biol
Chem. 278:2118–2123. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Luttrell LM, Daaka Y and Lefkowitz RJ:
Regulation of tyrosine kinase cascades by G-protein-coupled
receptors. Curr Opin Cell Biol. 11:177–183. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen ZJ, Wei W, Jiang GM, Liu H, Wei WD,
Yang X, Wu YM, Liu H, Wong CK, Du J, et al: Activation of GPER
suppresses epithelial mesenchymal transition of triple negative
breast cancer cells via NF-κB signals. Mol Oncol. 10:775–788. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ignatov A, Ignatov T, Weissenborn C,
Eggemann H, Bischoff J, Semczuk A, Roessner A, Costa SD and
Kalinski T: G-protein-coupled estrogen receptor GPR30 and tamoxifen
resistance in breast cancer. Breast Cancer Res Treat. 128:457–466.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Steiman J, Peralta EA, Louis S and Kamel
O: Biology of the estrogen receptor, GPR30, in triple negative
breast cancer. Am J Surg. 206:698–703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Girgert R, Emons G and Gründker C:
Inactivation of GPR30 reduces growth of triple-negative breast
cancer cells: Possible application in targeted therapy. Breast
Cancer Res Treat. 134:199–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vivacqua A, Lappano R, De Marco P, Sisci
D, Aquila S, De Amicis F, Fuqua SA, Andò S and Maggiolini M: G
protein-coupled receptor 30 expression is up-regulated by EGF and
TGF alpha in estrogen receptor alpha-positive cancer cells. Mol
Endocrinol. 23:1815–1826. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nielsen TO, Hsu FD, Jensen K, Cheang M,
Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler
L, et al: Immunohistochemical and clinical characterization of the
basal-like subtype of invasive breast carcinoma. Clin Cancer Res.
10:5367–5374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Girgert R, Bartsch C, Hill SM, Kreienberg
R and Hanf V: Tracking the elusive antiestrogenic effect of
melatonin: A new methodological approach. Neuro Endocrinol Lett.
24:440–444. 2003.PubMed/NCBI
|
|
19
|
Stanley ER, Palmer RE and Sohn U:
Development of methods for the quantitative in vitro analysis of
androgen-dependent and autonomous Shionogi carcinoma 115 cells.
Cell. 10:35–44. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen S, Zhou D, Yang C, Okubo T, Kinoshita
Y, Yu B, Kao YC and Itoh T: Modulation of aromatase expression in
human breast tissue. J Steroid Biochem Mol Biol. 79:35–40. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
von Minckwitz G, Jonat W, Fasching P, du
Bois A, Kleeberg U, Lück HJ, Kettner E, Hilfrich J, Eiermann W,
Torode J, et al: A multicentre phase II study on gefitinib in
taxane- and anthracycline-pretreated metastatic breast cancer.
Breast Cancer Res Treat. 89:165–172. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Green MD, Francis PA, Gebski V, Harvey V,
Karapetis C, Chan A, Snyder R, Fong A, Basser R and Forbes JF:
Australian New Zealand Breast Cancer Trials Group: Gefitinib
treatment in hormone-resistant and hormone receptor-negative
advanced breast cancer. Ann Oncol. 20:1813–1817. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Carey LA, Rugo HS, Marcom PK, Mayer EL,
Esteva FJ, Ma CX, Liu MC, Storniolo AM, Rimawi MF, Forero-Torres A,
et al: TBCRC 001: Randomized phase II study of cetuximab in
combination with carboplatin in stage IV triple-negative breast
cancer. J Clin Oncol. 30:2615–2623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bernsdorf M, Ingvar C, Jörgensen L, Tuxen
MK, Jakobsen EH, Saetersdal A, Kimper-Karl ML, Kroman N, Balslev E
and Ejlertsen B: Effect of adding gefitinib to neoadjuvant
chemotherapy in estrogen receptor negative early breast cancer in a
randomized phase II trial. Breast Cancer Res Treat. 126:463–470.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Girgert R, Emons G and Gründker C:
Inhibition of GPR30 by estriol prevents growth stimulation of
triple-negative breast cancer cells by 17β-estradiol. BMC Cancer.
14:9352014. View Article : Google Scholar : PubMed/NCBI
|