Introduction
Colorectal cancer (CRC) primarily occurs in the
colon and rectum. CRC is the third most commonly diagnosed cancer
in both men and women in the US based on the American Cancer
Society, 2016. According to the National Cancer Center of Korea, it
is the second most diagnosed cancer in men and the third in women.
Currently, the effective methods for treating CRC include
radiation, surgery, chemotherapy, and a combination of chemotherapy
with radiotherapy; however, most patients died from their disease
(1,2). If CRC is diagnosed and treated early,
then patients will have a higher survival rate, thus timely and
precise treatment is important (2,3). There
is continuous need to investigate and develop potential preventive
treatment approaches for CRC.
Carcinogenesis is a multistep process that involves
tumor initiation during which normal cells are genetically altered
into malignant cells (4,5). In addition, tumor promotion occurs
where the growth progression of small groups of malignant cells is
stimulated, and thereby the growing tumor becomes more aggressive
(4,5). The definition of chemoprevention is
the use of specific agents to interfere in the multistage
carcinogenesis (5,6). Furthermore, chemoprevention also
involves numerous types of cell death including apoptosis (7).
Apoptosis (programmed cell death), a mechanism that
is important for all multicellular organisms, controls cell
proliferation and maintains tissue homeostasis as well as
eliminates harmful or unnecessary cells from organisms (8). The evasion of apoptosis contributes to
carcinogenesis and tumor progression (9). There are two main apoptosis pathways,
including the extrinsic pathway, mediated by the activation of cell
surface receptors, which are death receptors. The death receptor
families are integrated into the plasma membrane and become
activated following ligation by their ligands (10). This leads to the formation of
aggregates of a series of proteins called death-inducing signaling
complexes that activate caspase-8.
Once caspase-8 is activated, its downstream
effector, caspase-3, directly cleaves and initiates the
mitochondrial pathway (intrinsic) of apoptosis by proteolytic
cleavage of BH3 interacting-domain death agonist (Bid) to initiate
t-Bid (10). The intrinsic
mitochondrial pathway is initiated by loss of mitochondrial
transmembrane potential and release of cytochrome c into the
cytosol, which then activates procaspase-9. This pathway is
initiated in the cells and regulated by proteins of the B-cell
lymphoma-2 (Bcl-2) family (11).
The members of the Bcl-2 family, which are key regulators of
apoptosis and overexpressed in numerous malignancies, are the most
crucial regulators of the intrinsic pathway (12). The extrinsic and intrinsic pathways
are regulated by influencing each other. Initiator caspases
(caspase-8 and −9) and active executioner caspases (caspase-3, −6
and −7) subsequently cleave key structural proteins and active
other enzymes (13).
The anthracycline doxorubicin (DOX) is one of the
most active drugs used in the treatment of a wide range of cancers
(14). This drug intercalates into
the DNA of living cells and causes cell death by inhibiting
topoisomerase II as well as the generation of reactive oxygen
species (ROS) and free radicals by redox reactions (15). DOX has been reported to induce
apoptosis through the intrinsic pathway by disrupting the
mitochondrial membrane potential with the release of cytochrome
c and subsequent activation of the caspase cascade (16,17).
Although DOX has strong anticancer activity and is approved by the
US Food and Drug Administration (FDA), it causes serious
side-effects in most of the major organs, especially the heart
(18–20). Numerous novel anthracycline
derivatives have been synthesized to overcome these side-effects
(21,22). Therefore, we studied a newly
synthesized DOX derivative, MHY451
[1-hydroxy-3-(oxiran-2-ylmethoxy)-5-(oxiran-2-ylmethyl)benzo[b]acridin-12(5H)-one].
The present study was designed to investigate the effects of MHY451
on HCT116 human colorectal cancer cells and the mechanism
underlying its induction of apoptosis and modulation of the cell
cycle in the same cell line.
Materials and methods
Chemicals
The simplified code name and structure of MHY451
[1-hydroxy-3-(oxiran-2-ylmethoxy)-5-(oxiran-2-ylmethyl)benzo[b]acridin-12(5H)-one]
used in the present study is shown in Fig. 1. MHY451 was dissolved in dimethyl
sulfoxide (DMSO), stored at −20°C before the experiments, and
working dilutions were prepared in culture medium. The maximum
concentration of DMSO did not exceed 0.1% (v/v) in the treatment
range of 1.25–20 µM, which did not affect cell growth. Antibodies
specific for Bcl-2-associated X protein (Bax), Bcl-2,
cyclin-dependent kinase 2 (Cdc2), Cdc25c, cyclin B1, p21, p53,
poly(adenosine diphosphate [ADP]-ribose) polymerase (PARP),
caspase-3, caspase-8 and caspase-9 were purchased from Santa Cruz
Biotechnology (Dallas, TX, USA). The polyclonal antibody against
Bid was purchased from Cell Signaling Technology (Danvers, MA,
USA).
Cell culture and viability assay
The human colorectal cancer cell lines HCT116 and
HT29 were cultured in Roswell Park Memorial Institute (RPMI)-1640
(GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10%
fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA,
USA), 100 units/ml penicillin (GE Healthcare Life Sciences) at 37°C
in a humidified atmosphere of 5% CO2. The effect of
MHY451 on cell viability was determined using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Amresco, Solon, OH, USA) assay. For the MTT assay, HCT116 cells
were seeded in a 24-well culture plate at a density of
5.5×104 cells/well, cultured for 24 h in the growth
medium, and then treated with or without various reagents at the
indicated concentrations. The cells were incubated with 0.5 mg/ml
MTT at 37°C for 2 h. The formazan granules generated by the live
cells were dissolved in DMSO, and the absorbance at 540 nm was
monitored using a multi-well reader (Thermo Fisher Scientific).
Nuclear staining with Hoechst
33342
Cells were seeded in 6-well culture plate at a
density of 2.7×105 cells/well and cultured for 24 h.
Cells were washed with phosphate-buffered saline (PBS) and fixed
with 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) in PBS
for 10 min at room temperature. Fixed cells were washed with PBS,
and stained with 4 µg/ml Hoechst 33342 (Life Technologies Corp.,
Grand Island, NY, USA) for 20 min at room temperature. The cells
were analyzed using fluorescent microscopy.
Cell cycle analysis using flow
cytometry
Cells were harvested by trypsinization and washed
once with PBS. After centrifugation, the cells were fixed in 70%
ethanol at 4°C overnight. The fixed cells were pelleted and stained
in cold propidium iodide (PI) solution (50 µg/ml in PBS) at room
temperature in the dark. The stained cells were analyzed using an
FC500 (Beckman Coulter, Istanbul, Turkey).
Western blot analysis
After MHY451 treatment, the cells were harvested and
washed with cold PBS. Total cells were lysed in lysis buffer [40 mM
Tris (pH 8.0), 120 mM sodium chloride (NaCl), 0.5% NP-40, 2 µg/ml
leupeptin and 100 µg/ml phenylmethylsulfonyl fluoride (PMSF)].
After centrifugation, the supernatant was collected, and protein
concentration was measured using protein assay reagents (Bio-Rad
Laboratories, Hercules, CA, USA). Equal amounts of protein were
denatured by boiling at 100°C for 5 min in 4X Laemmli sample buffer
(Bio-Rad Laboratories). Equal amounts of protein were separated
using 10–15% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred to polyvinylidene
difluoride (PVDF). The membranes were blocked with 5% non-fat dry
milk in Tris-buffered saline with Tween-20 buffer (TBS-T, 20 mM
Tris, 100 mM NaCl, pH 7.5 and 0.1% Tween-20) for 1 h at room
temperature. Then, the membranes were incubated with different
primary antibodies at 4°C overnight. After thorough washing with
TBS-T buffer, the membranes were incubated for 1 h with horseradish
peroxidase-conjugated secondary antibodies (Santa Cruz
Biotechnology). The membranes were washed with TBS-T buffer, and
the antigen-antibody complexes were detected using the enhanced
chemiluminescence (ECL) detection system (GE Healthcare Life
Sciences).
Annexin V staining
Annexin V-fluorescein isothiocyanate (FITC) was used
to quantitatively investigate the percentage of cells that
underwent apoptosis. Cells were seeded in a 6-well culture plate at
a density of 2.7×05 cells/well, cultured for 24 h,
harvested, trypsinized and washed once with cold PBS, and then
suspended the cells 1X binding buffer (Becton-Dickinson, San Jose,
CA, USA). The cells were stained with PI and Annexin solution
(Becton-Dickinson) for 15 min at room temperature in the dark. The
stained cells were analyzed using an Accuri C6 (BD Biosciences, Ann
Arbor, MI, USA) within 1 h.
Intracellular ROS measurement
For the assessment of intracellular ROS generation,
cells were seeded in a 96-well culture plate at a density of
0.5×104 cells/well and cultured for 24 h. After 1 day,
the cells were treated with MHY451 (2.5 µM) for 2, 4 and 6 h; the
medium was replaced with ROS buffer; and then 10 µM
2′,7′-dichlorofluorescein diacetate (DCFDA; Molecular Probes,
Eugene, OR, USA) was added. After 30 min, changes in fluorescence
intensity were measured seven times for 30 min using a fluorescence
plate reader (GENios; Tecan Instruments, Salzburg, Austria) and the
absorbance was measured at 485 and 530 nm.
Statistical analysis
Data were expressed as the mean ± standard deviation
(SD) of three separate experiments and analyzed using the Students
t-test. The acceptable level of significance was established at
P<0.05.
Results
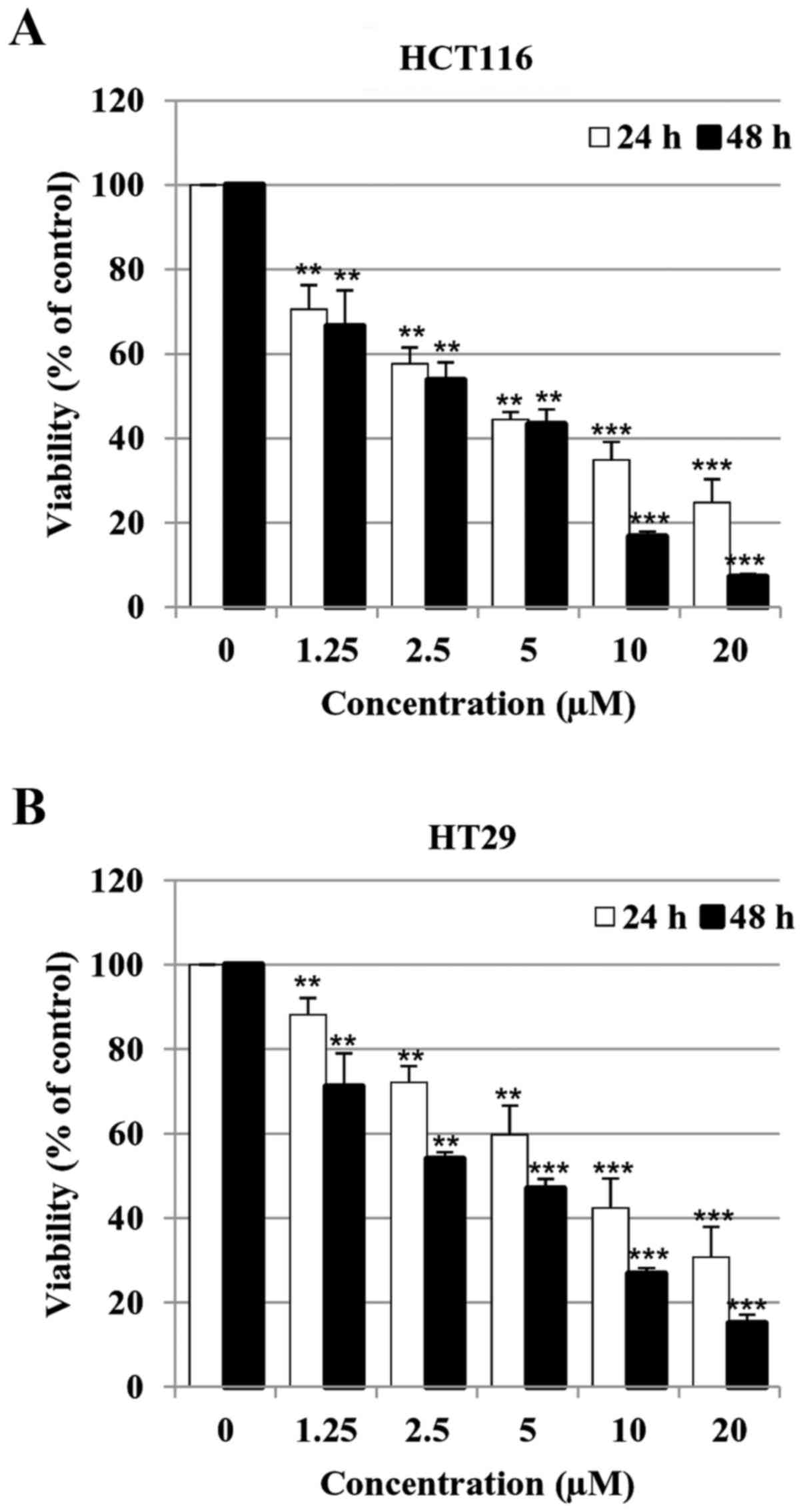
MHY451 suppresses the growth of HCT116
cells
To investigate the effect of MHY451 on the viability
of HCT116 and HT29 cells, the MTT assay was performed. Cell
viability was reduced by treatment with MHY451 in a concentration-
and time-dependent manner. As shown in Fig. 2, the half-maximal inhibitory
concentrations (IC50) of MHY 451 at 24 and 48 h were
3.14 and 2.87 µM in HCT116 cells (Fig.
2A) and 4.47 and 3.14 µM in HT29 cells (Fig. 2B), respectively. These results
indicate that the HCT116 cell line was more sensitive to treatment
with MHY451 than HT29 cells. Thus, HCT116 cells were chosen for
further experiments.
MHY451 modulates cell cycle
progression in HCT116 cells
To investigate whether MHY451 influenced cell cycle
distribution, HCT116 cells were treated with various concentrations
of MHY451 for 24 h and then the cell cycle was analyzed using flow
cytometry. As shown in Fig. 3A,
cells exposed to MHY451 exhibited G2/M cell cycle accumulation. A
total of 43.47% of the cells cultured with 5 µM MHY451 were in the
G2/M phase compared to 29.93% of control cells in the G2/M phase.
In addition, sub-G1 populations were increased from 2.09 in control
to 10.54% in cells treated with 10 µM MHY451. Next, we examined
whether MHY451 modulated the expression of G2/M cell cycle
regulators. Cells were treated with various concentrations of
MHY451 for 24 h, and then the level of G2/M cell cycle regulatory
proteins was examined using western blot analysis. As shown in
Fig. 3B, the expression levels of
cyclin B1, its regulatory protein Cdc2 and Cdc25c were decreased in
HCT116 cells by MHY451 treatment in a concentration-dependent
manner. The induction of p21WAF/CIP1 causes arrest in
the G1/G0 or G2/M phase of the cell cycle binding of the
cyclin-cyclin-dependent kinase (CDK) complex. Thus, further studies
were performed to determine whether p21WAF/CIP1 was
induced by MHY451 either by p53-dependent or p53-independent
pathways in HCT116 cells. The results showed that the expression
level of p53 was slightly increased and consequently the level of
p21WAF/CIP1 was also increased by MHY451 treatment in
HCT116 cells. These data suggest that the MHY451-induced G2/M phase
arrest was mediated by a p53-dependent pathway.
MHY451 treatment induces apoptosis in
HCT116 cells
To determine whether the growth inhibitory effects
of MHY451 were due to apoptosis, microphotographs were acquired.
HCT116 cells treated with MHY451 showed distinct morphological
changes compared with the control cells (Fig. 4A, upper panel). They were
round-shaped, which is a typical feature of apoptosis. Furthermore,
morphological changes in the cellular structures were assessed
using Hoechst 33342. As shown in Fig.
4A (lower panel), after MHY451 exposure, HCT116 cells showed
distinct morphological changes such as cell shrinkage and
compaction of the nuclei compared with control cells. In addition,
to confirm the apoptotic effect of MHY451 in the HCT116 cells, flow
cytometry was performed. As shown in Fig. 4B, the early apoptotic proportion
(lower right quadrant) increased from 3.5 to 25.4%, and the late
apoptotic proportion (upper right quadrant) increased from 3.0 to
27.8% after treatment with 10 µM MHY451. The effect of MHY451 on
apoptosis was further verified using western blot analysis. As
shown in Fig. 4C, treatment with
MHY451 caused proteolytic degradation of PARP, a molecular marker
of apoptosis. In addition, pro-caspase-9 decreased after treatment
with MHY451. Moreover, the expression level of pro-apoptotic Bax
was upregulated while the anti-apoptotic Bcl-2 protein was
downregulated in HCT116 cells. These results suggest that MHY451
induced apoptosis of HCT116 cells.
 | Figure 4.Effect of MHY451 on the induction of
apoptosis in HCT116 cells. (A) Morphological changes in
MHY451-treated cells. Nuclei of HCT116 cells stained with
fluorescent DNA-binding dye (Hoechst 33342). (B) Effect of MHY451
on cell death in cells stained with Annexin V-FITC/PI and analyzed
using flow cytometry. (C) Immunoblot analysis with pro-caspase-3,
−8, −9, Bax, Bcl-2, Bid and PARP (116 kDa) following treatment with
MHY451. Actin was used as a protein loading control. (D) Cells
stained with PI and analyzed using flow cytometry (**P<0.01 vs.
MHY451-treated cells). (E) Effects of 40 µM Z-VAD-FMK (Z-VAD) in
total cell lysate using immunoblot analysis with actin as loading
control. Representative results of three independent experiments
are shown. FITC, fluorescein isothiocyanate; PI, propidium iodide;
Bid, BH3-interacting domain death agonist; Bax, Bcl-2-associated X
protein; Bcl-2, B-cell CLL/lymphoma 2; PARP, poly(ADP-ribose)
polymerase. |
Caspases are involved in
MHY451-induced apoptosis in HCT116 cells
To confirm the role of caspase in MHY451-induced
apoptosis, the effects of Z-VAD-FMK, a pan caspase inhibitor, were
examined in MHY451-treated cells. As shown in Fig. 4D, pretreatment with Z-VAD-FMK
reduced the proportion of cells that underwent MHY451-induced
apoptosis in the HCT116 cells. These results indicate that
Z-VAD-FMK partially inhibited the apoptotic effect of MHY451 in
HCT116 cells. To further confirm this result, western blot analysis
was performed. As shown in Fig. 4E,
pretreatment with Z-VAD-FMK markedly inhibited the MHY451-induced
downregulation of pro-caspase-3 and PARP cleavage. These results
demonstrate that activation of the caspase cascade is involved in
the induction of apoptosis by MHY451.
MHY451 induces apoptosis in HCT116
cells via ROS generation
The regulation of intracellular ROS levels is vital
to cellular homeostasis and different ROS levels can cause
different biological responses. At low and moderate levels, ROS act
as signaling molecules while at high levels they induce cell damage
and death. Cancer cells show elevated ROS levels compared to those
of normal cells due to the accumulation of intrinsic or
environmental factors or both (23). It is currently becoming increasingly
clear that the generation of ROS can be used therapeutically in the
treatment of cancer (24).
Therefore, we examined the generation of ROS by MHY451 to explore
whether this was its mechanism of apoptosis induction. The
intracellular ROS level was measured using the ROS-detecting
fluorescent dye DCFDA in HCT116 cells. The cells were treated with
2.5 µM of MHY451 for various times. As shown in Fig. 5A, the cells exposed to MHY451 showed
increased levels of intracellular ROS in a time-dependent manner.
Next, HCT116 cells were pretreated with N-acetylcysteine
(NAC), a well-known ROS scavenger, and then treated with MHY451. At
the end of the incubation period, the levels of intracellular ROS
were measured. As shown in Fig. 5B,
pretreatment with NAC considerably blocked ROS generation in
MHY451-treated HCT116 cells. To further identify the relationship
between ROS generation and apoptosis, the effects of NAC were
examined in MHY451-treated cells. As shown in Fig. 5C, the presence of cells with sub-G1
DNA content following treatment with MHY451 with or without NAC was
evaluated using flow cytometry to quantify the onset of apoptosis.
Pretreatment of cells with NAC significantly inhibited cell death
in MHY451-treated cells (P<0.01). In agreement with these
observations, sequestration of ROS by NAC effectively suppressed
the decrease in pro-caspase-3 levels and prevented MHY451-induced
PARP cleavage in HCT116 cells (Fig.
5D). These results demonstrate that ROS generation plays an
important role in the MHY451-mediated apoptotic pathway in HCT116
cells.
Discussion
The present study was conducted to investigate the
anticancer effects of MHY451 in HCT116 cells. MHY451 is a novel
molecule based on DOX, which has anticancer mechanisms that might
be mediated by DNA intercalation, cell cycle distribution, or
apoptosis. In the present study, MHY451 induced apoptosis and cell
cycle arrest in HCT116 cells. Flow cytometric analysis revealed
that MHY451 induced G2/M cell cycle arrest. G2/M transition is
regulated by cyclin B/Cdc2, which has been identified as a
principal component of the mitosis-promoting factor (25).
p21WAF/CIP1, a member of the CDK
interacting protein/kinase inhibitory protein (CIP/KIP) family,
inhibits a variety of CDK proteins and is controlled by p53
(26). Furthermore, p53, a tumor
suppressor gene, induces cell cycle regulation and apoptosis. In
this study, the protein level of p53 was not significantly changed
while that of p21WAF/CIP1 increased in a
concentration-dependent manner in HCT116 cells. These results
demonstrate that MHY451 increased p21WAF/CIP1 levels and
decreased cyclin B1. Furthermore, its activating partners, Cdc25c
and Cdc2 (Fig. 3B), may be involved
in the p53-independent pathway by which MHY451 inhibited HCT116
cell growth and induced G2/M arrest.
Apoptosis, which is programmed cell death that
occurs after exposure to specific stimuli, is an important
component of cell growth control (27). There are two main apoptotic
pathways, which are the intrinsic and extrinsic pathways that are
initiated by the cleavage of caspase-3 and the action of caspase-9
and −8, respectively (28). The
intrinsic pathway involves mitochondrial disruption by
pro-apoptotic Bcl-2 family members and Bax oligomerization, which
consequently releases cytochrome c. Expression of Bcl-2
decreased whereas that of Bax increased in MHY451-treated HCT116
cells. In addition, Bid was remarkably decreased.
MHY451 effectively inhibited HCT116 cell
proliferation and induced apoptosis in a concentration-dependent
manner. To confirm the MHY451-induced apoptosis, cell cycle and
Annexin V/PI double staining was performed. MHY451 also increased
the sub-G1 populations in HCT116 cells (Fig. 3A). As shown in Fig. 4B, the increase in early and late
apoptosis was clearly observed in a concentration-dependent manner.
Further studies indicated that MHY451, in the presence of
Z-VAD-FMK, a pan-caspase inhibitor, prevented the cleavage of PARP.
Therefore, MHY451-induced apoptosis is likely mediated, at least in
part, by the caspase cascade.
This study is first to investigate the ROS-mediated
apoptotic effects of MHY451 in HCT116 human colorectal cancer
cells. In the biological system, ROS are constantly generated and
eliminated and play crucial roles in both normal and abnormal
condition. ROS, which are produced in the mitochondria, play a
crucial role in the regulation of cell death when accumulated in
excessive amounts (29). In cancer,
increased ROS serves as an endogenous source of DNA-damaging agents
that facilitate genetic instability and development of drug
resistance. Despite the negative impact of elevated ROS in cancer
cells, it is possible to exploit this characteristic to develop
novel therapeutic strategies to preferentially kill cancer cells
through an ROS-mediated mechanism (30). Several anticancer agents such as
cisplatin, oxaliplatin, and mitomycin C exert their effects, at
least in part, by the induction of ROS generation (31). In the present study, to reveal if
MHY451 affected the level of ROS, MHY451-treated HCT116 cells were
stained with DCFDA. MHY451-induced ROS generation increased in a
time-dependent manner (Fig. 5A).
Moreover, pretreatment with the ROS scavenger NAC significantly
decreased MHY451-induced ROS levels (Fig. 5B) and inhibited MHY451-induced
activation of caspase-3 and PARP cleavage, as well as subsequent
cell death. This observation suggests that ROS were involved in the
upstream events of caspase induced by MHY451 in HCT116 cells.
Although the effect of NAC on the MHY451-induced modulation of
pro-apoptotic proteins have not been elucidated, the apoptotic
effect of MHY451 is closely related to the generation of ROS.
In conclusion, MHY451 suppressed the growth of
HCT116 cells in a concentration-dependent manner by inducing G2/M
phase arrest. The present data also suggest that MHY451 induced
apoptosis through caspase and ROS-mediated pathways (Fig. 6). Moreover, the mechanism of action
of MHY451 should be studied further. Overall, these results
demonstrate that MHY451 may be a promising therapeutic agent for
treating colorectal cancer.
Acknowledgements
The present study was supported by the National
Research Foundation of Korea (NRF) Grant funded by the Korea
government (MSIP, no. 2009-0083538). This study was also supported
by the Department of Social Enterprise, Pusan National University.
We would like to thank the Aging Tissue Bank for providing research
information.
References
|
1
|
Roncucci L and Mariani F: Prevention of
colorectal cancer: How many tools do we have in our basket? Eur J
Intern Med. 26:752–756. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu KC, Shih TY, Kuo CL, Ma YS, Yang JL,
Wu PP, Huang YP, Lai KC and Chung JG: Sulforaphane induces cell
death through G2/M Phase arrest and triggers apoptosis in HCT 116
human colon cancer cells. Am J Chin Med. 44:1289–1310. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nautiyal J, Banerjee S, Kanwar SS, Yu Y,
Patel BB, Sarkar FH and Majumdar AP: Curcumin enhances
dasatinib-induced inhibition of growth and transformation of colon
cancer cells. Int J Cancer. 128:951–961. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin WW and Karin M: A cytokine-mediated
link between innate immunity, inflammation, and cancer. J Clin
Invest. 117:1175–1183. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
George VC, Dellaire G and Rupasinghe HPV:
Plant flavonoids in cancer chemoprevention: Role in genome
stability. J Nutr Biochem. 45:1–14. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mukhtar H: Chemoprevention: Making it a
success story for controlling human cancer. Cancer Lett.
326:123–127. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lim HS, Kang YJ, Sung B, Kim SH, Kim MJ,
Kim HR, Kim SJ, Choi YH, Moon HR, Chung HY, et al: Novel
dihydrobenzofuro[4,5-b][1,8]naphthyridin-6-one derivative, MHY-449,
induces cell cycle arrest and apoptosis via the downregulation of
Akt in human lung cancer cells. Oncol Rep. 34:2431–2438.
2015.PubMed/NCBI
|
|
8
|
Goldar S, Khaniani MS, Derakhshan SM and
Baradaran B: Molecular mechanisms of apoptosis and roles in cancer
development and treatment. Asian Pac J Cancer Prev. 16:2129–2144.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fulda S: Tumor resistance to apoptosis.
Int J Cancer. 124:511–515. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fulda S: Targeting apoptosis for
anticancer therapy. Semin Cancer Biol. 31:84–88. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee Y, Sung B, Kang YJ, Kim DH, Jang JY,
Hwang SY, Kim M, Lim HS, Yoon JH, Chung HY, et al: Apigenin-induced
apoptosis is enhanced by inhibition of autophagy formation in
HCT116 human colon cancer cells. Int J Oncol. 44:1599–1606.
2014.PubMed/NCBI
|
|
12
|
Ghobrial IM, Witzig TE and Adjei AA:
Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin.
55:178–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
McIlwain DR, Berger T and Mak TW: Caspase
functions in cell death and disease. Cold Spring Harb Perspect
Biol. 5:a0086562013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu G, Zheng J, Song E, Donovan M, Zhang
K, Liu C and Tan W: Self-assembled, aptamer-tethered DNA nanotrains
for targeted transport of molecular drugs in cancer theranostics.
Proc Natl Acad Sci USA. 110:7998–8003. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gonçalves C, Martins-Neves SR,
Paiva-Oliveira D, Oliveira VE, Fontes-Ribeiro C and Gomes CM:
Sensitizing osteosarcoma stem cells to doxorubicin-induced
apoptosis through retention of doxorubicin and modulation of
apoptotic-related proteins. Life Sci. 130:47–56. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tsang WP, Chau SP, Kong SK, Fung KP and
Kwok TT: Reactive oxygen species mediate doxorubicin induced
p53-independent apoptosis. Life Sci. 73:2047–2058. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dembinski JL and Krauss S:
Characterization and functional analysis of a slow cycling stem
cell-like subpopulation in pancreas adenocarcinoma. Clin Exp
Metastasis. 26:611–623. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shi Y, Moon M, Dawood S, McManus B and Liu
PP: Mechanisms and management of doxorubicin cardiotoxicity. Herz.
36:296–305. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu
YL, Liu LF and Yeh ET: Identification of the molecular basis of
doxorubicin-induced cardiotoxicity. Nat Med. 18:1639–1642. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vong LB and Nagasaki Y: Combination
treatment of murine colon cancer with doxorubicin and redox
nanoparticles. Mol Pharm. 13:449–455. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
De U, Chun P, Choi WS, Lee BM, Kim ND,
Moon HR, Jung JH and Kim HS: A novel anthracene derivative, MHY412,
induces apoptosis in doxorubicin-resistant MCF-7/Adr human breast
cancer cells through cell cycle arrest and downregulation of
P-glycoprotein expression. Int J Oncol. 44:167–176. 2014.PubMed/NCBI
|
|
22
|
Szwed M, Laroche-Clary A, Robert J and
Jozwiak Z: Efficacy of doxorubicin-transferrin conjugate in
apoptosis induction in human leukemia cells through reactive oxygen
species generation. Cell Oncol (Dordr). 39:107–118. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marengo B, Nitti M, Furfaro AL, Colla R,
Ciucis CD, Marinari UM, Pronzato MA, Traverso N and Domenicotti C:
Redox homeostasis and cellular antioxidant systems: Crucial players
in cancer growth and therapy. Oxid Med Cell Longev.
2016:62356412016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Renschler MF: The emerging role of
reactive oxygen species in cancer therapy. Eur J Cancer.
40:1934–1940. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Arion D, Meijer L, Brizuela L and Beach D:
cdc2 is a component of the M phase-specific histone H1 kinase:
Evidence for identity with MPF. Cell. 55:371–378. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bates S, Ryan KM, Phillips AC and Vousden
KH: Cell cycle arrest and DNA endoreduplication following
p21Waf1/Cip1 expression. Oncogene. 17:1691–1703. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Amendola A, Fesus L, Piacentini M and
Szondy Z: ‘Tissue’ transglutaminase in AIDS. J Immunol Methods.
265:145–159. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zamzami N, Marchetti P, Castedo M,
Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B and
Kroemer G: Sequential reduction of mitochondrial transmembrane
potential and generation of reactive oxygen species in early
programmed cell death. J Exp Med. 182:367–377. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pelicano H, Carney D and Huang P: ROS
stress in cancer cells and therapeutic implications. Drug Resist
Updat. 7:97–110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Santoro V, Jia R, Thompson H, Nijhuis A,
Jeffery R, Kiakos K, Silver AR, Hartley JA and Hochhauser D: Role
of reactive oxygen species in the abrogation of oxaliplatin
activity by cetuximab in colorectal cancer. J Natl Cancer Inst.
108:djv3942015. View Article : Google Scholar : PubMed/NCBI
|