Introduction
X-linked juvenile retinoschisis (XLRS, phenotype MIM
312700), first described in 1898 by Joseph Haas, represents the
leading cause of early macular degeneration in males (1,2). Its
prevalence ranges from 1:5,000 to 1:25,000 (3). Bilateral splitting of the inner retina is
the most common clinical finding and has been reported in 68–100%
of affected males. In ophthalmoscopy, the macula presents stellate
spoke-wheel pattern. Female carriers rarely present with retinal
abnormalities (4). As the disease
progresses, retinal cysts often coalesce with subsequent retinal
flattening and macular atrophy in older patients. Peripheral
schisis, usually located in the inferotemporal retina, is evident
in ~50% of the affected individuals (5). Although this condition is considered to
be congenital, symptoms generally present in the first decade of
life with visual failure, squint or nystagmus. Visual damage
usually progresses in the first two decades of life and remains
approximately stationary until the fifth or sixth decade, when the
development of macular atrophy induces additional visual loss
(3).
Patients with XLRS present a characteristic pattern
on the electroretinogram (ERG). A reduction in the amplitude of the
b-wave (generated by the activity of depolarising bipolar cells)
and a relative preservation of the negative a-wave (generated by
photoreceptors) give rise to, the so-called electronegative ERG
(6). The absence of negative ERG does
not necessarily exclude the presence of the pathology: in a series
of 24 XLRS patients, only 56.5% presented with typical negative ERG
(7).
Optical coherence tomography (OCT) is a key
diagnostic test for XLRS. Schisis seems to occur predominantly at
the inner nuclear layer (INL), occasionally at the outer nuclear
layer/outer plexiform layer (ONL/OPL), and only rarely at the
retinal nerve fiber layer (RNFL). The schisis cavity may extend
beyond the retinal vascular arcades (8).
The gene responsible for XLRS, retinoschisin 1
(RS1), was identified by Sauer et al (2) in 1997. The RS1 gene, on chromosome Xp22,
encodes for retinoschisin, a discoidin-domain containing protein,
which is secreted by photoreceptors and bipolar cells as a
homo-oligomeric complex (2,9,10).
Retinoschisin complex binds tightly to the surface of
photoreceptors and bipolar cells, contributing to maintain the
structural organization of the retina and of the
photoreceptor-bipolar synapse (4). In
people with XLRS, several missense and nonsense mutations,
insertion and deletion mutations, intragenic deletions and splice
site mutations have been identified (11).
The aim of the present study is to describe clinical
features of a family affected by the novel missense mutation I212N
of the RS gene and to provide a detailed follow-up based on
spectral domain optical coherence tomography (SD-OCT).
Materials and methods
Patients
The present study included a Caucasian Italian
family with two brothers clinically affected by XLRS. Both patients
underwent a complete ophthalmological assessment and were followed
up for ten years, including best-corrected visual acuity (BCVA) and
fundus examination. Detailed multimodal retinal imaging (fundus
photography, OCT, fundus autofluorescence (FAF) and retinal
angiography) was collected. Moreover, retinal function was
evaluated with ERG and Goldmann perimetry. Clinical and genetic
testing was extended to other family members. The current study was
conducted in accordance with The Declaration of Helsinki.
XLRS1 gene analysis
Written informed consent for genetic analysis was
obtained from the all subjects. Whole blood samples were then
collected. DNA was extracted using the Qiagen Biorobot DNA
extraction kit (Qiagen, Inc., Valencia, CA, USA) according to
manufacturer's instructions and quantified by Nanodrop spectral
analysis (Thermo Fisher Scientific, Inc., Waltham, MA, USA). DNA
fragmentation and degradation were evaluated by standard agarose
gel electrophoresis. A total of 100 ng DNA were amplified by
standard polymerase chain reaction (PCR) procedures with a PCR
mixture containing 2.5 µl 10X concentrated PCR buffer (Solis
BioDyne, Tartu, Estonia), 0.7 µl 50 mM MgCl2 (Solis
BioDyne), 0.75 µl 10 mM deoxyribonucleotide triphosphates (Solis
BioDyne), 0.3 µl 100 µM forward primer and 0.3 µl of 100 µM reverse
primer (primer sequences are listed in Table I) (Integrated DNA Technologies,
Coralville, IA, USA) and 0.5 µl 5 U/µl Hot Start DNA Polymerase
(Solis BioDyne). Thermocycling consisted of one cycle of enzyme
activation (at 95°C for 15 min), followed by 35 cycles of DNA
amplification (at 95°C for 45 sec, at 59°C for 45 sec and at 72°C
for 1 min). PCR products were then separated by agarose gel
electrophoresis (1.5% agarose gel in tris-borate-EDTA;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and purified with
Invisorb spin columns (Invitek, Inc., Hayward, CA, USA).
PCR-purified products were re-amplified with terminating
nucleotides using Big Dye Terminator v3.1 (Applied Biosystems;
Thermo Fisher Scientific, Inc.). Sequencing analysis was performed
with an ABI Prism 3100 Avant automated sequencer (Thermo Fisher
Scientific, Inc.) equipped with 36 cm capillary array filled with
POP6 polymer (Thermo Fisher Scientific, Inc.). Electropherograms
were analyzed using Sequencing Analysis software (Applied
Biosystems, version 5.1; Thermo Fisher Scientific, Inc.).
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Primer | Sequence (5′-3′) |
|---|
| RS1_exon 1F |
GGAAAGCCATCCACACAAAG |
| RS1_exon 1R |
GGTTAACTTGATGGGGCTCA |
| RS1_exon 2F |
TCCTGACCTCAAGTGATCTGC |
| RS1_exon 2R |
TTCTTCCAGAAGGGGTGTTG |
| RS1_exon 3F |
GGAGAAAACCCGCATTAACA |
| RS1_exon 3R |
GACGATGCATAAGGACTGAGTG |
| RS1_exon 4F |
CCACCACGCCAGTTAATTTT |
| RS1_exon 4R |
GCAAAGCAGATGGGTTTGTT |
| RS1_exon 5F |
ACAGAGGGCAGTGACAGGAG |
| RS1_exon 5R |
GGAGACAAGGCTCAGACTGC |
| RS1_exon 6F |
ACCCAGCACTGCAGTTACAA |
| RS1_exon 6R |
GGGCTAGCTCCAGAAAGGAA |
Results
Patients
Three members of a family with two brothers affected
by XLRS were studied. The first proband was a 41-year-old male and
complained of mild visual disturbance since the age of 9, where
medical records reported a spoke-wheel appearance of the macula and
a BCVA of 0.8. His medical history was unremarkable. At the first
presentation, BCVA was 0.4 in oculus uterque and remained stable
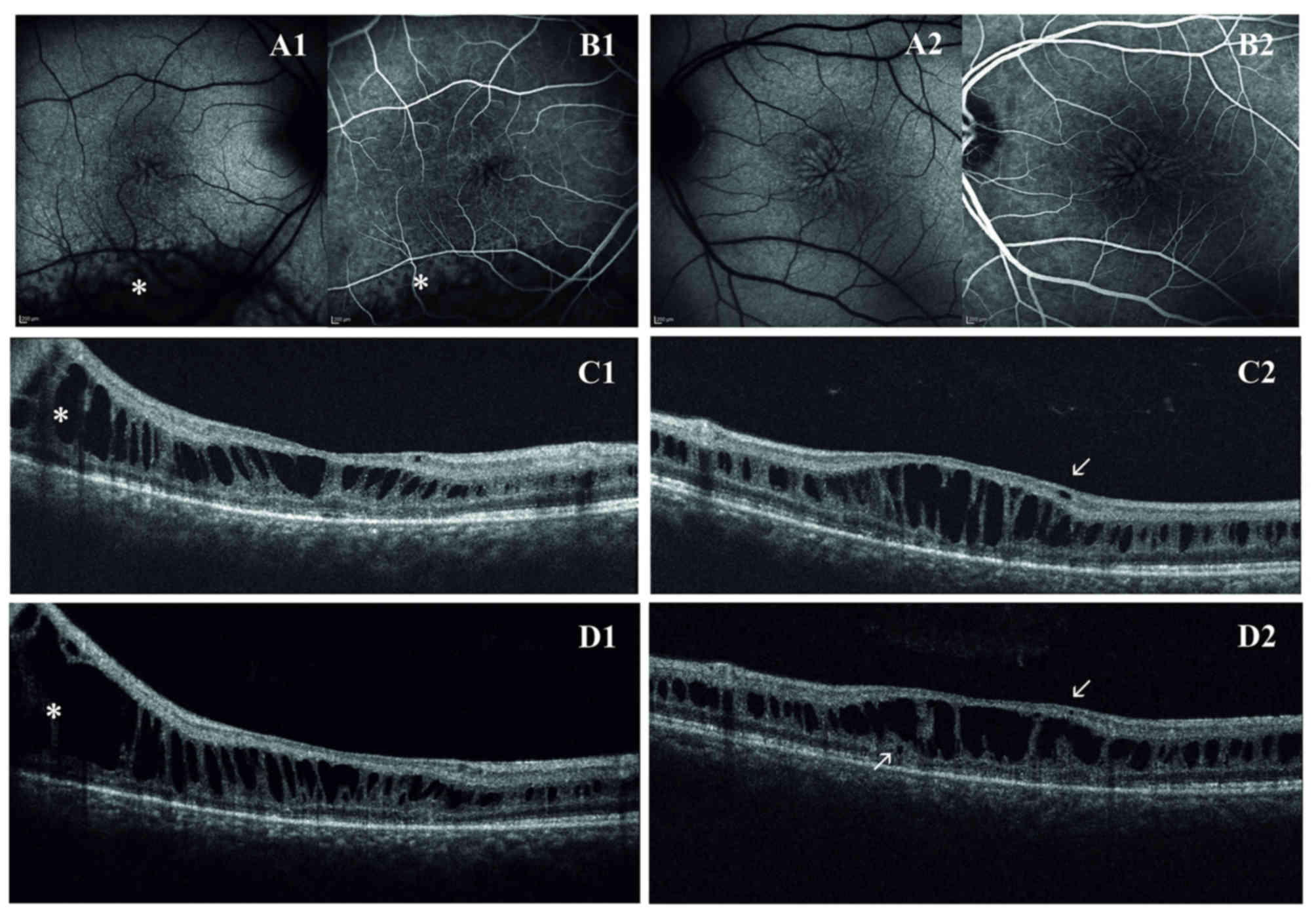
for all the follow-up (2007 through to 2016). Fundus examination
reported the typical spoke wheel appearance of the macula in both
eyes. In the right eye, a peripheral schisis in the
inferior-temporal retina reaching the vascular arcade was also
observed. Macular abnormalities were best recognized with FAF that
indicated hyper- and hypo-FAF arranged in a radial pattern.
Fluorescein angiography did not reveal any late leakage and
confirmed the non-exudative origin of macular cysts.
On first examination with OCT, macular thickness was
significantly increased. OCT scans presented a cystic degeneration,
primarily involving the INL, though some small cysts were detected
in the outer plexiform layer (OPL) and in the ganglion cell layer
(GCL). In RE, peripheral scans documented retinal schisis extending
inferiorly. The first OCT was acquired when the patient was ~31
years old and BCVA was already reduced. In the right eye, the
ten-year follow-up demonstrated that there were no noticeable
variations in retinal morphology or in the overall width of the
peripheral schisis. However, the progressive straining of retinal
layers breaks up the wall of macular cysts, weakening the retinal
structure. Indeed, in the left eye, there was a moderate reduction
in central retinal thickness (from 448 µm to 402 µm) that precedes
the late, atrophic stage of XLRS. Outer retinal layers (external
limiting membrane, inner segment/outer segment junction and
photoreceptor layer) demonstrated diffuse atrophic changes with no
significant progression during the follow-up (Fig. 1). Goldmann perimetry indicated in RE an
absolute scotoma superiorly, that matched the peripheral retinal
schisis. The dark-adapted 0.01 ERG (rod response) was reduced. The
dark-adapted 3.0 ERG (combined rod-cone response) showed a b/a
ratio <1, which matched the definition of an electronegative
ERG. The patient reported a delay in implicit time and a decrease
in the amplitude of the b-wave in the light adapted 3.0 ERG. A
delayed light adapted 30 Hz flicker peak time and decreased
amplitude was also present. The diagnosis of XLRS was proposed and
all family members were then invited for eye examination. His
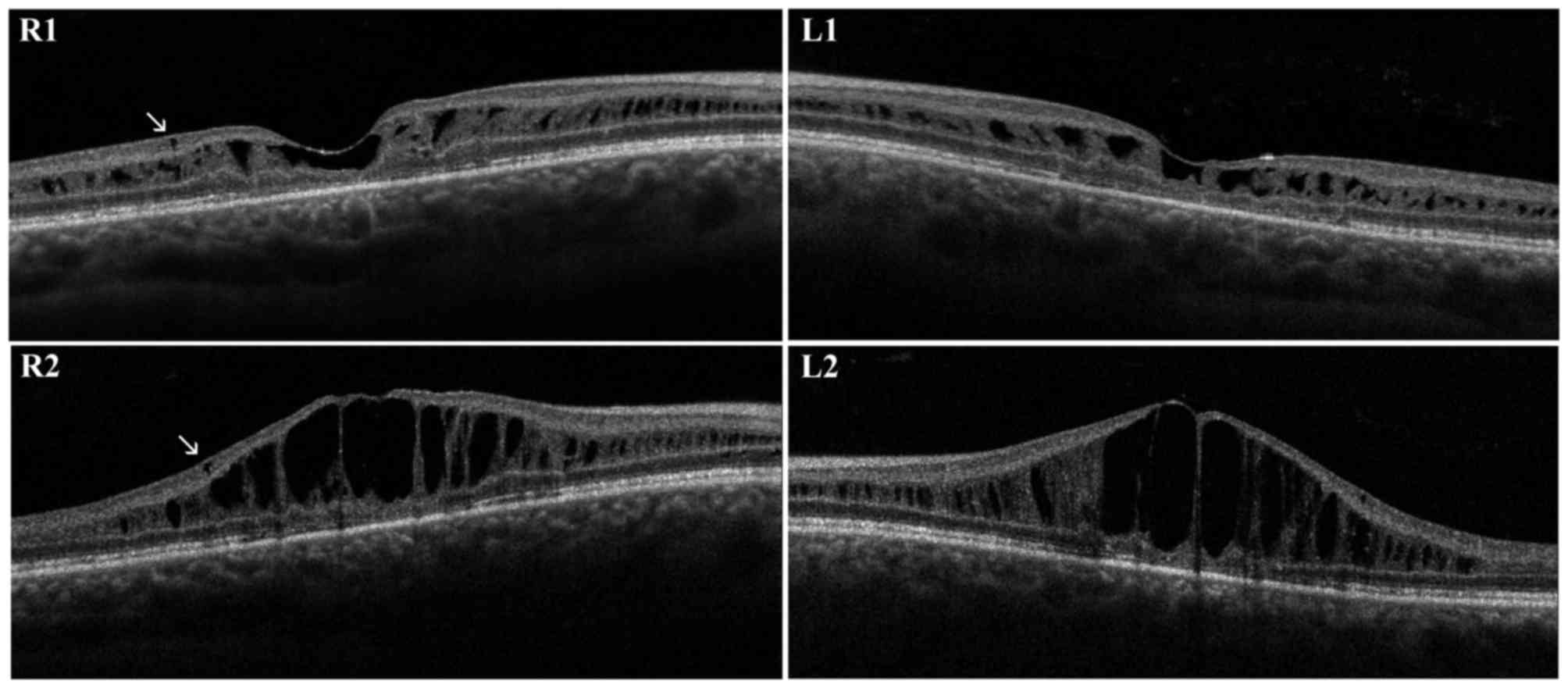
brother was a 30-year-old male. The diagnosis of unspecified
retinal dystrophy was made when he was 8 years old and he presented
with a initial visual loss associated with characteristic spoke
wheel appearance of the macula. At the examination, BCVA was 0.5 in
RE and 0.6 in LE and progressively decreased to 0.4 in RE and 0.3
in LE during follow-up. Fundus examination, OCT and retinal
angiography revealed the typical features of XLRS. An
electronegative ERG further supported the diagnosis. First OCT
scans presented irregular empty spaces in the INL. Some small cysts
could be observed in the GCL. Atrophic changes involving outer
retinal layers (external limiting membrane, inner segment/outer
segment junction and photoreceptor layer) were recognizable. During
the follow-up, the OCT scans revealed a progressive enlargement and
coalescence of the cysts located in the INL, with subsequent
retinal thickening. Small cysts in the GCL did not show further
enlargement. Atrophic changes of outer retinal layers remained
stable during follow-up (Fig. 2).
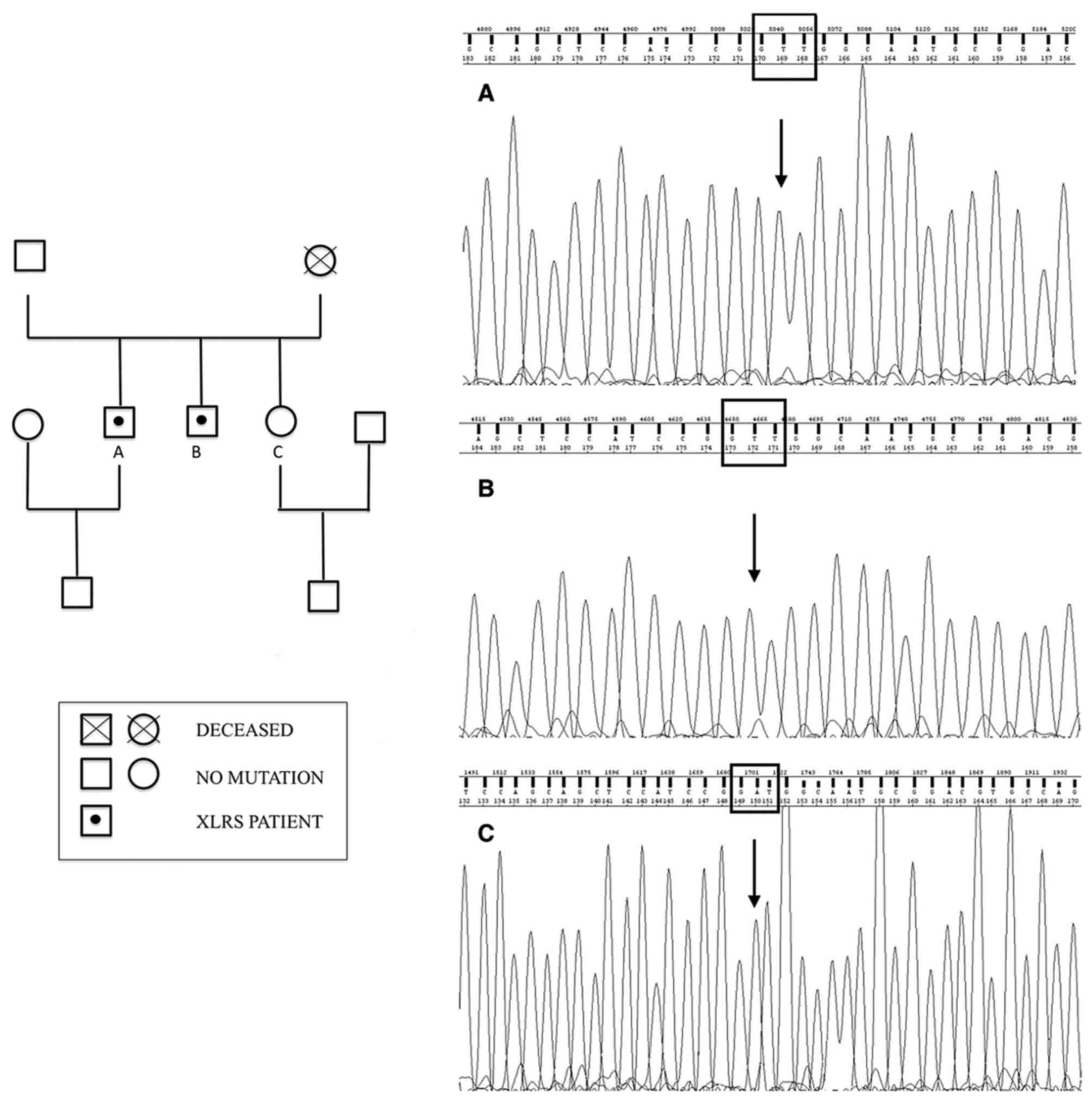
Ophthalmologic examination of their sister (21 years
old) and of their father (69 years old) was normal. Their mother
was deceased, but no significant visual disturbances were reported,
even though it was not possible to ascertain if she was a healthy
carrier or if she developed a de novo mutation. Indeed, ocular
diseases in their maternal grandfather were not reported. Then, the
authors drew a genealogical tree based on available information
(Fig. 3).
Genetic testing
Screening of RS1 by sequencing of PCR-amplified DNA
identified a novel missense mutation in exon 6 in both XLRS
patients. This mutation from A to T at nucleotide position 635
changes the nonpolar isoleucine to positively charged asparagine
(p.Ile212Asn).
This substitution was predicted to be harmful to
protein function by in silico analysis (Polyphen2=0.989;
Fig. 3).
Discussion
Mutations in the RS1 gene are responsible for
inherited and sporadic XLRS. To date 191 causative genetic
variations have been identified. Among all described variations,
missense mutations represent the most recurrent subtype (100
mutations of 191). Mutations can affect all regions of the RS gene,
although a substantial clustering is observed within the region
coding for the discoidin domain (85 of 191) (4). The novel missense mutation that we found
in the patients is capable to produce the typical clinical
phenotype of XLRS, as it affects the discoidin domain of
retinoschisin.
Retinoschisin is a 24-kDa protein expressed
exclusively by photoreceptors and bipolar cells in the retina and
pineal gland. RS1 is secreted as a soluble disulfide
bond-stabilized octamer. Most of the monomer (157 amino acids)
comprises a discoidin domain, a globular fold that is highly
conserved in a family of extracellular or transmembrane proteins
implicated in cell adhesion or cell-cell interactions. The high
number of mutations within the discoidin domain indicates that it
is essential for the normal function of this protein. Although the
role of retinoschisin in the retina is not well understood, it has
been hypothesized that it works as a cell adhesion protein to
maintain the structural organization of the retina and of the
photoreceptor-bipolar synapse (4,5,10,12).
Differential diagnosis of XLRS comprises cystic
changes of the macula that could arise from exudative and
degenerative disorders or abnormalities of the vitreoretinal
interface. Degenerative cystoid maculopathies may be hereditary
[XLRS, Enhanced S-Cone/Goldmann Favre Syndrome (ESCS/GFS)] or
acquired [microcystic macular edema (MME)]. All these conditions do
not show late leakage on fluorescein angiography.
ESCS/GFS is a recessive disorder caused by mutations
in the NR2E3 gene. This gene encodes for a nuclear receptor
expressed in the outer nuclear layer of the retina. It suppresses
cone differentiation during embryogenesis, therefore loss of NR2E3
results in retinas with a decreased number of rod photoreceptors
and an increase in cones, predominantly expressing the S-cone opsin
(13). Time trend of macular cysts
development in ESCS/GFS resemble what happens in XLRS. The
progressive enlargement and coalescence of macular cysts in young
adulthood is followed by resolution of the schisis and reduction of
macular thickness (14). However,
location of cystic spaces seems different from XLRS. The analysis
of published images shows large confluent cysts in the outer
retinal layer associated with small and well-demarcated cysts in
the INL (15,16).
MME is a recently described OCT entity,
characterized by the appearance of small retinal cysts in patients
with optic neuritis and optic atrophy of various aetiology
(multiple sclerosis, neuromielitis optica, glaucoma) probably due
to retrograde synaptic degeneration. As in XLRS, MME cysts involve
the INL, however they do not seem to enlarge significantly with
time. Moreover in MME cysts are usually circumscribed to the
parafoveal region (17,18), whereas in XLRS cystic degeneration may
extend beyond the vascular arcades.
Diagnosis of XLRS in the patients of the present
study was strongly supported by clinical and instrumental findings.
Indeed both patients presented with the classical phenotype of
XLRS, with spoke-wheel maculopathy, association with peripheral
schisis and ERG b-wave suppression. Ophthalmological history,
regarding age of onset of symptoms and visual decline follows the
typical course described in literature (7). Visual symptoms begin in the first decade,
and then visual function progressively decreases, remaining stable
in the following 10 years.
SD-OCT images collected in the two patients
documented the natural history of the disease, characterized by the
formation of small cysts located in the INL that progressively
enlarge and coalesce. Gradually, the stretching of the wall of
macular cysts weakens the retinal structure leading to the collapse
of the cysts. Small isolated cysts were also observed in the GCL
and OPL that, differently from those located in the INL, do not
seem to increase over time. The possible presence of small cysts
not determining macular splitting have been previously described by
Gregori et al (19), who also
found similar empty spaces in the ONL and in the GCL. The same
paper reported that macular schisis may occur also in the OPL
(19). An immunochemistry study
(20) in the normal mouse eye detected
retinoschisin protein in all retinal layers; this may explain why
retinal schisis can develop in different layers, even if some
layers (GCL and RNFL) seem relatively resistant. The foveomacular
retinoschisis is usually located in the INL, while the extramacular
schisis could be equally found in the INL, ONL and GCL/NFL
(21). In the present study, cystic
degeneration at the level of peripheral schisis in RE of the first
proband was located in the INL (Fig.
1).
Since clinical and instrumental findings were highly
suggestive for XLRS, the authors performed genetic analysis. The
sequencing of the proband DNA, revealed the hemizygous missense
mutation p.lle212Asn (c.635A>T) in the RS1 gene. The same
mutation was identified in the proband's brother and has never been
reported before. It consists in a substitution of the non-polar
amino acid, isoleucine, with the polar amino acid, asparagine. The
mutation is located in a domain in which several genetic
alterations associated with XLRS has been found (positions p.206,
p.209, p.211, p.213, p.2015 and p.219). Furthermore, the PolyPhen-2
predictor has given to this mutation a high likelihood of protein
dysfunction.
Given the high number of RS1 mutations, several
studies investigated a possible correlation between genotype and
phenotype. Even though some reports describe a correlation between
specific variations and the severity of clinical phenotype
(22,23), the majority of published studies found
that XLRS patients had relatively uniform clinical manifestations,
although with great intra-familial variation in age at onset and
progression (1,24–26).
Interestingly, the deletion of exon 1 and promoter
region, which causes complete absence of retinoschisin, is
associated with a wide variability of the phenotype, suggesting
that other genetic and/or epigenetic factors are likely to act as
significant phenotypic modifiers in XLRS (27,28).
The absence of leakage on fluorescein angiography in
XLRS patients suggests that vascular hyperpermeability serves a
minor role, if any, in the pathogenesis of the disease. However,
both oral and topical carbonic anhydrase inhibitors (CAIs) have
been used successfully in the management of XLRS. CAIs act both on
retinal and RPE cell function by acidifying the subretinal space,
decreasing the standing potential as well as raising retinal
adhesiveness, probably by increasing RPE fluid transport (29).
A promising approach for XLRS treatment is gene
therapy. The disease is an excellent candidate for gene therapy as
the majority of mutations have been shown to lead to a complete
deficiency of the secreted protein in the retinal structures.
Genetic treatment studies, conducted in rabbits and mouse, with
intravitreal administration of adeno-associated viral vector coding
the human RS1 gene demonstrated an improvement in retinal structure
and function (30,31). Other strategies comprise the
intravitreal administration of adipose-derived mesenchymal stem
cells, genetically modified to secrete the human RS1 gene, or solid
lipid nanoparticles inducing the expression of retinoschisin in
photoreceptors. Both treatments demonstrated morphological and
functional improvements in mouse models (32,33). Two
phase I/II trials are testing safety and efficacy of
adeno-associated viral vectors, called AAV8-scRS/IRBPhRS and
rAAV2tYF-CB-hRS1, as a gene therapy approach in XLRS patients.
Viral vectors have been demonstrated to be able to shuttle normal
RS1 DNA into retinal cells. Results are expected from 2017
(34,35).
The present study describes long-term morphological
and functional changes of XLRS patients affected by a novel RS1
mutation. Although the correspondence between genotype and
phenotype is still under debate, is reasonable that siblings
affected by XLRS could share other genetic and/or epigenetic
factors capable to influence clinical course of the disease and
perhaps treatment response to upcoming genetic therapies.
References
|
1
|
The Retinoschisis Consortium: Functional
implications of the spectrum of mutations found in 234 cases with
X-linked juvenile retinoschisis. Hum Mol Genet. 7:1185–1192. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sauer CG, Gehrig A, Warneke-Wittstock R,
Marquardt A, Ewing CC, Gibson A, Lorenz B, Jurklies B and Weber BH:
Positional cloning of the gene associated with X-linked juvenile
retinoschisis. Nat Genet. 17:164–170. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mooy CM, Van Den Born LI, Baarsma S,
Paridaens DA, Kraaijenbrink T, Bergen A and Weber BH: Hereditary
X-linked juvenile retinoschisis: A review of the role of Müller
cells. Arch Ophthalmol. 120:979–984. 2002.PubMed/NCBI
|
|
4
|
Molday RS, Kellner U and Weber BH:
X-linked juvenile retinoschisis: Clinical diagnosis, genetic
analysis, and molecular mechanisms. Prog Retin Eye Res. 31:195–212.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tantri A, Vrabec TR, Cu-Unjieng A, Frost
A, Annesley WH Jr and Donoso LA: X-linked retinoschisis: A clinical
and molecular genetic review. Surv Ophthalmol. 49:214–230. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sikkink SK, Biswas S, Parry NRA, Stanga PE
and Trump D: X-linked retinoschisis: An update. J Med Genet.
44:225–232. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Renner AB, Kellner U, Fiebig B, Cropp E,
Foerster MH and Weber BH: ERG variability in X-linked congenital
retinoschisis patients with mutations in the RS1 gene and the
diagnostic importance of fundus autofluorescence and OCT. Doc
Ophthalmol. 116:97–109. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu J, Ni Y, Keane PA, Jiang C, Wang W and
Xu G: Foveomacular schisis in juvenile X-linked retinoschisis: An
optical coherence tomography study. Am J Ophthalmol.
149:973–978.e2. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang T, Waters CT, Rothman AMK, Jakins TJ,
Römisch K and Trump D: Intracellular retention of mutant
retinoschisin is the pathological mechanism underlying X-linked
retinoschisis. Hum Mol Genet. 11:3097–3105. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu WW, Wong JP, Kast J and Molday RS: RS1,
a discoidin domain-containing retinal cell adhesion protein
associated with X-linked retinoschisis, exists as a novel
disulfide-linked octamer. J Biol Chem. 280:10721–10730. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Molday LL, Hicks D, Sauer CG, Weber BHF
and Molday RS: Expression of X-linked retinoschisis protein RS1 in
photoreceptor and bipolar cells. Invest Ophthalmol Vis Sci.
42:816–825. 2001.PubMed/NCBI
|
|
12
|
Tolun G, Vijayasarathy C, Huang R, Zeng Y,
Li Y, Steven AC, Sieving PA and Heymann JB: Paired octamer rings of
retinoschisin suggest a junctional model for cell-cell adhesion in
the retina. Proc Natl Acad Sci USA. 113:5287–5292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yzer S, Barbazetto I, Allikmets R, van
Schooneveld MJ, Bergen A, Tsang SH, Jacobson SG and Yannuzzi LA:
Expanded clinical spectrum of enhanced S-cone syndrome. JAMA
Ophthalmol. 131:1324–1330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sohn EH, Chen FK, Rubin GS, Moore AT,
Webster AR and MacLaren RE: Macular function assessed by
microperimetry in patients with enhanced S-cone syndrome.
Ophthalmology. 117:1199–1206.e1. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bušić M, Bjeloš M, Bosnar D, Ramić S and
Bušić I: Cystoid macular lesions are resistant to topical
dorzolamide treatment in enhanced S-cone syndrome child. Doc
Ophthalmol. 132:67–73. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Salvatore S, Fishman GA and Genead MA:
Treatment of cystic macular lesions in hereditary retinal
dystrophies. Surv Ophthalmol. 58:560–584. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sigler EJ: Microcysts in the inner nuclear
layer, a nonspecific SD-OCT sign of cystoid macular edema. Invest
Ophthalmol Vis Sci. 55:3282–3284. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Murata N, Togano T, Miyamoto D, Ochiai S
and Fukuchi T: Clinical evaluation of microcystic macular edema in
patients with glaucoma. Eye (Lond). 30:1502–1508. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gregori NZ, Berrocal AM, Gregori G, Murray
TG, Knighton RW, Flynn HW Jr, Dubovy S, Puliafito CA and Rosenfeld
PJ: Macular spectral-domain optical coherence tomography in
patients with X linked retinoschisis. Br J Ophthalmol. 93:373–378.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Prenner JL, Capone A Jr, Ciaccia S, Takada
Y, Sieving PA and Trese MT: Congenital X-linked retinoschisis
classification system. Retina. 26 Suppl:S61–S64. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gregori NZ, Lam BL, Gregori G, Ranganathan
S, Stone EM, Morante A, Abukhalil F and Aroucha PR: Wide-field
spectral-domain optical coherence tomography in patients and
carriers of X-linked retinoschisis. Ophthalmology. 120:169–174.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Atchaneeyasakul LO, Trinavarat A,
Pituksung A, Jinda W, Thongnoppakhun W and Limwongse C: Mutations
in the XLRS1 gene in Thai families with X-linked juvenile
retinoschisis. Jpn J Ophthalmol. 54:89–93. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li X, Ma X and Tao Y: Clinical features of
X linked juvenile retinoschisis in Chinese families associated with
novel mutations in the RS1 gene. Mol Vis. 13:804–812.
2007.PubMed/NCBI
|
|
24
|
Kim SY, Ko HS, Yu YS, Hwang JM, Lee JJ,
Kim SY, Kim JY, Seong MW, Park KH and Park SS: Molecular genetic
characteristics of X-linked retinoschisis in Koreans. Mol Vis.
15:833–843. 2009.PubMed/NCBI
|
|
25
|
Lesch B, Szabó V, Kánya M, Somfai GM,
Vámos R, Varsányi B, Pámer Z, Knézy K, Salacz G, Janáky M, et al:
Clinical and genetic findings in Hungarian patients with X-linked
juvenile retinoschisis. Mol Vis. 14:2321–2332. 2008.PubMed/NCBI
|
|
26
|
Riveiro-Alvarez R, Trujillo-Tiebas MJ,
Gimenez-Pardo A, Garcia-Hoyos M, Lopez-Martinez MA, Aguirre-Lamban
J, Garcia-Sandoval B, Vazquez-Fernandez del Pozo S, Cantalapiedra
D, Avila-Fernandez A, et al: Correlation of genetic and clinical
findings in Spanish patients with X-linked juvenile retinoschisis.
Invest Ophthalmol Vis Sci. 50:4342–4350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eksandh LC, Ponjavic V, Ayyagari R,
Bingham EL, Hiriyanna KT, Andréasson S, Ehinger B and Sieving PA:
Phenotypic expression of juvenile X-linked retinoschisis in Swedish
families with different mutations in the XLRS1 gene. Arch
Ophthalmol. 118:1098–1104. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Iannaccone A, Mura M, Dyka FM, Ciccarelli
ML, Yashar BM, Ayyagari R, Jablonski MM and Molday RS: An unusual
X-linked retinoschisis phenotype and biochemical characterization
of the W112C RS1 mutation. Vision Res. 46:3845–3852. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Verbakel SK, van de Ven JP, Le Blanc LMP,
Groenewoud JMM, de Jong EK, Klevering BJ and Hoyng CB: Carbonic
anhydrase inhibitors for the treatment of cystic macular lesions in
children with X-linked juvenile retinoschisis. Invest Ophthalmol
Vis Sci. 57:5143–5147. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Marangoni D, Bush RA, Zeng Y, Wei LL,
Ziccardi L, Vijayasarathy C, Bartoe JT, Palyada K, Santos M,
Hiriyanna S, et al: Ocular and systemic safety of a recombinant
AAV8 vector for X-linked retinoschisis gene therapy: GLP studies in
rabbits and Rs1-KO mice. Mol Ther Methods Clin Dev. 5:160112016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zeng Y, Petralia RS, Vijayasarathy C, Wu
Z, Hiriyanna S, Song H, Wang YX, Sieving PA and Bush RA: Retinal
structure and gene therapy outcome in retinoschisin-deficient mice
assessed by spectral-domain optical coherence tomography. Invest
Ophthalmol Vis Sci. 57:OCT277–OCT287. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bashar AE, Metcalfe AL, Viringipurampeer
IA, Yanai A, Gregory-Evans CY and Gregory-Evans K: An ex vivo gene
therapy approach in X-linked retinoschisis. Mol Vis. 22:718–733.
2016.PubMed/NCBI
|
|
33
|
Apaolaza PS, Del Pozo-Rodríguez A, Solinís
MA, Rodríguez JM, Friedrich U, Torrecilla J, Weber BH and
Rodríguez-Gascón A: Structural recovery of the retina in a
retinoschisin-deficient mouse after gene replacement therapy by
solid lipid nanoparticles. Biomaterials. 90:40–49. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Study of RS1 ocular gene transfer for
X-linked retinoschisis. https://www.clinicaltrials.govDecember. 2016
|
|
35
|
Safety and efficacy of rAAV-hRS1 in
patients with X-linked retinoschisis (XLRS). https://www.clinicaltrials.govDecember. 2016
|