Introduction
Colorectal cancer (CRC) ranks third in terms of high
incidence and as the fourth leading cause of cancer-related deaths
worldwide (1). It is estimated that
~1.3 million new cases are diagnosed and nearly 0.7 million succumb
to CRC each year worldwide (1). CRC
incidence rates are highest in Alaska Natives and African Americans
and lowest in Asian/Pacific Islanders, and they are 30–40% higher
in men than in women (2). The
incidence and mortality of CRC increase with age, and ~50% of CRC
patients are older than 70 years (1). Currently, screening is one of the most
effective diagnostic methods, and treatments mainly include
surgery, chemotherapy, radiation therapy, targeted therapy and some
combination therapy. The incidence and death rates have declined in
both men and women in recent years, as a result of improved
screening and advances in clinical treatment (1). However, the potential mechanism of CRC
has not been clarified clearly, especially at the molecular level
(3).
Circular RNAs (circRNAs), small (~21 nt) non-coding
RNAs, that form a covalently closed continuous loop, are
characterized by high expression levels, stability, and a large
number of miRNA binding sites (4).
Due to the fact that circRNAs do not have 5′ or 3′ ends, they are
resistant to exonuclease-mediated degradation and share high
evolutionary conservation in cells (5). They base-pair directly target
messenger RNAs (mRNAs), and can trigger cleavage of the mRNA
depending on the degree of complementarity. circRNAs are considered
to be involved in a large, diverse set of biological processes
(BPs), such as gene regulation (6).
Research has demonstrated that some circRNAs are associated with
the bioprocesses of proliferation, metastasis, prognosis and a
curative effect in CRC (1).
At present, the biological functions of most
circRNAs are unclear, and little is known about circRNAs and their
relationship with CRC. It is worthwhile to examine how circRNAs can
be used to study the pathogenesis and some BPs of CRC. In the
present study, the differentially expressed circRNAs, as well as
their related mRNAs and miRNAs, were screened and analyzed in CRC
tissue compared with adjacent tissue, in order to explore the
potential molecular mechanism from the circRNA expression
levels.
Materials and methods
Patients and sample collection
Four samples from CRC patients, aged 50 to 62 years,
were collected from Tianjin Union Medical Center, Nankai University
Affiliated Hospital in 2016. The patients had not been treated with
radiotherapy or chemotherapy before the surgery. The CRC tissue and
their adjacent tissue samples were obtained after CRC surgeries,
and recorded as treatment and control groups, respectively. The
experimental study was approved by the hospital ethics committee,
and the informed consent forms were obtained when the patients were
accepted for the study by the hospital. All procedures performed in
the studies involving human participants were in accordance with
the ethical standards of Committee of the Tianjin Union Medical
Center (Tianjin, China) and with the 1964 Helsinki declaration and
its later amendments or comparable ethical standards.
Sample processing and circRNA
acquisition
The samples from the two groups were immediately
washed with 0.9% NaCl (RNase-free) and quickly dipped in RNase
inhibitor (Epicentre: Illumina, Inc., San Diego, CA, USA) according
to the manufacturer's instructions. Then, total RNA was extracted
from the CRC tissues using TRIzol (Invitrogen: Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to the manufacturer's
instructions. Total RNA was digested with RNase R (Epicentre:
Illumina, Inc.) to remove linear RNAs, and was then reversed
transcribed as cDNA. The cDNA was purified with
NucleoSpin® Extract II kits (Macherey-nagel, Düren,
Germany), and was labeled with a fluorescent probe (Arraystar Super
RNA Labeling kit; Arraystar, Inc., Rockville, MD, USA). The
fluorescent cRNA was purified with the the NucleoSpin®
Extract II kits. The concentration and specific activity of the
labeled cRNAs (pmol Cy3/µg cRNA) were assessed using NanoDrop
ND-1000 (Thermo Fisher Scientific, Inc.). A total of 1 µg of each
labeled cRNA was fragmented by adding 5 µl of 10X blocking agent
and 1 µl of 25X fragmentation buffer. Finally, 25 µl of 2X
hybridization buffer was added to dilute the labeled cRNA. Then, a
total of 50 µl mixtures were heated at 95°C for 3 min, and cooled 2
min on ice. The hybridization solution was dispensed into the
gasket slide and assembled to construct the circRNA expression
microarray slide. The slides were centrifuged for 16 h at 40 × g at
65°C in an Agilent Hybridization Oven (Agilent Technologies, Inc.,
Santa Clara, CA, USA). Finally, the hybridized arrays were washed,
fixed and scanned using the Agilent scanner G2565CA (Agilent
Technologies, Inc.).
Single array analysis and cluster
analysis
The Agilent Feature Extraction 10.7 software
(Agilent Technologies, Inc.) graph chip was used to read the values
and to obtain the original data. The GeneSpring GX 12.5 software
(Agilent Technologies, Inc.) was used for quintile normalization
and subsequent data processing of the original data. After
standardization, the cluster analyses of the eight samples were
performed with hierarchical and average linkage algorithms of R
package.
Identification of the differential
expressed circRNAs
After single array and cluster analysis, the
differentially expressed circRNAs were identified in CRC tissue
samples compared with the adjacent tissue samples using P<0.05
and |log2(fold-change)|>1. Moreover, the volcano plot was
plotted according to the P-value and log2(fold change). In
addition, the Circos plot also was drawn.
Screening related mRNAs and enrichment
analysis
KEGG orthology based annotation system (KOBAS) is a
web server for gene/protein functional annotation (annotate module)
and functional gene set enrichment (enrichment module) (1). In the present study, the related mRNAs
of the differentially expressed circRNAs were annotated with the
molecule annotation system (MAS) 3.0, and the diseases, pathways
and functional enrichment analysis, in which they were enriched,
were performed using KOBAS 3.0. The terms of enriched diseases
included KEGG disease, FunDO, NHGRI GWAS catalog and OMIM; the
enriched pathways contained Reactome, KEGG pathways, PANTHER and
BioCyc; and BPs, molecular function (MF) and cellular component
(CC) were involved in the Gene Ontology (GO) terms.
Screening related target miRNAs and
construction of the circRNA/miRNA network
A circRNA can bind to a miRNA and indirectly
regulate the translation of an mRNA, and miRanda is an algorithm
used to find genomic targets for miRNAs. The related target miRNAs
for the differentially expressed circRNAs were predicted with the
miRanda algorithm. Subsequently, the key circRNAs with high
|log2(fold-change)|, which were involved in the most circRNA/miRNA
pairs were chosen. Ultimately, the circRNA/miRNA network for the
key circRNAs was constructed and visualized using Cytoscape
(7) software (Cytoscape 3.4.0,
http://www.cytoscape.org/).
Verification of related circRNAs and
its target genes
The tumor tissues and paracancerous specimens from
four CRC patients were collected, after the patients had signed
informed consent. The target gene of hsa_circ_0126897_CBC1
was found to be SLC4A4 from circBase (http://circrna.org/cgi-bin/singlerecord.cgi?id=hsa_circ_0126897).
The SYBR® Premix Ex Taq™ kit (Takara Biotechnology Co.,
Ltd., Shiga, Japan) and the Applied Biosystems™ QuantStudio™ 5
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Foster City, CA, USA) were used to detect the mRNA levels of
hsa_circ_0126897_CBC1 and SLC4A4 according to the
manufacturer's instructions. All the primers were designed and
synthesized by Takara Biotechnology Co., Ltd. (Beijing, China). The
primers of hsa_circ_0126897_CBC1:
3′-CTCATGGTGATGCTGAACCTTCTTA-5′ and 3′-CTCATGGTGATGCTGAACCTTCTTA-5′
(139 bp); and SLC4A4: 3′-AGCACTCTATACGCCCCAAG-5′ and
3′-TTCCTTTTCTCTCACGCCCT-5′ (540 bp). In addition, β-actin
was used as a reference, and the primer sequences were
5′-CTACAATGAGCTGCGTGTGG-3′ and 5′-AGGCATACAGGGACAACACA-3′ (308
bp).
Statistical analysis
SPSS v17.0 (SPSS, Inc., Chicago, IL, USA) was used
for all statistical analyses, and data were expressed as the mean ±
SD. To compare both two groups t-test was used, and P<0.05 was
considered statistically significant.
Results
Differentially expressed circRNAs
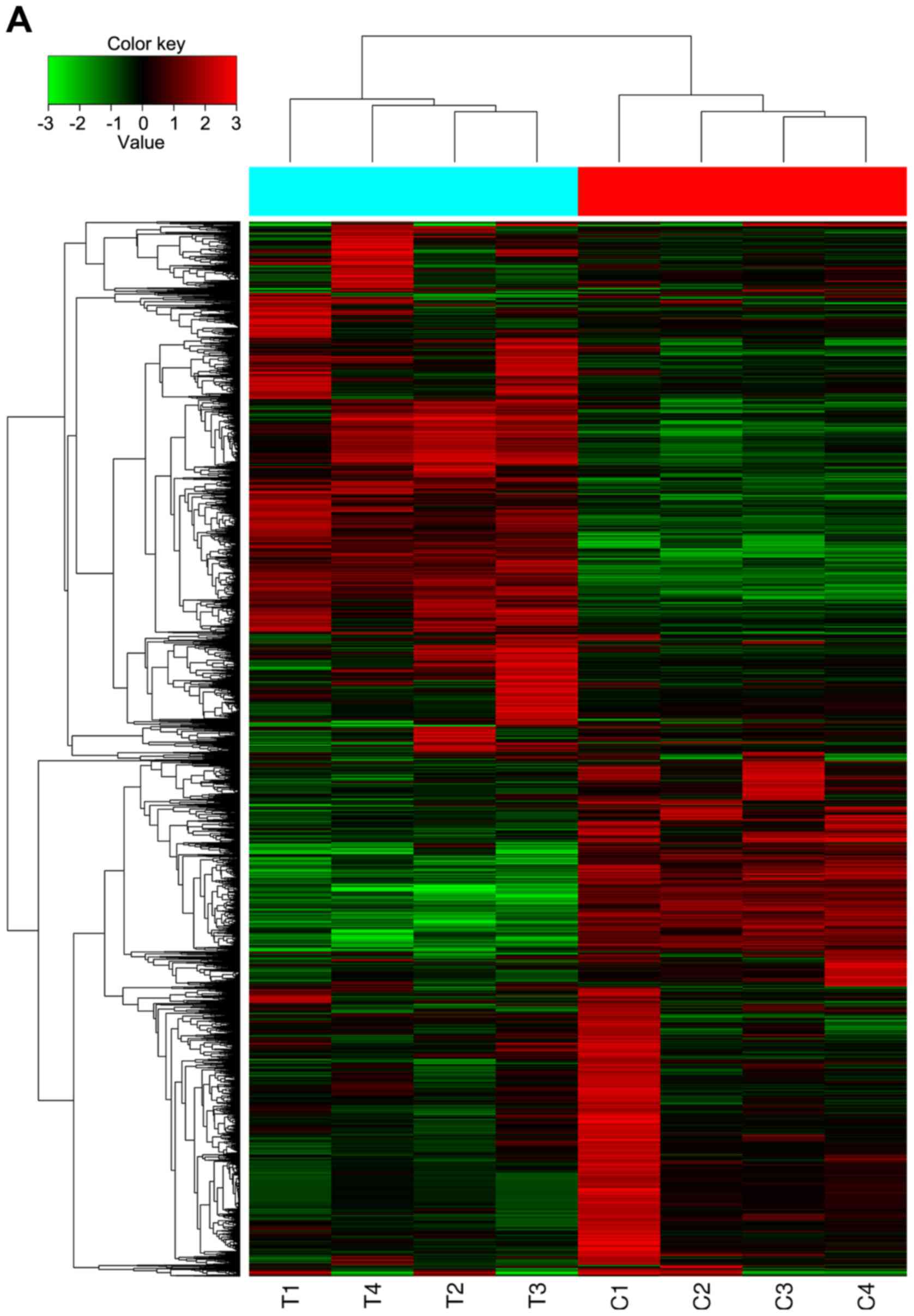
After single array analysis, the cluster analysis
between the two groups was performed. The cluster graph is
displayed in Fig. 1A, which
revealed that 4 CRC tissue samples (T1-T4) were clustered into one
group, and the 4 adjacent tissue samples (C1-C4) were clustered
into one class. Differentially expressed circRNAs were identified
according to fluorescence signal intensity (Fig. 1B), and the volcano plot and the
Circos plot are presented in Fig. 1C
and D, respectively. Fig. 1
revealed that the differentially expressed circRNAs could separate
tumor samples from paracancerous tissues, commendably. A total of
10,245 (6,264 up- and 3,981 downregulated) differentially expressed
circRNAs were identified, and the top 30 most significant ones are
presented in Table I. It was
evident from Table I that
hsa_circ_0027997_CBC1, hsa_circ_0060967_CBC1 and
hsa_circ_0055546_CBC1 were the top 3 differentially
expressed circRNAs.
 | Table I.The top 30 most significant
differentially expressed circRNAs based on the P-value. |
Table I.
The top 30 most significant
differentially expressed circRNAs based on the P-value.
| circRNAs name | P-value | FC | Regulation |
|---|
|
hsa_circ_0027997_CBC1 | 0.000307 | 2.402326 | Down |
|
hsa_circ_0060967_CBC1 | 0.000315 | 2.264531 | Up |
|
hsa_circ_0055546_CBC1 | 0.000344 | 2.086458 | Down |
|
hsa-circRNA7788-9_CBC1 | 0.000514 | 2.821571 | Down |
|
hsa_circ_0004619_CBC1 | 0.00053 | 2.251397 | Down |
|
hsa-circRNA5955-2_CBC1 | 0.000618 | 2.216281 | Down |
|
hsa_circ_0089626_CBC1 | 0.000743 | 2.603937 | Down |
|
hsa_circ_0047851_CBC1 | 0.000887 | 2.100605 | Down |
|
hsa_circ_0109686_CBC1 | 0.001066 | 3.009866 | Down |
|
hsa_circ_0026344_CBC1 | 0.001555 | 5.709074 | Down |
|
hsa_circ_0073207_CBC1 | 0.001635 | 2.687123 | Down |
|
hsa_circ_0088103_CBC1 | 0.001688 | 2.40819 | Down |
|
hsa_circ_0094226_CBC1 | 0.001766 | 5.875593 | Down |
|
hsa_circ_0070283_CBC1 | 0.001785 | 4.989792 | Down |
|
hsa_circ_0103088_CBC1 | 0.001941 | 2.771014 | Down |
|
hsa_circ_0136666_CBC1 | 0.002348 | 3.634452 | Up |
|
hsa-circRNA1102_CBC1 | 0.00248 | 3.036569 | Down |
|
hsa-circRNA7032-14_CBC1 | 0.002612 | 2.136622 | Up |
|
hsa-circRNA6158-6_CBC1 | 0.002704 | 3.008672 | Down |
|
hsa_circ_0011276_CBC1 | 0.00276 | 2.926497 | Down |
|
hsa_circ_0031263_CBC1 | 0.003919 | 7.843025 | Up |
|
hsa_circ_0069013_CBC1 | 0.004342 | 2.26783 | Up |
|
hsa_circ_0011924_CBC1 | 0.004687 | 3.432366 | Up |
|
hsa-circRNA3677-10_CBC1 | 0.004807 | 2.059462 | Down |
|
hsa_circ_0005525_CBC1 | 0.005082 | 2.523352 | Down |
|
hsa_circ_0060745_CBC1 | 0.005263 | 2.027748 | Up |
|
hsa_circ_0042328_CBC1 | 0.005733 | 2.532399 | Down |
|
hsa_circ_0072715_CBC1 | 0.005974 | 2.193656 | Up |
|
hsa_circ_0014712_CBC1 | 0.006073 | 2.107837 | Up |
|
hsa-circRNA3698-20_CBC1 | 0.006194 | 2.07261 | Down |
The related mRNAs and enriched
terms
The related mRNAs of the differentially expressed
circRNAs were annotated by KOBAS 3.0, and they were enriched in
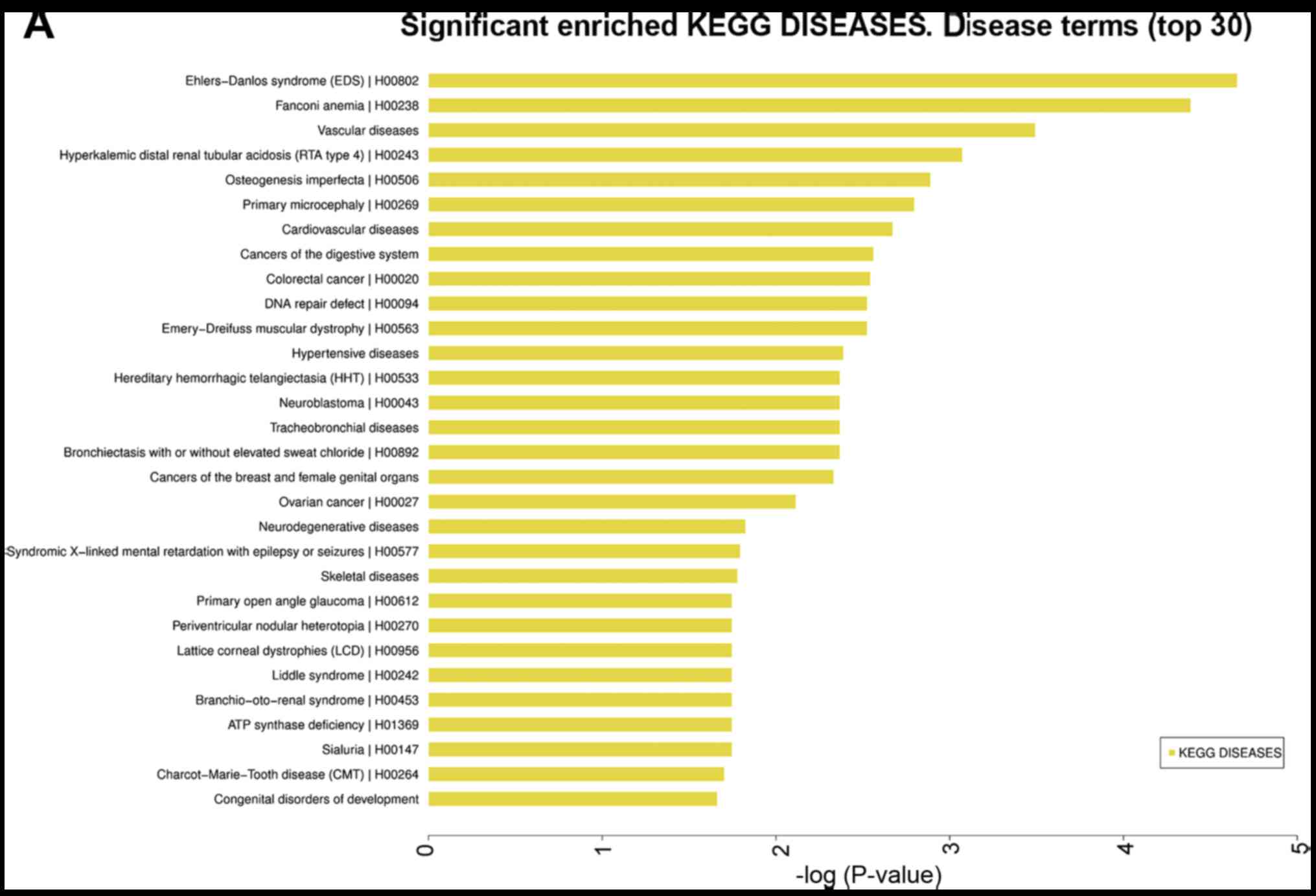
some disease terms, GO terms and pathways. Furthermore, the
enriched diseases included 462 KEGG diseases, 411 FunDO, 669 NHGRI
GWAS catalog, and 845 OMIM, and the top 30 disease terms are shown
in Fig. 2A-D. The KEGG diseases
contained breast and ovarian cancer as well as CRC. The FunDO
contained skin and stomach cancer as well as adenocarcinoma The
NHGRI GWAS catalog contained lung and bladder cancer, and OMIM
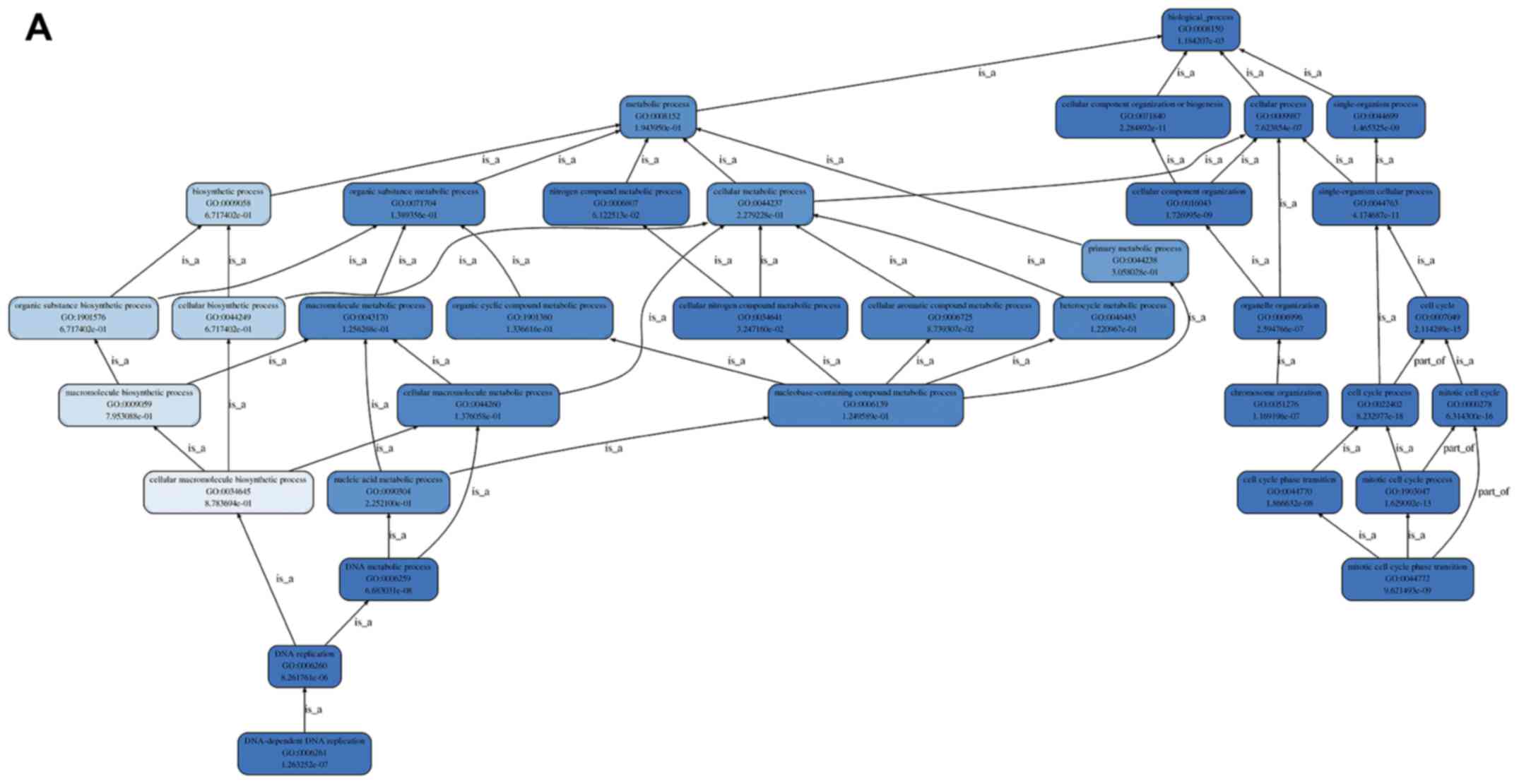
contained glioblastoma, gastric cancer and CRC. The enriched GO
terms contained 8,485 BP; 2,026 MF; and 1,095 CC, and their
hierarchy and top 30 GO terms are shown in Fig. 3A-D. The cell cycle process, the
mitotic cell cycle and the cell cycle were the top 3 most
significant BPs (Fig. 3D). A total
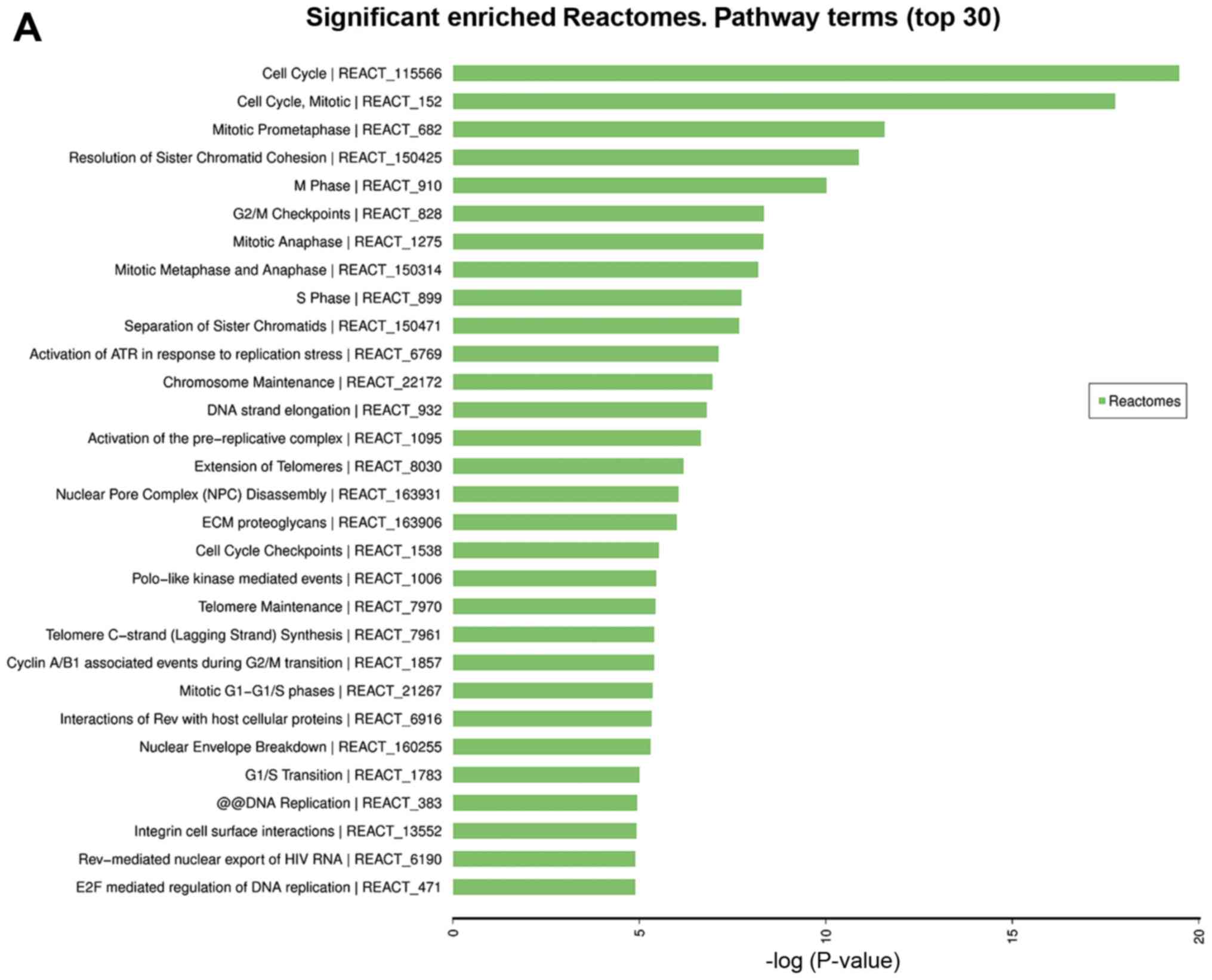
of 1,334 Reactomes, 281 KEGG pathways, 117 PANTHER and 193 BioCyc
were involved in the enriched pathways, and the top 30 terms of
each are shown in Fig. 4A-D. Cell
cycle, and ‘cell cycle, mitotic’ were the top 2 most significant
Reactome pathways, cell cycle was the most significant KEGG
pathway, and DNA replication was the most significant PANTHER
pathway.
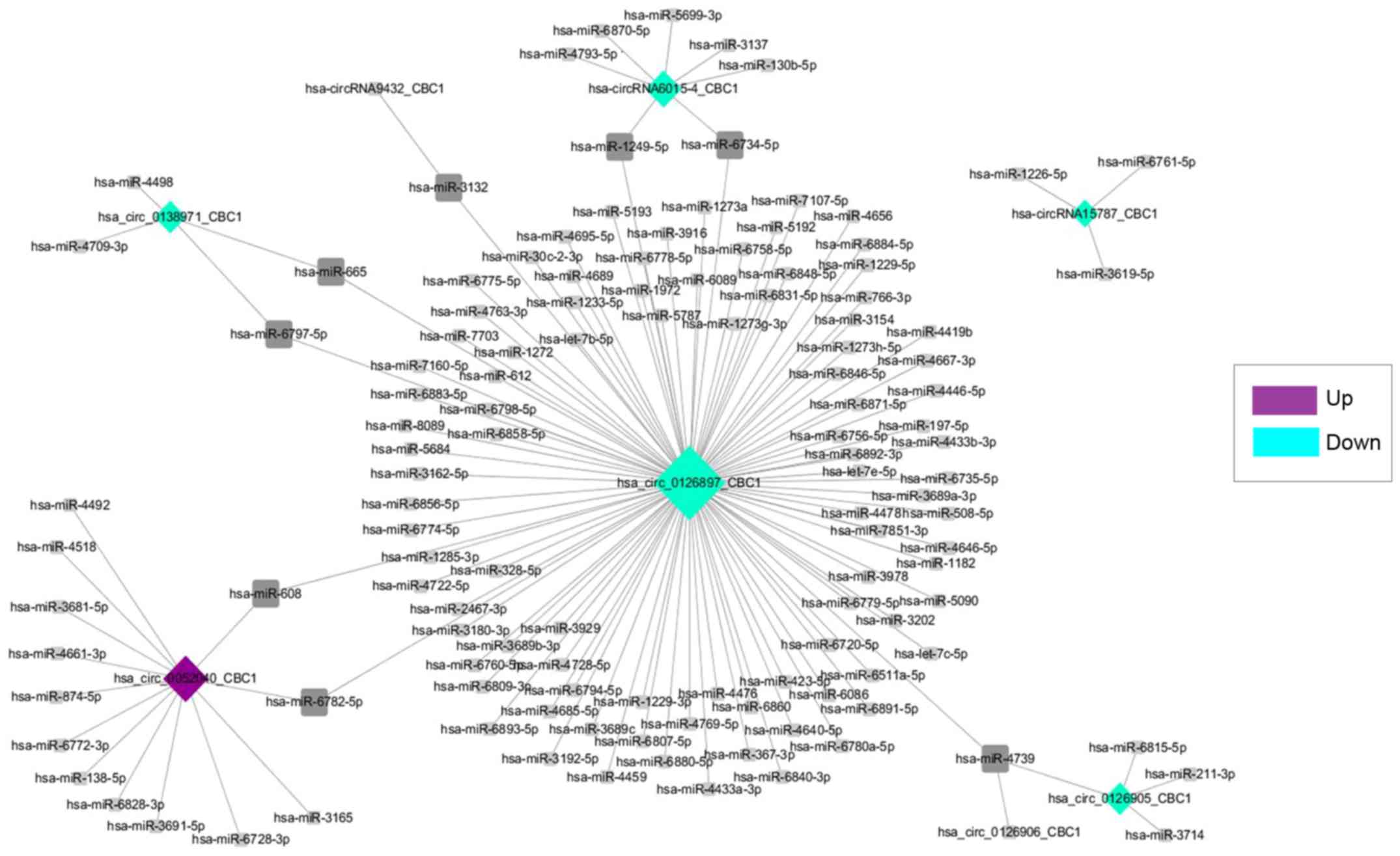
CircRNA/miRNA network
The related target miRNAs for the differentially
expressed circRNAs were predicted. Then, the eight key
differentially expressed circRNAs, which were associated with the
most target miRNAs were chosen. They were hsa_circ_0126897_CBC1,
hsa_circ_0052040_CBC1, hsa−circRNA6015−4_CBC1,
hsa_circ_0126905_CBC1, hsa−circRNA15787_CBC1,
hsa_circ_0138971_CBC1, hsa_circ_0126905_CBC1 and
hsa−circRNA9432_CBC1, and were involved in 133 circRNA/miRNA
pairs. Finally, the circRNA/miRNA network was established, and is
presented in Fig. 5. In the
network, hsa_circ_0126897_CBC1 was involved in the most
pairs, which was significantly downregulated in the CRC samples
compared with their adjacent tissue samples. In addition, the
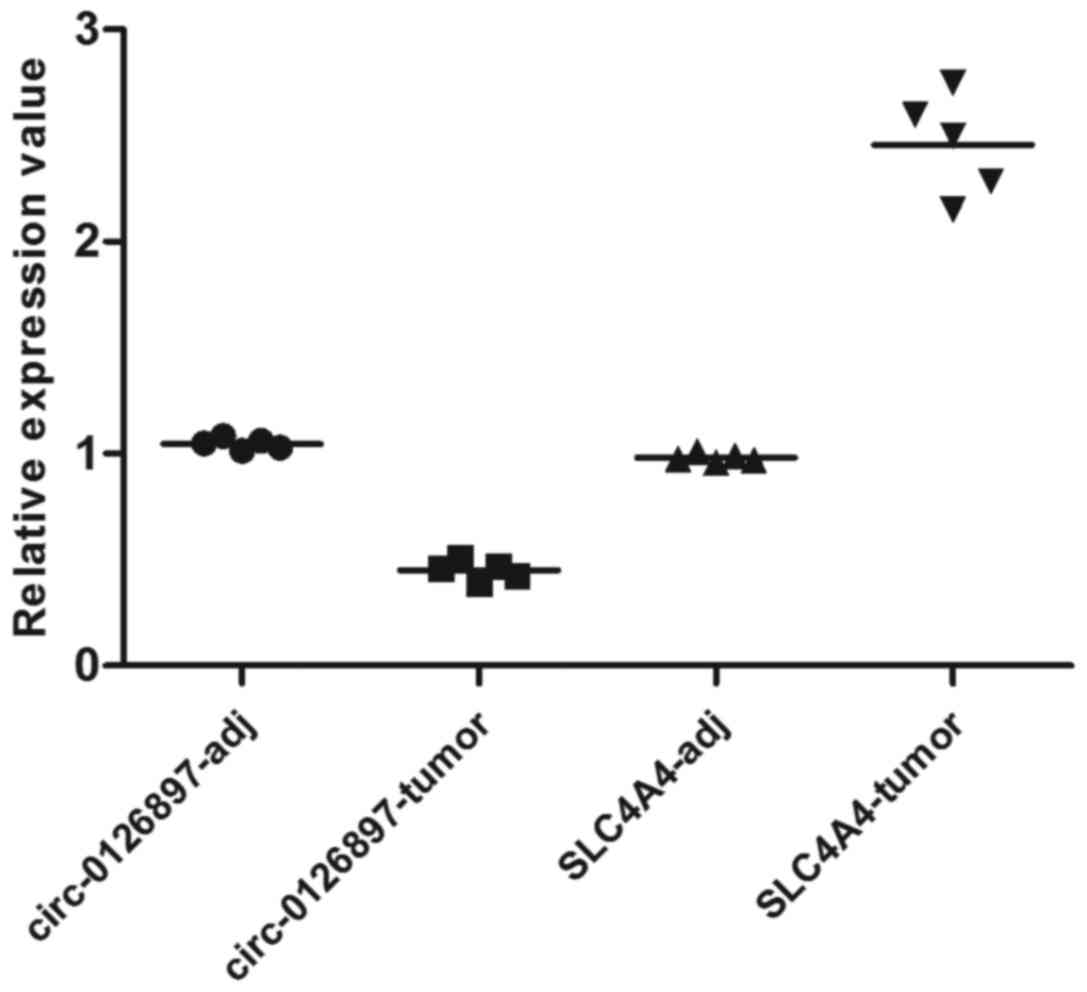
scatter plot figure of hsa_circ_0126897_CBC1 and its target
gene (SLC4A4) is presented in Fig. 6. The mRNA level of
hsa_circ_0126897_CBC1 was significantly decreased in the CRC
samples compared to their adjacent tissue samples (P<0.0001,
t=24.70). Moreover, the expression of target gene (SLC4A4)
was also decreased in the CRC samples (P<0.0001, t=13.72).
Discussion
circRNAs can act as competing endogenous RNAs or
miRNA sponges, regulating alternative splicing or transcription,
modulating the expression of the parental gene, managing the local
concentration of RBPs and playing an important role in oncogenesis
and the malignant behavior of cancer (8). Recent studies have reported that a
global reduction of circRNA abundance was observed in CRC samples
compared with healthy tissue, therefore allowing for the
proliferation of CRC cells (9,10). In
the present study, we not only found many downregulated circRNAs
but also many upregulated ones. However, the related molecular
mechanism was complex and associated with mRNA, miRNAs, protein
translation, and regulation of some GO terms and pathways.
Therefore, the related mRNAs and miRNAs were screened.
The enriched GO term results revealed that the top 3
BB terms were cell cycle, mitotic cell cycle and cell cycle process
(Fig. 3A), which were BPs
associated with the cell cycle. The cell cycle is a highly
organized process regulated by cyclins, cyclin-dependent kinases
(CDKs), and cyclin-dependent kinase inhibitors. Recent studies
suggested that altered expression of cyclins may be involved in
human cancer proliferation and development (11,12).
CRC could be controlled by intervening in the cell cycle, such as
targeting cyclin G (13). At
present, only a few studies have reported that circRNAs were
involved in the BPs of the cell cycle. A recent study demonstrated
that the circRNA circ-Foxo3 was highly expressed in
non-cancer cells, and suppressed cell cycle progression by binding
to CDK1 and CDK2 (14). Another
study concluded that circ100284, regulated EZH2 via
miR-217, and was involved in the arsenite-accelerated cell
cycle of human keratinocytes in carcinogenesis (15). Therefore, we suspected that circRNAs
may regulate some mRNAs, which were associated with the cell cycle,
thus having an effect on CRC. Moreover, the top 2 reactome pathways
were ‘cell cycle, mitotic’ and ‘cell cycle’ (Fig. 4A), and the most significant enriched
KEGG pathway was ‘cell cycle’ (Fig.
4B). These results further demonstrated the aforementioned
views, and that the cell cycle was closely associated with the
occurrence and development of CRC.
Upon completion of this study, the circRNA/miRNA
network was constructed, and hsa_circ_0126897_CBC1 was the
node involved in the most circRNA/miRNA pairs (Fig. 5). Currently, some circRNAs have been
identified to be related to the occurrence and development of CRC,
such as hsa_circ_001988, hsa_circ_001569 and
hsa_circ_0000069 (1). More
circRNAs were considered to be biomarkers for some diseases. Qin
et al (16) suspected that
hsa_circ_0001649 may serve as a novel potential biomarker
for HCC and function in the tumorigenesis and metastasis of HCC.
Peripheral blood circRNA hsa_circ_0124644 can be used as a
diagnostic biomarker of coronary artery disease (17). hsa_circRNA_103636 was a
potential novel diagnostic and therapeutic biomarker in major
depressive disorders (18).
circPVT1 may be a proliferative factor and prognostic marker in
gastric cancer (19).
hsa_circ_0005075 functioned as a potential biomarker in
hepatocellular carcinoma development (20). However, the study of circRNAs is
lacking basic research and is not as developed as mRNA and miRNA
reseach. At present, no study has been retrieved, reporting the
functions of hsa_circ_0126897_CBC1. Furthermore,
SLC4A4 is the target gene of hsa_circ_0126897_CBC1,
and a Na(+)/HCO3(−) co-transporter. Parks and Pouyssegur
reported that SLC4A4 knockdown had a strong impact on cell
proliferation, migration, and invasion of CRC and breast cancer
cells, and also altered the expression of other proteins including
CAIX (21). Hence, their results
indicated that SLC4A4 contributes to cell migration and
tumor cell phenotype. Gerber et al (22) revealed that SLC4A4 was
significantly upregulated in chronic myeloid leukemia (CML) cells
and persistent leukemia stem cells, which may be used to develop
novel therapies for CML. In the present study, we found that the
expression of hsa_circ_0126897_CBC1 and its target gene were
significantly decreased in CRC tissue (Fig. 6), and that this circRNA participated
in many regulated processes of miRNAs. Hence, it was suspected that
hsa_circ_0126897_CBC1 was a potential biomarker for CRC. In
future, the design of related studies to research the function of
hsa_circ_0126897_CBC1 in CRC cells and in animal models will
be undertaken, to elucidate the related mechanism involved.
In conclusion, hsa_circ_0126897_CBC1 may be a
potential biomarker for CRC, and circRNAs could affect the BPs or
pathways of the cell cycle thus influencing the occurrence and
development of CRC. In addition, fundamental research should focus
on circRNAs in the future.
Acknowledgements
We would like to thank all the members of our
research group for their enthusiastic participation in this
study.
Funding
No funding was received.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
XZ designed the experiments. SC and LZ performed
data analysis. YS interpretated the data. SC, LZ and YS edited the
manuscript. XZ reviewed and edited the manuscript. All authors read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
The experimental study was approved by the Ethics
Committee of Nankai University Affiliated Hospital, and the
informed consent forms were obtained when the patients were
accepted for the study by the hospital. All procedures performed in
the study involving human participants were in accordance with the
ethical standards of the Institutional and/or National Research
Committee and with the 1964 Helsinki declaration and its later
amendments or comparable ethical standards.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Song M, Garrett WS and Chan AT: Nutrients,
foods, and colorectal cancer prevention. Gastroenterology.
148:1244–60.e16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD, Fedewa SA, Ahnen DJ,
Meester RGS, Barzi A and Jemal A: Colorectal cancer statistics,
2017. CA Cancer J Clin. 67:177–193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mäkinen MJ: Colorectal serrated
adenocarcinoma. Histopathology. 50:131–150. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wilusz JE and Sharp PA: Molecular biology.
A circuitous route to noncoding RNA. Science. 340:440–441. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jeck WR, Sorrentino JA, Wang K, Slevin MK,
Burd CE, Liu J, Marzluff WF and Sharpless NE: Circular RNAs are
abundant, conserved, and associated with ALU repeats. RNA.
19:141–157. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ding XC, Weiler J and Grosshans H:
Regulating the regulators: Mechanisms controlling the maturation of
microRNAs. Trends Biotechnol. 27:27–36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Taborda MI, Ramírez S and Bernal G: MI T:
Circular RNAs in colorectal cancer: Possible roles in regulation of
cancer cells. World J Gastrointest Oncol. 9:62–69. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arvelo F, Sojo F and Cotte C: Biology of
colorectal cancer. Ecancermedicalscience. 9:5202015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bachmayr-Heyda A, Reiner AT, Auer K,
Sukhbaatar N, Aust S, Bachleitner-Hofmann T, Mesteri I, Grunt TW,
Zeillinger R and Pils D: Correlation of circular RNA abundance with
proliferation - exemplified with colorectal and ovarian cancer,
idiopathic lung fibrosis, and normal human tissues. Sci Rep.
5:80572015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gillett CE and Barnes DM: Demystified …
cell cycle. Mol Pathol. 51:310–316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartek J and Lukas J: Cell cycle. Order
from destruction. Science. 294:66–67. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Perez R, Wu N, Klipfel AA and Beart RW Jr:
A better cell cycle target for gene therapy of colorectal cancer:
Cyclin G. J Gastrointest Surg. 7:884–889. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Du WW, Yang W, Liu E, Yang Z, Dhaliwal P
and Yang BB: Foxo3 circular RNA retards cell cycle progression via
forming ternary complexes with p21 and CDK2. Nucleic Acids Res.
44:2846–2858. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xue J, Liu Y, Luo F, Lu X, Xu H, Liu X, Lu
L, Yang Q, Chen C, Fan W, et al: Circ100284, via miR-217 regulation
of EZH2, is involved in the arsenite-accelerated cell cycle of
human keratinocytes in carcinogenesis. Biochim Biophys Acta.
1863:753–763. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qin M, Liu G, Huo X, Tao X, Sun X, Ge Z,
Yang J, Fan J, Liu L and Qin W: hsa_circ_0001649: A circular RNA
and potential novel biomarker for hepatocellular carcinoma. Cancer
Biomark. 16:161–169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao Z, Li X, Gao C, Jian D, Hao P, Rao L
and Li M: Peripheral blood circular RNA hsa_circ_0124644 can be
used as a diagnostic biomarker of coronary artery disease. Sci Rep.
7:399182017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cui X, Niu W, Kong L, He M, Jiang K, Chen
S, Zhong A, Li W, Lu J and Zhang L: hsa_circRNA_103636: Potential
novel diagnostic and therapeutic biomarker in Major depressive
disorder. Biomarkers Med. 10:943–952. 2016. View Article : Google Scholar
|
|
19
|
Chen J, Li Y, Zheng Q, Bao C, He J, Chen
B, Lyu D, Zheng B, Xu Y, Long Z, et al: Circular RNA profile
identifies circPVT1 as a proliferative factor and prognostic marker
in gastric cancer. Cancer Lett. 388:208–219. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shang X, Li G, Liu H, Li T, Liu J, Zhao Q
and Wang C: Comprehensive circular RNA profiling reveals that
hsa_circ_0005075, a new circular RNA biomarker, is involved in
hepatocellular carcinoma development. Medicine (Baltimore).
95:e38112016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Parks SK and Pouyssegur J: The
Na(+)/HCO3(−) Co-transporter SLC4A4 Pplays a role in
growth and migration of colon and breast cancer cells. J Cell
Physiol. 230:1954–1963. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gerber JM, Gucwa JL, Esopi D, Gurel M,
Haffner MC, Vala M, Nelson WG, Jones RJ and Yegnasubramanian S:
Genome-wide comparison of the transcriptomes of highly enriched
normal and chronic myeloid leukemia stem and progenitor cell
populations. Oncotarget. 4:715–728. 2013. View Article : Google Scholar : PubMed/NCBI
|