Introduction
Primary immunodeficiency disorders (PID), also termed as inborn errors of immunity (IEI), encompass a heterogeneous array of conditions characterized by compromised immune system function involving essential components, such as neutrophils, macrophages, dendritic cells (DC), complement proteins and natural killer cells, as well as T and B lymphocytes (1).
To date, a variety of >400 disorders have been listed under the term IEI and many of them have been described only in a small number of patients and families (2-4). While PIDs are primarily hereditary, acquired deficiencies have also been identified (5). It is crucial to differentiate IEI from secondary immunodeficiencies, which arise more frequently due to various factors such as infections, medications, metabolic disease and environmental conditions. By contrast, IEI stem from genetic anomalies resulting in impaired immune cell functionality (5).
Immunoglobulin A deficiency is the most common IEI, estimated to affect 1 in 300 to 1 in 500 individuals, whereas other IEI are rarer, with an approximate prevalence of 1 in 1,200 live births (6). The clinical indications of IEI are highly diverse and patients have a higher infection susceptibility. IEI may be diagnosed as a normal infection (ears, lungs and the sinuses), although they may be undetected if misdiagnosed (7). The diagnosis of such disorders requires specialized testing, accurate and timely diagnosis, as well as a relatively high suspicion (8). An experienced clinical immunologist in the evaluation of immunodeficiencies should be consulted for an accurate and early diagnosis, as well as precise treatment, which could prevent the rate of morbidity related to such disorders and it should improve the patient outcomes (9). Furthermore, Amaya-Uribe et al (10) found that there is a high prevalence of autoimmune disease (AD) among cases of IEI, indicating a link between the occurrence of both conditions. The authors found that a defect in autoimmune regulator and T-regulatory cells of the central and peripheral tolerance is associated with AD development. In addition, the study found that chronic and recurrent infection through molecular mimicry, as well as activation of super antigens, are vital for the progression of AD, which are characteristics of IEI (10).
The basic classification of IEI depends upon the type of immune system defects; they are broadly classified as adaptive or innate immunity disorders, respectively (1). The advent of whole-exome sequencing (WES) has significantly contributed to identifying novel causative genes in IEI cases (11). Particularly in regions with elevated rates of parental consanguinity, such as Jordan, where ~40% of marriages are consanguineous, WES has proven effective in identifying disease-associated genes and variants. The prevalence of consanguinity before 1980 in Amman was ~30%, leading to a heightened predisposition to IEI (according to www.consang.net). However, identifying new mutations requires robust evidence of their clinical relevance, necessitating functional genomic studies. These investigations delve into the impact of novel mutations on gene transcription, translation, transcriptional regulation and protein structure and function, ultimately shedding light on the precise effects of these mutations on proteins. Thus, genetic testing is a must when a family history of IEI has been detected in one of the family members (12). The present study, for the first time, investigated a cohort of Jordanian patients in whom signal transducer and activator of transcription 1 (STAT1), elastase, neutrophil expressed (ELANE) and interferon induced with helicase C domain1 (IFIH1) mutations were identified, and a new association with variants in NLR family, pyrin domain-containing 12 (NLRP12), G-protein-signaling modulator 1 (GPSM1) and TNFα-induced protein 3-interacting protein 1 (TNIP1) was discovered.
Patients and methods
Patient samples
Blood samples of members from 9 families (total of 37 samples) were collected at the Primary Immunodeficiency Department at Queen Rania Al Abdullah Hospital for Children, King Hussein Medical Center (Amman, Jordan). All patients with IEI and their family members, for whom immunologic or genetic testing was carried out, signed a consent form. Genomic DNA was extracted using the DNeasy blood & tissue kit (Qiagen, Inc.) and stored at -80˚C. The quality and quantity of the DNA samples was measured using a NanoPhotometer® N50 spectrophotometer (i IMPLEN).
Each patient had been noted to have recurrent infection, fever, sinusitis, lymphadenopathy and a family history of IEI or autoimmunity, and had undergone extensive lab tests, including complete blood count (CBC) and flow cytometry to determine any immune malfunction. Patients with abnormal immune results, such as agammaglobulinemia, low lymphocyte count or high immunoglobulin count, were considered patients with IEI. The samples used in the present study were chosen at random with no set criteria or procedure. The probands and their families presented at the Department of Immunology at Queen Rania Children's Hospital in September 2021. By October 2021, the blood samples had been drawn at the Department of Immunology and the consent forms had been signed.
Patients and pedigrees
A total of 37 IEI samples from 9 Jordanian families (patients and their father, mother and/or siblings), who had poorer response to conventional treatments and were in need of an alternative therapeutic strategy were chosen randomly for this study. A total of four patients were from consanguineous families (patient ID-26, ID-32, ID-34 and ID-35], while the remaining probands had unrelated parents. In addition, three patients had a positive family history of PID (ID-26, ID-29 and ID-32), while one (ID-29) had a strong family history of familial Mediterranean fever (FMF) and three others (ID-28, ID-30 and ID-33) had a negative family history. The father of proband PID-31 had chronic kidney disease and vision problems.
Proband ID-26 was a 2.5-year-old male who, at the age of 14 months, had developed lymphoproliferation, autoimmune cytopenia and a decreased level of consciousness. The patient had negative culture yields, Epstein-Barr virus viremia and was treated with high-dose intravenous immunoglobulin, rapamycin. The laboratory results showed that the patient had hypogammaglobulinemia (after rituximab). In addition, the proband had a negative next-generation sequencing (NGS) result for the immunodeficiency panel and an elevated double-negative T-cell level.
Patient ID-28 was a one-year-old male with recurrent admissions with fever and severe neutropenia. Once admitted with a submental abscess, the patient required surgical drainage. The patient suffered from severe neutropenia. The proband had no indication of maturation arrest in a bone marrow study performed at the age of 3 months. The patient also had normal immunoglobulin levels and a normal lymphocyte subset.
Patient ID-29 was a 12-year-old female diagnosed with refractory Juvenile dermatomyositis since the age of 6 years. The patient did not have any history of breakthrough infections. The proband had hypergammaglobulinemia and steroid-induced lymphopenia, and also had a heterozygous M694V mutation in the familial Mediterranean fever gene.
Patient ID-30 was a 7-year-old male with recurrent admission with cough and fever since the early infancy period. The patient suffered from chronic mycosis of the skin, developmental delay and hypothyroidism. The proband's immunoglobulin levels were normal, as well as the dihydrorhodamine test and CBC.
Patient ID-31 was a 3-year-old female diagnosed with biliary stasis at the age of 2 months. The patient had a history of recurrent fever since the early infantile period and a negative culture yield. Furthermore, the patient had a history of short liver synovitis, microscopic hematuria and prompt response to steroids. The laboratory analysis results showed that the patient suffered from neutrophilic leukocytosis. Inflammatory markers were also high, as well as the urine levels of mevalonate kinase. The patient's immunoglobulin levels were normal.
Patient ID-32 was a 6-month-old male patient with a history of chronic cough, failure to thrive and chronic diarrhea. Lab tests showed decreased naïve T cells, no lymphopenia, a reduced number of switched memory B cells and normal immunoglobulin levels. The family history indicated positivity for similar illness (the proband's brother) and the parents are consanguineous.
Patient ID-33 was a 4-year-old female diagnosed with secondary alveolar proteinosis at the age 8 months, improved on antimicrobials and systemic steroids also, and was recurrently admitted with chest infections. The patient had a negative culture yield, developmental delay, as well as epilepsy disorder at the age of 2 years.
Patient ID-34 was a 14-year-old male diagnosed with unclassified arthritis at the age of 11 years, managed by anti-TNF. The patient had been diagnosed with esophageal candidiasis and at the age of 14 years, he had a prolonged fever. The initial lab results showed normal immunodeficiency work-up and the patient also had a lymph node biopsy with atypical lymphoblasts. In addition, the patient had a positive family history of malignancy.
Patient ID-35 was a 9-month-old male diagnosed with complicated meningitis at the age of 7 months. In addition, the patient has a polio vaccine-associated paralysis and the candidate's diagnosis was based on major histocompatibility complex II deficiency. The patient has agammaglobulinemia and absent CD4 lymphocytes. The proband also had a positive family history of IEI with an affected brother. A summary of the cases is provided in Table I.
 |
Table I
Clinical history, immunological laboratory findings and the mutated genes found for each patient with inborn errors of immunity in the cohort.
|
Table I
Clinical history, immunological laboratory findings and the mutated genes found for each patient with inborn errors of immunity in the cohort.
| Patient |
Patient's history |
Immunological Lab findings |
Mutated genes identified |
| ID-26 |
• 2.5-year-old male patient |
• Hypogammaglobulinemia (after rituximab) |
STAT2 |
| |
• Developed lymphoproliferation, autoimmune cytopenia and decreased level of consciousness at the age of 14 months |
• Negative NGS for immunodeficiency panel |
PIK3R4 (novel) |
| |
• Negative culture yields, EBV viremia |
• High DNT |
|
| |
• Managed by high dose IVIG, rapamycin |
|
|
| ID-28 |
• One-year-old male patient |
• Severe neutropenia |
ELANE |
| |
• Recurrent admissions with fever, severe neutropenia |
• No maturation arrest on bone marrow study performed at the age of 3 months |
|
| |
• Once admitted with submental abscess, required surgical drainage |
• Normal IG level, normal lymphocyte subset |
|
| ID-29 |
• 12-year-old female patient |
• Hypergammaglobulinemia |
IL17RA (novel) |
| |
• Diagnosis of refractory juvenile dermatomyositis since the age of 6 years |
• Steroid-induced lymphopenia |
|
| |
• No history of breakthrough infections |
• Heterozygous mutation for FMF m694 |
|
| ID-30 |
• 7-year-old male patient |
• Normal immunoglobulin level |
STAT1 (novel) |
| |
• Recurrent admission with cough and fever since early infancy period |
• Normal DHR |
CARMIL2 (novel) |
| |
• Chronic mycosis of the skin |
• Normal CBC |
|
| |
• Developmental delay, hypothyroidism |
|
|
| ID-31 |
• 3-year-old female patient |
• Neutrophilic leukocytosis |
TNIP1 (novel) |
| |
• Diagnosis of biliary stasis at the age of 2 months, recurrent history of fever since early infantile period, negative culture yield |
• High inflammatory markers |
|
| |
• History of short liver synovitis |
• High MVK in urine |
|
| |
• History of microscopic hematuria |
• Normal immunoglobulin level |
|
| |
• Prompt response to steroid |
|
|
| ID-32 |
• 6-month-old male patient |
• No lymphopenia |
GPSM1 |
| |
• History of chronic cough |
• Decreased naïve T cells, reduced number of switched memory B cells |
NLRP12 (deleterious) |
| |
• Failure to thrive |
• Normal immunoglobulin level |
|
| |
• Chronic diarrhea |
|
|
| ID-33 |
• 4-year-old female patient |
• No lymphopenia |
IFIH1 |
| |
• Diagnosed with secondary alveolar proteinosis at the age of 8 months, improved on antimicrobials and systemic steroids |
• Transient hypogammaglobulinemia, improved after the age of 2 years |
MBL2 (pathogenic) |
| |
• Recurrent admissions with chest infection |
• Low CD4 count |
|
| |
• Negative culture yields |
|
|
| |
• Developmental delay, epilepsy disorder at the age of 2 years |
|
|
| ID-34 |
• 14-year-old male patient |
• Initial lab showed normal immunodeficiency work-up |
TYK2 |
| |
• Diagnosed with unclassified arthritis at the age of 11 years, managed by anti-TNF |
• LN biopsy with atypical lymphoblast |
UNG |
| |
• Diagnosed with esophageal candidiasis |
|
|
| |
• Prolonged fever, disseminated intravascular coagulation at the age of 14 years |
|
|
| ID-35 |
• 9-month-old male patient |
• Agammaglobulinemia absent |
IL17RC |
| |
• Diagnosis of complicated meningitis at the age of 7 months |
• CD4 lymphocyte subset |
CDCA7 |
| |
• Polio vaccine-associated paralysis |
|
|
Pedigree charts were constructed using Progeny Genetics (https://pedigree.progenygenetics.com/), to reveal the occurrence and appearance of the PID phenotypes of particular patients and their ancestors from one generation to the next.
WES
Genomic DNA from each sample (3 µg) was randomly fragmented using the Covaris Acoustic System with the following settings: Frequency, 500 kHz; temperature, 4˚C; and duration, 5 min. The fragmented DNA was then linked to adapters. The DNA was purified using Agencourt AMPure SPRI beads (Beckman Coulter, Inc.) according to the manufacturer's protocol, extracted, and amplified by ligation-mediated PCR. For the ligation-mediated PCR, KAPA HiFi HotStart DNA Polymerase (KAPA Biosystems) was used. The thermocycling conditions were as follows: Initial denaturation at 98˚C for 2 min; 15 cycles of denaturation at 98˚C for 20 sec, annealing at 60˚C for 30 sec and elongation at 72˚C for 1 min; and final extension at 72˚C for 5 min. It was then hybridized to Agilent's SureSelect Human All Exon V8 (Agilent Technologies, Inc.) for enrichment and the captured PCR products were subjected to quantitative PCR to estimate the magnitude of enrichment. Each captured library was subsequently sequenced using an Illumina NovaSeq sequencer (Illumina, Inc.). The sequenced reads were aligned to the human genome reference [University of California Santa Cruz (UCSC) hg38; https://genome.ucsc.edu/] using SOAP aligner (soap2.21) and duplicated reads were filtered out. SOAPsnp (v.1.03) was used to assemble the consensus sequence and call genotypes in target regions, and integrative genomic viewer (IGV) 2.17.4 (https://igv.org) was utilized to visually inspect all reported variants. Patients born in consanguineous families were expected to show an autosomal recessive (AR) inheritance pattern. Therefore, the synonymous mutations were filtered out and the variants with a >0.5% frequency in dbSNP143 (https://vatlab.github.io/vat-docs/applications/annotation/variants/dbsnp/), 1000 genomes (internationalgenome.org) and HapMap (coriell.org) were excluded. The new single nucleotide polymorphisms (SNPs) detected were confirmed using Sanger sequencing. For this purpose, exon-specific PCR primers were designed using Primer3 (https://primer3.ut.ee/), the annealing location of which was tested using in-silico PCR (https://genome.ucsc.edu/cgi-bin/hgPcr). Subsequently, the genomic DNA was amplified using FastGene Taq DNA polymerase (5 U/µl; 0.5 µl), 10 µM forward primer (1 µl), 10 µM reverse primer (1 µl), 10 µM dNTP mix (1 µl), 10X Buffer A (15 mM) (5 µl), 100 ng of genomic DNA, with the mixture topped up to 50 µl with nuclease-free water, in a BioRad (T100) thermal cycler (Bio-Rad Laboratories, Inc.) The thermocycling program was as follows: Initial denaturation at 95˚C for 5 min, 35 cycles of 95˚C for 15 sec, 60˚C for 30 sec and 72˚C for 1 min, followed by a final extension at 72˚C for 5 min. The PCR products were electrophoresed on a 2% agarose gel and purified with a QIAquick gel extraction kit (Qiagen, Inc.). They were then Sanger sequenced at Macrogen Europe BV and the sequencing electrophoregrams were analyzed using Lasergene17 (DNAStar, Inc.).
Pathway analysis
The genes harboring non-silent mutations were assigned to gene sets using the Molecular Signatures Database Human Collection [MSigDB 2023.1.Hs (https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp)], where the ‘canonical pathways’ were explored. Only 1 nonsynonymous mutation per gene was accounted for in each IEI sample.
Databases
ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) was used to confirm the SNVs' associations with IEI. The Online Mendelian Inheritance in Man (OMIM) database (https://www.omim.org/) was used to acquire information on the genes of interest and their relationship with the phenotype.
Results
WES results
WES was performed and the candidate genes were selected according to the patients' symptoms and diagnosis. Nearly 200 genes were chosen from a PID gene panel and visualized for each patient in order to cover all the possible disease-causing genes. Only deleterious and damaging SNPs were considered for downstream analysis, while benign and likely benign SNPs were excluded.
The detected SNPs were in relevant genes to each patient's symptoms. These were heterozygous in the following genes: Interleukin 17 receptor A (IL17RA) (c.1345C>G; p.Arg449Gly), STAT1 (c.1061T>C; p.Leu354Pro), phosphoinositide 3-kinase regulatory subunit 4 (PIK3R4) (c.574C>T; p.Leu192Phe), TNIP1 (c.460C>G; p.Asp154His), STAT2 (c.1466C>G; p.Pro489Arg), ELANE (c.659G>A; p.Arg220Gln), capping protein regulator and myosin 1 linker 2 (CARMIL2) (c.3683C>T; p.Pro1228Leu), NLRP12 (c.2591T>A; p.Leu863Gln), mannose binding lectin 2 (MBL2) (c.154C>T; p.Arg52Cys), TYK2 (c.1342C>T; p.Arg448Trp), uracil DNA glycosylase (UNG) (c.262C>T; p.Arg88Cys) and cell division cycle associated 7 (c.679C>T; p.Arg227Cys). Furthermore, two homozygous SNPs were detected, one in GPSM1 (c.1283C>A, stop gained; p.Ser428Ter) and another in IFIH1 (c.2891G>A; p.Cys964Tyr). In addition, a CTC/CT frameshift mutation (L/X) was detected in GPSM1 in patient PID-32 and a TCTGGTCTT in-frame insertion (p.V42VWSF) in IL17RC in patient PID-35. Of note, five novel SNPs were identified in CARMIL2, TNIP1, STAT1, IL17RA and PIK3R4. Of these, c.1061T>C in STAT1 was de novo and further confirmed using Sanger sequencing (Fig. 1, Fig. 2, Fig. 3, Fig. 4 and Fig. 5 and Tables II and III).
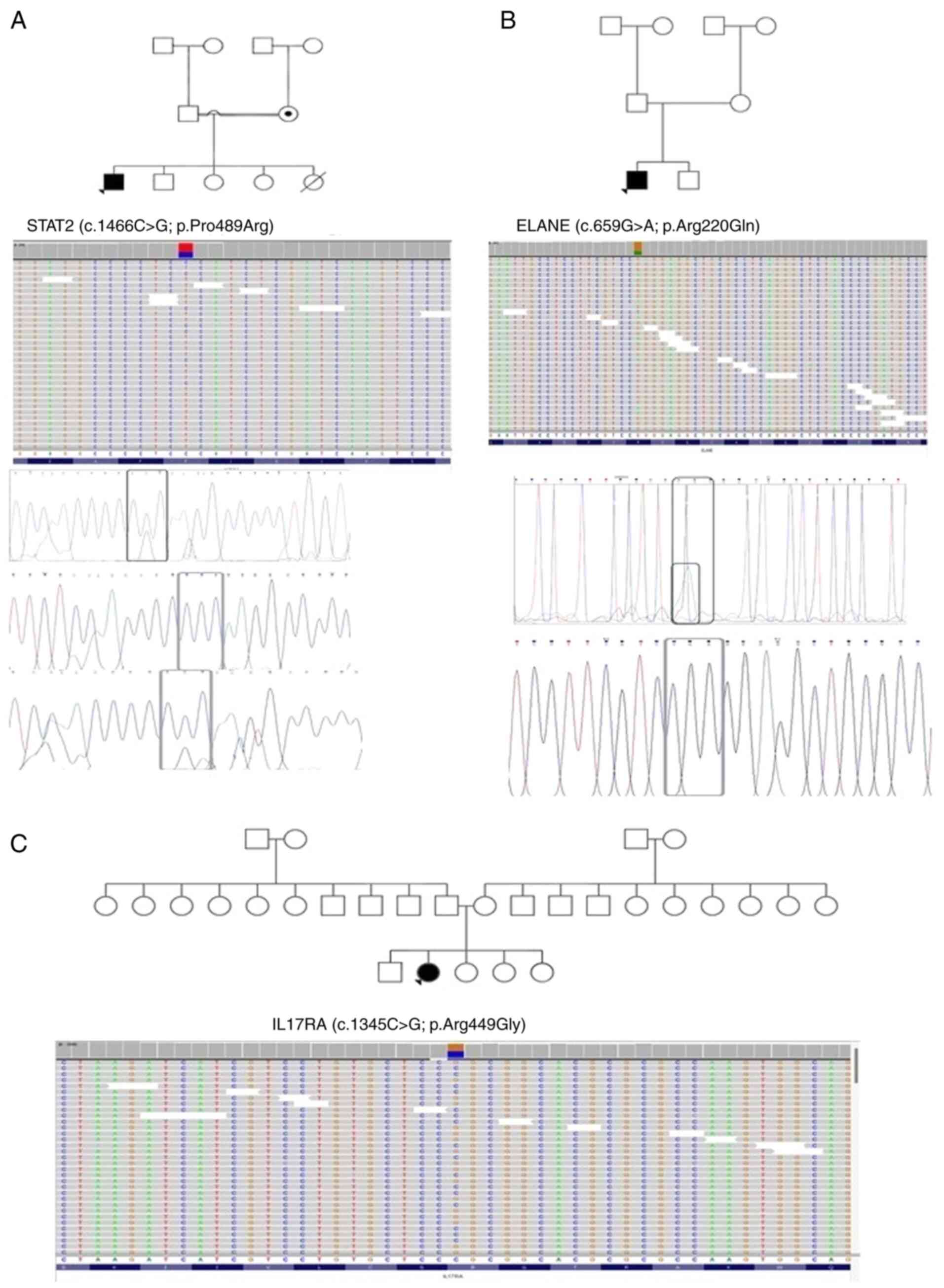 |
Figure 1
Pedigrees of patients (A) ID-26, (B) ID-28 and (C) ID-29, along with the single nucleotide polymorphisms detected in STAT2, ELANE and IL17RA, respectively, through exome-sequencing and Sanger sequencing. Probands are indicated with arrowheads. Circles with a black dot indicate the mother or sister being a carrier of the corresponding SNV. A double line between parents represents consanguinity. A square with a black dot indicates the father or brother being a carrier of the corresponding SNV. A circle/square with a strike-through line indicates a deceased subject. The different colors in the next-generation sequencing reads from IGV depict the different nucleotides. Red is for ‘T’, blue is for ‘C’, green is for ‘A’ and brown is for ‘G’. STAT2, signal transducer and activator of transcription 2; ELANE, elastase, neutrophil expressed; IL17RA, interleukin 17 receptor A; SNV, single nucleotide variant.
|
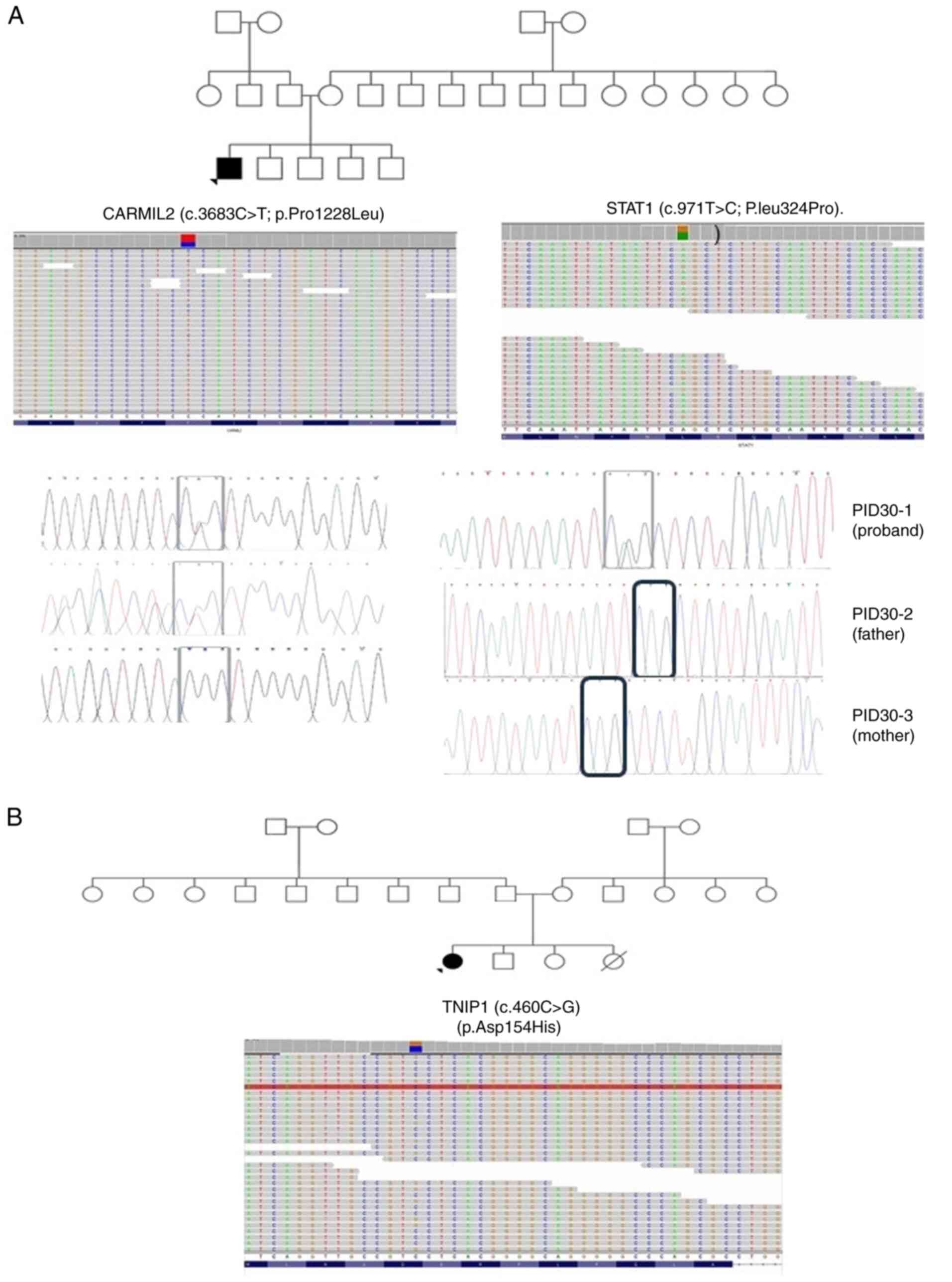 |
Figure 2
Pedigrees of patients (A) ID-30 and (B) ID-31, along with the single nucleotide polymorphisms detected in (A) CARMIL2 and STAT1 and (B) TNIP1 through exome-sequencing and Sanger sequencing. Probands are indicated with arrowheads. Circles with a black dot indicate the mother or sister being a carrier of the corresponding SNV. A double line between parents represents consanguinity. A square with a black dot indicates the father or brother being a carrier of the corresponding SNV. A circle/square with a strike-through line indicates a deceased subject. The different colors in the next-generation sequencing reads from IGV depict the different nucleotides. Red is for ‘T’, blue is for ‘C’, green is for ‘A’ and brown is for ‘G’. CARMIL2, capping protein regulator and myosin 1 linker 2; STAT1, signal transducer and activator of transcription 1; TNIP1, TNFα-induced protein 3-interacting protein 1; SNV, single nucleotide variant.
|
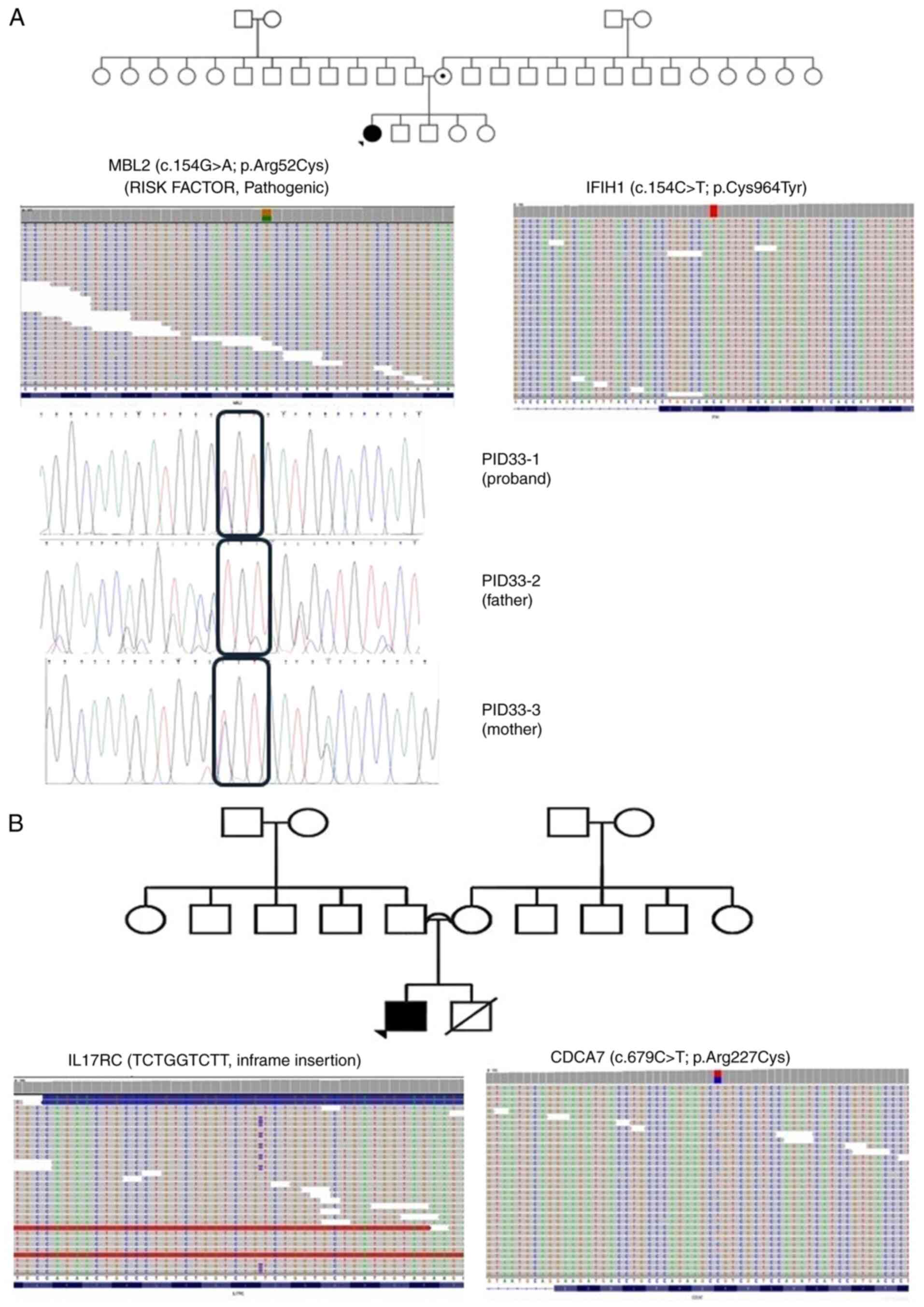 |
Figure 3
Pedigrees of patients (A) ID-33 and (B) ID-35, along with the single nucleotide polymorphisms detected in (A) MBL2 and IFIH1 and (B) IL17RC and CDCA7 through exome-sequencing and Sanger sequencing. Probands are indicated with arrowheads. Circles with a black dot indicate the mother or sister being a carrier of the corresponding SNV. A double line between parents represents consanguinity. A square with a black dot indicates the father or brother being a carrier of the corresponding SNV. A circle/square with a strike-through line indicates a deceased subject. The different colors in the next-generation sequencing reads from IGV depict the different nucleotides. Red is for ‘T’, blue is for ‘C’, green is for ‘A’ and brown is for ‘G’. MBL2, mannose binding lectin 2; IFIH1, interferon induced with helicase C domain1; IL17RC, interleukin 17 receptor C; CDCA7, cell division cycle associated 7; cell division cycle associated 7; SNV, single nucleotide variant.
|
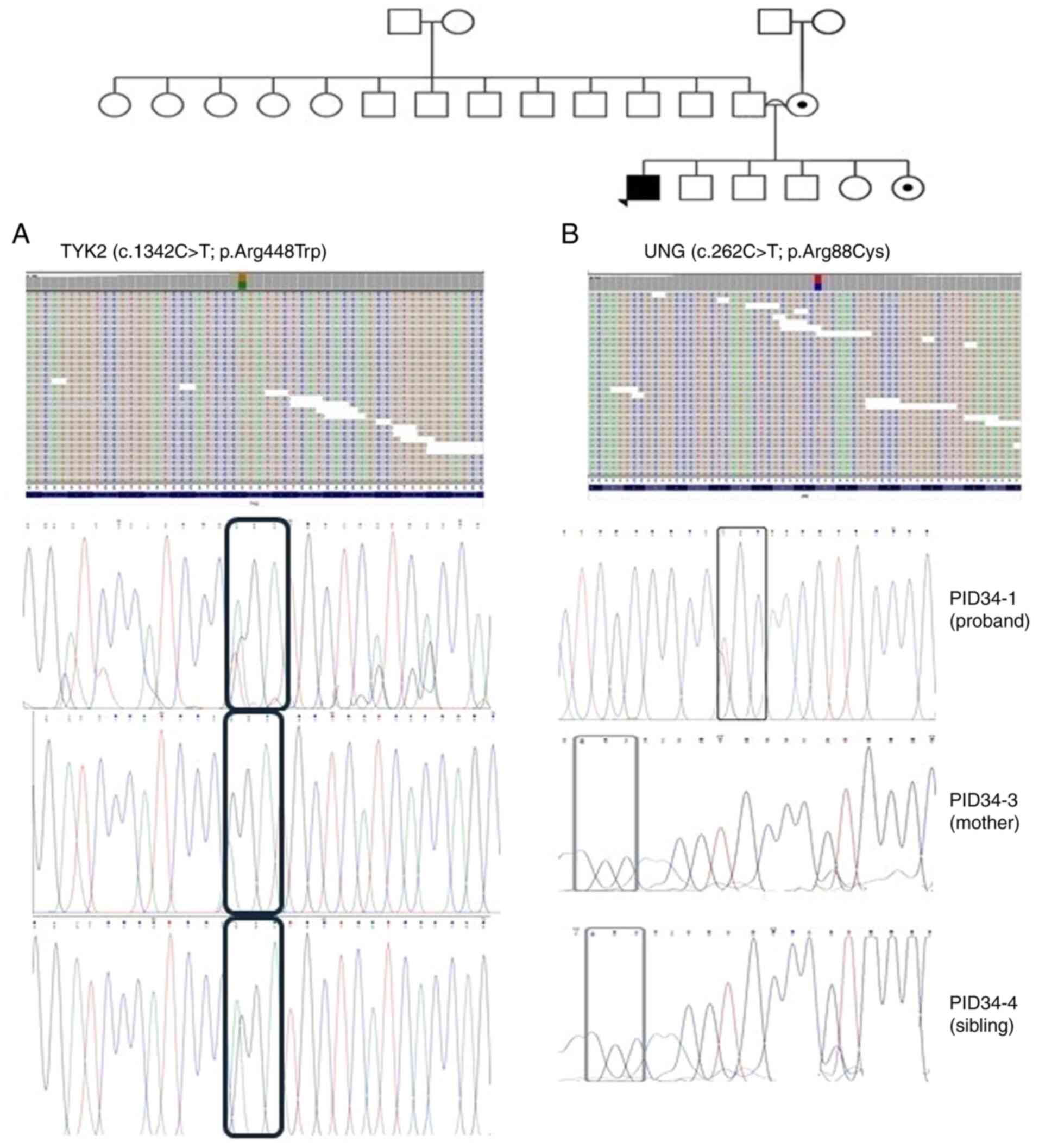 |
Figure 4
Pedigree of patient ID-34, along with the single nucleotide polymorphisms detected in TYK2 (A) and UNG (B) through exome-sequencing and Sanger sequencing. Probands are indicated with arrowheads. Circles with a black dot indicate the mother or sister being a carrier of the corresponding SNV. A double line between parents represents consanguinity. A square with a black dot indicates the father or brother being a carrier of the corresponding SNV. A circle/square with a strike-through line indicates a deceased subject. The different colors in the next-generation sequencing reads from IGV depict the different nucleotides. Red is for ‘T’, blue is for ‘C’, green is for ‘A’ and brown is for ‘G’. TYK2, tyrosine kinase 2; UNG, uracil DNA glycosylase; SNV, single nucleotide variant.
|
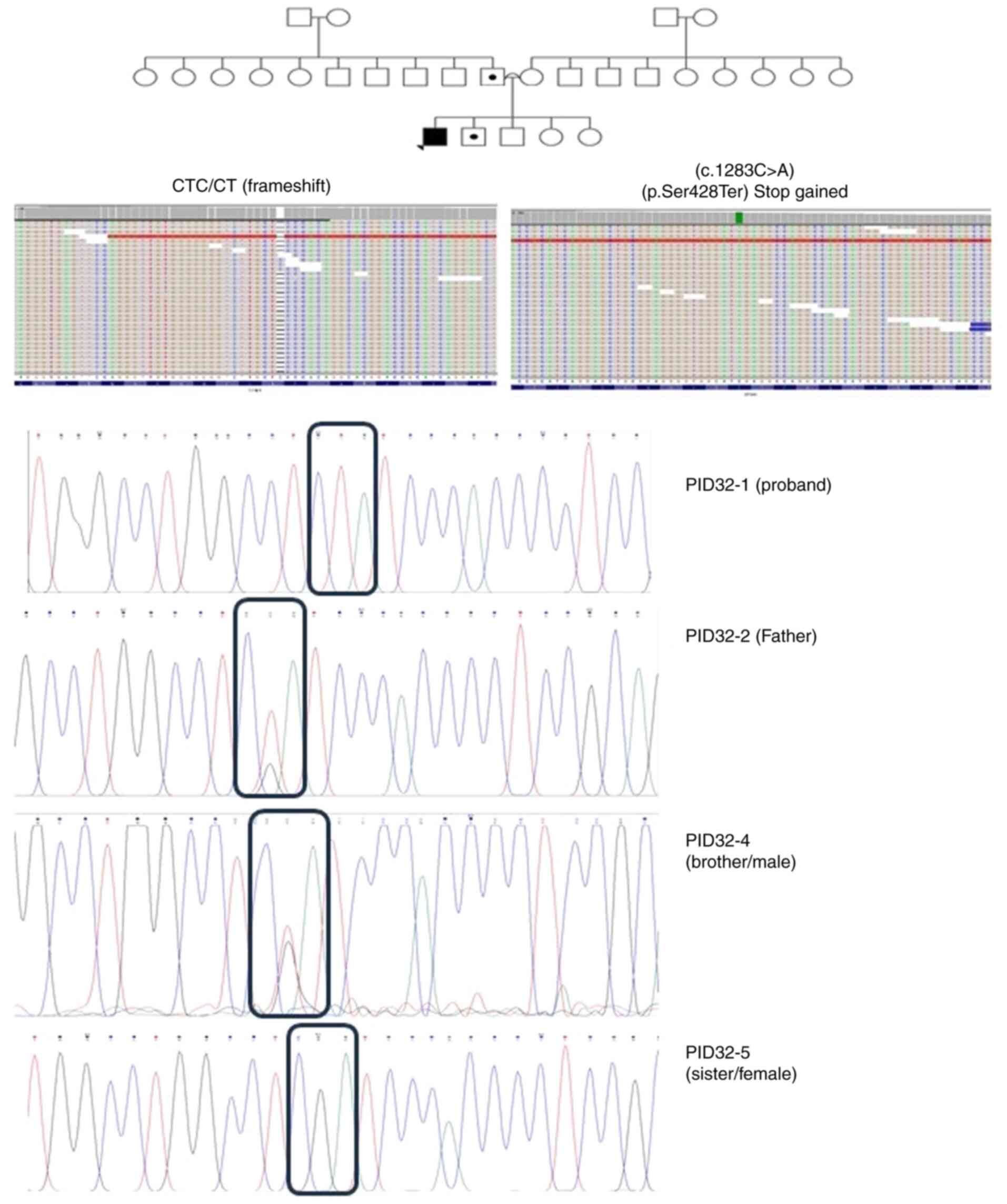 |
Figure 5
Pedigree of patient ID-32, along with the single nucleotide polymorphisms detected in GPSM1 (frameshift and stop gained), through exome-sequencing and Sanger sequencing. Probands are indicated with arrowheads. Circles with a black dot indicate the mother or sister being a carrier of the corresponding SNV. A double line between parents represents consanguinity. A square with a black dot indicates the father or brother being a carrier of the corresponding SNV. A circle/square with a strike-through line indicates a deceased subject. The different colors in the next-generation sequencing reads from integrative genomic viewer depict the different nucleotides. Red is for ‘T’, blue is for ‘C’, green is for ‘A’ and brown is for ‘G’. GPSM1, G-protein-signaling modulator 1; SNV, single nucleotide variant.
|
|
Table II
Variants for each gene, mode of inheritance, CDS and protein change, consanguinity and family history for each patient with inborn errors of immunity.
|
Table II
Variants for each gene, mode of inheritance, CDS and protein change, consanguinity and family history for each patient with inborn errors of immunity.
| Patient |
Gene |
Zygosity |
Inheritance |
CDS change |
Protein change |
Consanguinity |
Family history |
| ID-26 |
i) STAT2 |
i) HET |
i) AR |
i) c.1345C>G |
i) p.Arg449Gly |
Consanguineous |
• Positive family history of similar phenotype |
| |
ii) PIK3R4 |
ii) HET |
ii) - |
ii) c.574C>T |
ii) p.Leu192Phe |
|
• Sister died due to uncontrolled autoimmunity |
| ID-28 |
ELANE |
HET |
AD |
c.659G>A |
p.Arg220Gln |
Non-consanguineous |
Negative family history |
| ID-29 |
IL17RA |
HET |
AR |
c.1345C>G |
p.Arg449Gly |
Non-consanguineous |
• Strong family history of FMF |
| |
|
|
|
|
|
|
• Brother diagnosed with sacroiliitis |
| ID-30 |
i) STAT1 |
i) HET |
i) AR/AD |
i) c.1061T>C |
i) p.Leu354Pro |
Non-consanguineous |
Negative family history |
| |
ii) CARMIL2 |
ii) HET |
ii) AR |
ii) c.3683C>T |
ii) p.Pro1228Leu |
|
|
| ID-31 |
TNIP1 |
HET |
- |
c.460C>G |
p.Asp154His |
Non-consanguineous |
Father has chronic kidney disease and vision problems |
| ID-32 |
i) GPSM1 |
i) HOM |
i) - |
i) c.1283C>A |
i) p.Ser428Ter |
Consanguineous |
Positive family history of similar illness (proband's brother) |
| |
ii) NLRP12 |
ii) HET |
ii) AD |
ii) c.2591T>A |
ii) p.Leu863Gln |
|
|
| ID-33 |
i) IFIH1 |
i) HOM |
i) AR |
i) c.2891G>A |
i) p.Cys964Tyr |
Consanguineous |
Negative family history |
| |
ii) MBL2 |
ii) HET |
ii) AD |
ii) c.154C>T |
ii) p.Arg52Cys |
|
|
| ID-34 |
i) TYK2 |
i) HET |
i) AR |
i) c.1342C>T |
i) p.Arg448Trp |
Consanguineous |
• Positive family history of malignancy (father, uncle and brother) |
| |
ii) UNG |
ii) HET |
ii) AR |
ii) c.262C>T |
ii) p.Arg88Cys |
|
• Proband's aunt died due to SLE |
| ID-35 |
i) IL17-RC |
i) HET |
i) AR |
i) TCTGGTCTT |
i) p.V42VWSF |
Non-consanguineous |
Positive family history (proband's brother) |
| |
ii) CDCA7 |
ii) HET |
ii) AR |
ii) c.679C>T |
ii) p.Arg227Cys |
|
|
|
Table III
SNP and protein change for each detected gene per patient with inborn errors of immunity, along with the SIFT, PolyPhen-2, CADD score, ClinVar and OMIM phenotype.
|
Table III
SNP and protein change for each detected gene per patient with inborn errors of immunity, along with the SIFT, PolyPhen-2, CADD score, ClinVar and OMIM phenotype.
| Patient |
Gene ID |
SNP change |
Protein change |
SIFT |
PolyPhen-2 |
CADD score |
ClinVar |
OMIM phenotype |
| ID-26 |
STAT2 |
c.1466C>G |
p.Pro489Arg |
0 (deleterious) |
0.966 (damaging) |
22.5 |
Uncertain significance |
Immunodeficiency 44 |
| |
PIK3R4 |
c.574C>T |
p.Leu192Phe |
0 (deleterious) |
0.959 (damaging) |
26.9 |
- |
- |
| ID-28 |
ELANE |
c.659G>A |
p.Arg220Gln |
0.56 (tolerated) |
0.96 (probably damaging) |
21.8 |
Pathogenic |
Severe congenital neutropenia |
| ID-29 |
IL17RA |
c.1345C>G |
p.Arg449Gly |
0 (deleterious) |
0.997 (probably damaging) |
25.8 |
- |
IMD-51 |
| ID-30 |
STAT1 |
c.1061T>C |
p.Leu354Pro |
0 (deleterious) |
0.989 (probably damaging) |
25.2 |
- |
IMD 31 A,B,C |
| |
CARMIL2 |
c.3683C>T |
p.Pro1228Leu |
0 (deleterious) |
0.995 (probably damaging) |
25.0 |
- |
IMD 58 |
| ID-31 |
TNIP1 |
c.460C>G |
p.Asp154His |
0.01 (deleterious) |
0.996 (possibly damaging |
24.2 |
- |
- |
| ID-32 |
GPSM1 |
CTC/CT frameshift |
L/X |
- |
- |
0.07 |
- |
- |
| |
|
c.1283C>A (Stop gained) |
p.Ser428Ter |
- |
- |
0.22 |
- |
- |
| |
NLRP12 |
c.2591T>A |
p.Leu863Gln |
0 (deleterious) |
0.934 (possibly damaging) |
24.3 |
- |
Familial cold autoinflammatory syndrome 2 |
| ID-33 |
IFIH1 |
c.2891G>A |
p.Cys964Tyr |
0 (deleterious) |
1 (probably damaging) |
26.1 |
- |
Immunodeficiency 95 |
| |
MBL2 |
c.154C>T |
p.Arg52Cys |
0 (deleterious) |
0.997 (Probably damaging) |
24.3 |
Risk factor (pathogenic) |
Chronic infection (due to MBL2 deficiency) |
| ID-34 |
TYK2 |
c.1342C>T |
p.Arg448Trp |
0.01 (deleterious) |
0.462 (possibly damaging) |
24.8 |
Uncertain significance |
Immunodeficiency 35 |
| |
UNG |
c.262C>T |
p.Arg88Cys |
0 (deleterious) |
0.714 (possibly damaging) |
31.0 |
Uncertain significance; conflicting interpretations of pathogenicity |
Immunodeficiency with hyper IgM, type 5 |
| ID-35 |
CDCA7 |
c.679C>T |
p.Arg227Cys |
0.01 (deleterious) |
0.899 (possibly damaging) |
23.6 |
- |
ICF3 |
| |
IL17RC |
TCTGGTCTT (inframe insertion) |
p. v42VWSF |
- |
- |
- |
- |
Candidiasis |
Kyoto Encyclopedia of Genes and Genomes (KEGG; https://www.genome.jp/kegg/pathway.html) pathway analysis also showed involvement of the IL-17 signaling pathway [IL-17 receptor A (IL17RA), c.1345C>G], T helper type 17 (Th17) cell differentiation (STAT1, c.1061T>C), the JAK-STAT signaling pathway [STAT1 and tyrosine kinase 2 (TYK2)], neutrophil extracellular trap (NET) formation (ELANE, c.659G>A), cocaine addiction (GPSM1, c.1283C>A) and cytokine-cytokine receptor interaction (IL17RC, TCTGGTCTT in-frame insertion) (Fig. 6).
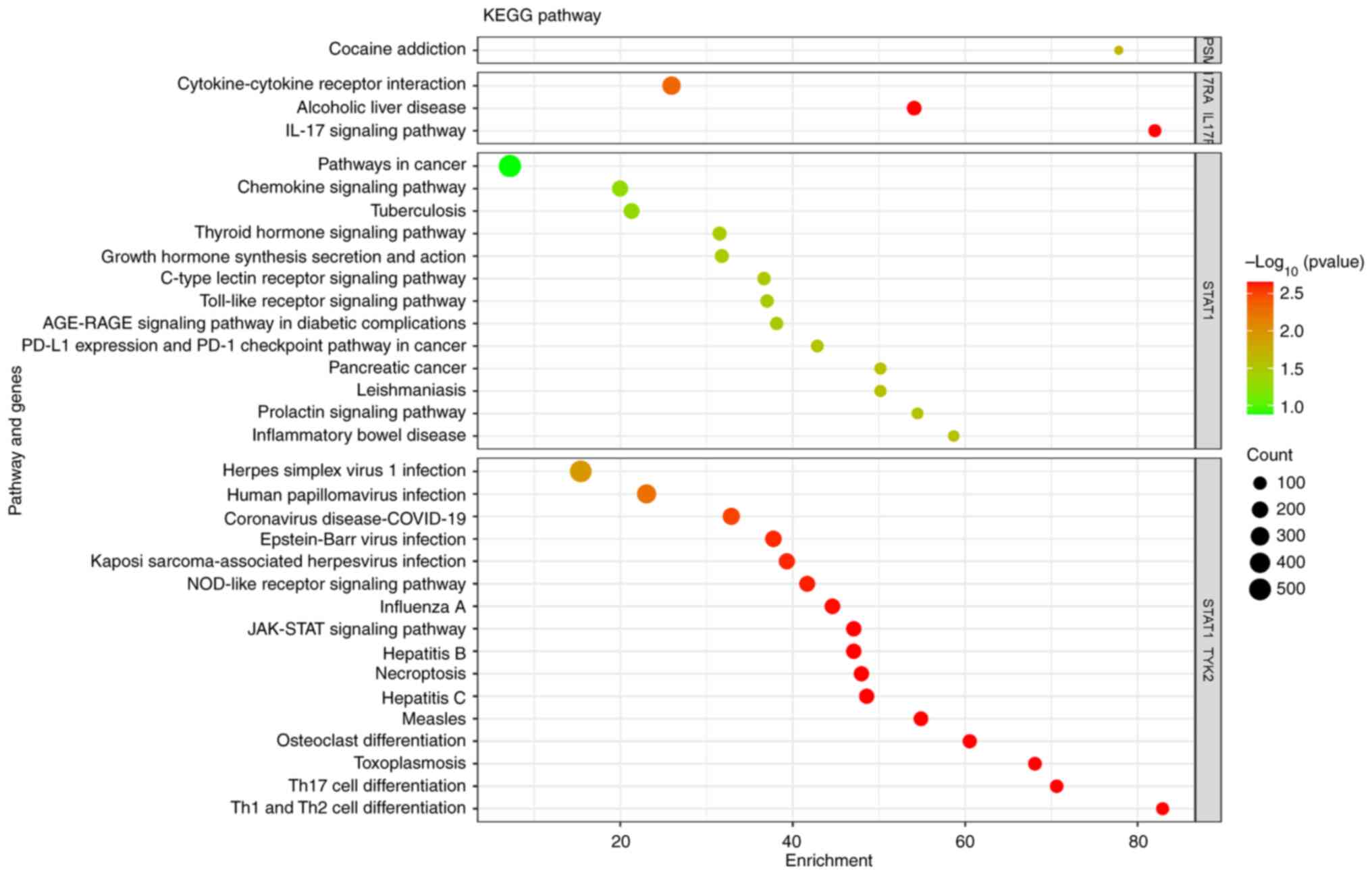 |
Figure 6
Bubble plot of KEGG pathway enrichment analysis of the genes with the detected single nucleotide polymorphisms in the Jordanian cohort with inborn errors of immunity. ‘Count’ indicates the number of genes enriched in each pathway. KEGG, Kyoto Encyclopedia of Genes and Genomes.
|
Discussion
Primary immunodeficiencies encompass a diverse spectrum of disorders that necessitate precise diagnostic strategies. The emergence of NGS techniques has revolutionized clinical immunology. These methodologies not only aid in pinpointing the causative genetic variations but also inform therapeutic interventions, tailoring treatments to individual patient's genetic profiles (13,14).
A salient factor influencing the incidence of novel mutations is consanguinity. In Jordan, consanguinity is more prevalent in rural areas (~40%) than in its capital, Amman, where these levels are ~25.5%. The historical trend of consanguinity in Amman also seems to have influenced the prevalence of PID. Marriages before 1980 had a consanguinity rate of ~30%. (15)
Consanguinity does not seem to be a relevant bias in the present cohort of patients with PID. A novel homozygous mutation of uncertain significance was found in GPSM1, only in patient ID-32. In addition, the homozygous IFIH1 variant was found in a family where consanguinity was not previously reported. In all other patients, only heterozygous variants/mutations were found.
Arunachalam et al (14) elucidated that PIDs are typically monogenic, arising from an SNV. By contrast, polygenic diseases often manifest as autoimmune conditions such as rheumatoid arthritis or systemic lupus erythematosus. Of note, certain genetic variations that are associated with PIDs also appear in autoimmune diseases, underscoring the interwoven nature of immunological disorders (14,15).
It is true that several studies have shed light on the genetic landscape of PIDs, but with different results. For instance, Arunachalam et al (14) identified numerous genetic variants across different PID subtypes. In addition, studies from Aghamohammadi et al (16,17), Abolhasssani et al (18,19) and Kilic et al (20) provided valuable insight into the distribution of PID subtypes in Iranian and Turkish populations, respectively.
In the present study, the genetic underpinnings of PIDs in 9 Jordanian patients were explored, identifying mutations in key immunological genes, including STAT1/2, MBL2, TYK2, UNG, TNIP1, CARMIL2 and ELANE, as well as a 9-bp insertion in IL17RC, and one frameshift and nonsense mutation in the same exon in GPSM1. Of note, a STAT2 mutation was found in patient ID-26 suffering from hypogammaglobulinemia (after treatment with rituximab). However, STAT2 heterozygosity may have no impact on the patient; thus, it should be investigated along with PIK3R4 in order to determine their effect on the patient.
Furthermore, mutations in TYK2 and UNG were found in patient ID-34, who had been diagnosed with unclassified arthritis and esophageal candidiasis.
Mutations in STAT1 have been reported in different ethnicities, affecting critical cellular processes, such as STAT1-mediated responses to IFNG and IL27 (21,22) In the present cohort, patient ID-30 had a heterozygous SNV in the CARMIL2 and STAT1 genes. CARMIL2 is associated with immunodeficiency-58 (IMD58). This is an AR primary immunologic disorder distinguished by early-onset skin lesions, such as eczematous dermatitis, infectious abscesses and warts, as well as recurrent respiratory infections or allergies, among others. CARMIL2 deficiency, as described by Wang et al (23), presents with a unique set of clinical features, with compromised T-cell subsets and antibody responses. STAT1 deficiency, on the other hand, is associated with three distinct phenotypes depending on whether it's loss of function (LOF) AD/AR or gain of function (GOF) AD, while LOF mutations correlate with compromised Th1 immunity. GOF mutations are associated with impaired Th17 immunity-in the case of patient ID-30, GOF mutations were more likely suggested based on the patient's clinical presentation. IMD31C is an immunologic dysregulation disorder with highly variable manifestations caused by AD GOF mutations in STAT1. Most patients present with chronic mucocutaneous candidiasis in infancy or early childhood (24). Other highly variable characteristics include persistent infections, enteropathy with villous atrophy and autoimmune disorders such as hypothyroidism and diabetes mellitus. In addition, IMD31C is inherited in an AD manner (25-27). Both genes (STAT1, CARMIL2) could be relevant to the patient's disease, STAT1 function (e.g. phosphorylation) should be investigated, in addition to IL-17A production testing post-stimulation by phorbol 12-myristate 13-acetate and ionomycin, are useful to activate transcription factors for intracellular signaling and production of cytokines of many different immune cell types in the presence of Brefeldin, as well as the enumeration of Th17 lymphocytes, feasible through C-C motif chemokine receptor 6 and C-X-C motif chemokine receptor 3 markers, to ascertain the diagnosis. In addition, CARMIL2 expression should be detected in the patient. Similarly, mutations in IL17RA and TYK2 have been associated with characteristic clinical phenotypes and cellular defects (28-30). In the present study, a new IL17RA mutation was detected in a patient suffering from hypergammaglobulinemia and steroid-induced lymphopenia, namely ID-29. Two new mutations, one in STAT1 and another in CARMIL2, were also detected in a patient suffering from chronic mycosis of the skin, developmental delay and hypothyroidism, namely ID-30. In addition, a new TNIP1 mutation was found in ID-31, a patient diagnosed with biliary stasis at the age of 2 months, with are current history of fever since the early infantile period, and a history of short liver synovitis and microscopic hematuria. Furthermore, studies have emphasized the clinical significance of mutations in genes such as UNG, MBL2 and IL17RC (14,31-35).
Furthermore, studies reported the existence of NLRP12 deficiency in Chinese patients, being a compelling case of the diverse clinical manifestations associated with PID mutations, ranging from erythema nodosa to systemic symptoms such as fever and lymphadenopathy (36-38).
ELANE encodes leukocyte elastase, being produced by neutrophil precursors, and its mutations have been associated with different immunodeficiencies, such as congenital neutropenia (14,39,40). In the present study, ELANE mutation was found in patient ID-28, who suffered from severe neutropenia. The patient was diagnosed with no maturation arrest in the bone marrow study performed at the age of 3 months.
IFIH1 (IMD95) encodes an RIG-I-like receptor involved in the sensing of viral RNA (41,42). IFIH1 variants are also related to hepatitis C virus clearance (43). Heterozygous gain-of-function IFIH1 mutations have also been associated with autoimmune diseases (44). Furthermore, rare gain-of-function mutations in IFIH1 significantly increase the production of type I IFN, leading to Aicardi-Goutières or Singleton-Merten syndromes (45,46). Nonetheless, IMD95 is an AR disorder characterized primarily by recurrent and severe viral respiratory infections beginning in infancy or early childhood (47). Infections such as human rhinovirus which is the most common viral infectious agent in humans and is the predominant cause of the common cold and respiratory syncytial virus, a contagious virus that causes infections of the respiratory tract, frequently necessitate hospitalization or respiratory support. Immunologic testing is usually normal, though some minor abnormalities may be detected. The disorder is caused by a loss of the innate immune system's ability to sense viral genetic information, resulting in a lack of interferon production, poor response to viral and immunologic stimulation and failure to control viral replication (48). The patient in whom the IFIH1 mutation was detected had a low CD4 count. She was diagnosed with secondary alveolar proteinosis at the age of 8 months, had recurrent admissions with chest infection, as well as developmental delay and epilepsy disorder at the age of 2 years, which were associated with the patient's symptoms and initial diagnosis (15).
Familial cold autoinflammatory syndrome-2 is an AD autoinflammatory disorder that causes episodic and recurring rash, urticaria, arthralgia, myalgia and headaches. In most patients, these episodes are accompanied by fever and serologic signs of inflammation. The majority of patients, but not all, report that exposure to colds triggers their episodes. Other symptoms may include abdominal pain, thoracic pain and sensorineural deafness. The age of onset varies, ranging from infancy to middle age, and the severity and clinical manifestations are diverse (49). In the present study, patient ID-32 had a mutation in NLRP12 and according to various databases, the variant found is pathogenic. The patient had a history of chronic cough, failure to thrive and chronic diarrhea, which explains the symptoms. Furthermore, a mutation in GPSM1 was detected for the same patient; he had decreased naïve T cells and a reduced number of switched memory B cells.
Despite the high rate of consanguinity (4 out of 9 patients), only two homozygous variants were identified, with only one potentially linked to an IEI. This can be explained by considering several factors related to genetic diversity, the complexity of inheritance patterns and the nature of the diseases involved. IEIs are often genetically heterogeneous, meaning they can be caused by mutations in different genes. Despite the high rate of consanguinity, the patients may carry mutations in different genes that lead to similar clinical phenotypes. In addition, not all individuals with a pathogenic variant will necessarily manifest the disease (incomplete penetrance) and the severity of the disease can vary among those who do (variable expressivity) (38,50). This could result in only some of the patients showing symptoms of an IEI despite having related genetic backgrounds. Certain genetic conditions are caused by having two different mutations in a gene (one on each allele) rather than two identical mutations (homozygosity). In consanguineous populations, there may still be a presence of compound heterozygous variants that are not immediately apparent as homozygous but that lead to disease (51). Besides homozygous variants, other mechanisms, such as copy number variations, epigenetic changes or mutations in regulatory regions may be responsible for IEIs and may not be easily detected by standard genetic screening focused on homozygous variants (52-54). Finally, IEIs may be influenced by multiple genes and environmental factors. Therefore, not all individuals with consanguinity and clinical symptoms will have a clear, single-gene cause that can be identified as homozygous mutations.
The IL-17 family of cytokines plays crucial roles in both acute and chronic inflammatory responses and IL-17A, the hallmark cytokine of the Th17 cells, has important roles in protecting the host against extracellular pathogens, but also promotes inflammatory pathology in autoimmune disease (55).
The mutation in STAT1 affects the differentiation of Th17 cells, which are a subset of CD4+ T cells involved in epithelial cell- and neutrophil-mediated immune responses against extracellular microbes and in the pathogenesis of autoimmune diseases. The same mutation along with the c.1342C>T mutation in TYK2 also affects the JAK-STAT signaling pathway. This is the principal signaling mechanism for a wide array of cytokines and growth factors. Following the binding of cytokines to their cognate receptor, STATs are activated by members of the JAK family of tyrosine kinases. Once activated, they dimerize and translocate to the nucleus and modulate the expression of target genes. In addition to the activation of STATs, JAKs mediate the recruitment of other molecules such as the MAPKs and PI3K. These molecules process downstream signals via the Ras/Raf/MAPK and PI3K pathways, which results in the activation of additional transcription factors.
Our analysis also hints towards a deregulation in the NET formation through the mutation in ELANE. Neutrophils play a central role in innate immune defense. One of the mechanisms of neutrophil action is the formation of NETs, the extracellular structures composed of chromatin coated with histones, proteases and granular and cytosolic proteins that help catch and kill microorganisms.
Finally, two more pathways affected by mutations in GPSM1 and IL17RC were those of cocaine addiction and cytokine-cytokine receptor interaction, respectively.
The present study has certain limitations, which should be acknowledged. It focuses on the Jordanian population, thus limiting the generalizability of the present findings. Furthermore, the sample size was relatively small (nine families), potentially affecting the statistical power of the study. In addition, the present study lacks functional validation of the main findings. However, it still provides a direction for future research.
In conclusion, the intricate tapestry of primary immunodeficiencies is gradually being unraveled with the aid of advanced genetic techniques. Understanding the genetic basis of these disorders among consanguineous Jordanian families holds promise for the development of personalized therapeutic interventions and improved patient outcomes.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be requested from the corresponding author. All sequencing data have been uploaded to the Figshare repository (10.6084/m9.figshare.26183549).
Authors' contributions
LO, AZ and MAH proposed and designed the study. LO and AZ carried out the molecular genetic studies. LO, RA and AZ analyzed the results, performed immunological description of the patients and statistical analysis. RA, EA and MAH prepared patient samples and extracted DNA for genetic studies. AZ and LO drafted the manuscript. AZ edited the manuscript. All authors have read and agreed to the final version of the manuscript. LO and AZ confirm the authenticity of all the raw data.
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethical Committee of the Jordanian Royal Medical Services (11/11/2021), and was supervised by Dr Raed Al-Zyoud (Consultant Paediatric Allergist, Immunologist and Rheumatologist at Royal Medical services, Amman, Jordan). Written informed consent to participate in the study was obtained from all subjects and family members involved in the study. Parents/guardians provided consent for minor probands.
Patient consent for publication
The patients and their family members provided written consent for their medical data to be published. The parents/guardians provided consent for minor probands.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Geha RS, Notarangelo LD, Casanova JL, Chapel H, Conley ME, Fischer A, Hammarström L, Nonoyama S, Ochs HD, Puck JM, et al: Primary immunodeficiency diseases: An update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 120:776–794. 2007.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, Klein C, Morio T, Oksenhendler E, Picard C, et al: Human inborn errors of immunity: 2022 update on the classification from the International Union of immunological societies expert committee. J Clin Immunol. 42:1473–1507. 2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, Klein C, Morio T, Oksenhendler E, Picard C, et al: The ever-increasing array of novel inborn errors of immunity: An interim update by the IUIS committee. J Clin Immunol. 41:666–679. 2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Erdős M, Mironska K, Kareva L, Stavric K, Hasani A, Lányi Á, Kállai J and Maródi L: A novel mutation in SLC39A7 identified in a patient with autosomal recessive agammaglobulinemia. The impact of the J Project. Pediatr Allergy Immunol. 33(e13805)2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Notarangelo LD: Primary immunodeficiencies. J Allergy Clin Immunol. 125 (2 Suppl 2):S182–S194. 2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Boyle JM and Buckley RH: Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 27:497–502. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bonilla FA, Khan DA, Ballas ZK, Chinen J, Frank MM, Hsu JT, Keller M, Kobrynski LJ, Komarow HD, Mazer B, et al: Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 136:1186–1205.e1-78. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Oliveira JB and Fleisher TA: Laboratory evaluation of primary immunodeficiencies. J Allergy Clin Immunol. 125 (2 Suppl 2):S297–S305. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Shehata N, Palda V, Bowen T, Haddad E, Issekutz TB, Mazer B, Schellenberg R, Warrington R, Easton D, Anderson D and Hume H: The use of immunoglobulin therapy for patients with primary immune deficiency: An evidence-based practice guideline. Transfus Med Rev. 24 (Suppl 1):S28–S50. 2010.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Amaya-Uribe L, Rojas M, Azizi G, Anaya JM and Gershwin ME: Primary immunodeficiency and autoimmunity: A comprehensive review. J Autoimmun. 99:52–72. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lam HY, Clark MJ, Chen R, Chen R, Natsoulis G, O'Huallachain M, Dewey FE, Habegger L, Ashley EA, Gerstein MB, et al: Performance comparison of whole-genome sequencing platforms. Nat Biotechnol. 30:78–82. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
El Hawary RE, Meshaal SS, Abd Elaziz DS, Alkady R, Lotfy S, Eldash A, Erfan A, Chohayeb EA, Saad MM, Darwish RK, et al: Genetic testing in Egyptian patients with inborn errors of immunity: A single-center experience. J Clin Immunol. 42:1051–1070. 2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Rios JJ and Delgado MR: Using whole-exome sequencing to identify variants inherited from mosaic parents. Eur J Hum Genet. 23:547–550. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Arunachalam AK, Maddali M, Aboobacker FN, Korula A, George B, Mathews V and Edison ES: Primary immunodeficiencies in India: Molecular diagnosis and the role of next-generation sequencing. J Clin Immunol. 41:393–413. 2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lamborn I, Jing H, Zhang Y, Drutman SB, Abbott JK, Munir S, Bade S, Murdock HM, Santos CP, Brock LG, et al: Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J Exp Med. 214:1949–1972. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Aghamohammadi A, Rezaei N, Yazdani R, Delavari S, Kutukculer N, Topyildiz E, Ozen A, Baris S, Karakoc-Aydiner E, Kilic SS, et al: Consensus Middle East and North Africa Registry on inborn errors of immunity. J Clin Immunol. 41:1339–1351. 2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Aghamohammadi A, Mohammadinejad P, Abolhassani H, Mirminachi B, Movahedi M, Gharagozlou M, Parvaneh N, Zeiaee V, Mirsaeed-Ghazi B, Chavoushzadeh Z, et al: Primary immunodeficiency disorders in Iran: Update and new insights from the third report of the national registry. J Clin Immunol. 34:478–490. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Abolhassani H, Kiaee F, Tavakol M, Chavoshzadeh Z, Mahdaviani SA, Momen T, Yazdani R, Azizi G, Habibi S, Gharagozlou M, et al: Fourth update on the Iranian National registry of primary immunodeficiencies: Integration of molecular diagnosis. J Clin Immunol. 38:816–832. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Abolhassani H, Azizi G, Sharifi L, Yazdani R, Mohsenzadegan M, Delavari S, Sohani M, Shirmast P, Chavoshzadeh Z, Mahdaviani SA, et al: Global systematic review of primary immunodeficiency registries. Expert Rev Clin Immunol. 16:717–732. 2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Kilic SS, Ozel M, Hafizoglu D, Karaca NE, Aksu G and Kutukculer N: The prevalances (Correction) and patient characteristics of primary immunodeficiency diseases in Turkey-two centers study. J Clin Immunol. 33:74–83. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Cheng J, Myers TG, Levinger C, Kumar P, Kumar J, Goshu BA, Bosque A and Catalfamo M: IL-27 induces IFN/STAT1-dependent genes and enhances function of TIGIT+ HIVGag-specific T cells. iScience. 25(103588)2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kamiya S, Owaki T, Morishima N, Fukai F, Mizuguchi J and Yoshimoto T: An indispensable role for STAT1 in IL-27-induced T-bet expression but not proliferation of naive CD4+ T cells. J Immunol. 173:3871–3877. 2004.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang Y, Ma CS, Ling Y, Bousfiha A, Camcioglu Y, Jacquot S, Payne K, Crestani E, Roncagalli R, Belkadi A, et al: Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J Exp Med. 213:2413–2435. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tolomeo M, Cavalli A and Cascio A: STAT1 and its crucial role in the control of viral infections. Int J Mol Sci. 23(4095)2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, Noel RJ, Verbsky JW, Freeman AF, Janssen E, et al: Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J Allergy Clin Immun. 31:1611–1623. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Sampaio EP, Hsu AP, Pechacek J, Bax HI, Dias DL, Paulson ML, Chandrasekaran P, Rosen LB, Carvalho DS, Ding L, et al: Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol. 131:1624–1634. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Tsumura M, Okada S, Sakai H, Yasunaga S, Ohtsubo M, Murata T, Obata H, Yasumi T, Kong XF, Abhyankar A, et al: Dominant-negative STAT1 SH2 domain mutations in unrelated patients with Mendelian susceptibility to mycobacterial disease. Hum Mutat. 33:1377–1387. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Frede N, Rojas-Restrepo J, Caballero Garcia de Oteyza A, Buchta M, Hübscher K, Gámez-Díaz L, Proietti M, Saghafi S, Chavoshzadeh Z, Soler-Palacin P, et al: Genetic analysis of a cohort of 275 patients with Hyper-IgE syndromes and/or chronic mucocutaneous candidiasis. J Clin Immunol. 41:1804–1838. 2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lv G, Sun G, Wu P, Du X, Zeng T, Wen W, Zhou L, An Y, Tang X, He T, et al: Novel mutations of TYK2 leading to divergent clinical phenotypes. Pediatr Allergy Immunol. 33(e13671)2022.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Levy R, Okada S, Béziat V, Moriya K, Liu C, Chai LY, Migaud M, Hauck F, Al Ali A, Cyrus C, et al: Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc Natl Acad Sci USA. 113:E8277–E8285. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Bonilla FA and Geha RS: Update on primary immunodeficiency diseases. J Allergy Clin Immunol. 117:S435–S441. 2006.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Okada S, Puel A, Casanova JL and Kobayashi M: Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin Transl Immunology. 5(e114)2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, et al: Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol. 4:1023–1028. 2003.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Garcia-Laorden MI, Sole-Violan J, Rodriguez de Castro F, Aspa J, Briones ML, Garcia-Saavedra A, Rajas O, Blanquer J, Caballero-Hidalgo A, Marcos-Ramos JA, et al: Mannose-binding lectin and mannose-binding lectin-associated serine protease 2 in susceptibility, severity, and outcome of pneumonia in adults. J Allergy Clin Immunol. 122:368–374, 374.e1-2. 2008.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ling Y, Cypowyj S, Aytekin C, Galicchio M, Camcioglu Y, Nepesov S, Ikinciogullari A, Dogu F, Belkadi A, Levy R, et al: Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med. 212:619–631. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Miao J, Zhang J, Huang X, Wu N, Wu D and Shen M: NLRP12-associated autoinflammatory disease in Chinese adult patients: A single-centre study. RMD Open. 9(e003598)2023.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wang W, Zhou Y, Zhong LQ, Li Z, Jian S, Tang XY and Song HM: The clinical phenotype and genotype of NLRP12-autoinflammatory disease: A Chinese case series with literature review. World J Pediatr. 16:514–519. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kingdom R and Wright CF: Incomplete penetrance and variable expressivity: From clinical studies to population cohorts. Front Genet. 25(920390)2022.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhang D, Su G, Hao S, Lai J and Feng S: Paediatric autoimmune diseases with ELANE mutations associated with neutropenia. Pediatr Rheumatol Online J. 21(41)2023.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Skokowa J, Germeshausen M, Zeidler C and Welte K: Severe congenital neutropenia: Inheritance and pathophysiology. Curr Opin Hematol. 14:22–28. 2007.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Ferreira RC, Pan-Hammarström Q, Graham RR, Gateva V, Fontán G, Lee AT, Ortmann W, Urcelay E, Fernández-Arquero M, Núñez C, et al: Association of IFIH1 and other autoimmunity risk alleles with selective IgA deficiency. Nat Genet. 42:777–780. 2010.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Song J, Li M, Li C, Liu K, Zhu Y and Zhang H: Friend or foe: RIG-I like receptors and diseases. Autoimmun Rev. 21(103161)2022.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Huang P, Wu JJ, Zhang JW, Hou YQ, Zhu P, Yin R, Yu RB, Zhang Y, Yue M and Hou W: Genetic variants of IFIH1 and DHX58 affect the chronicity of hepatitis C in the Chinese Han population. PeerJ. 11(e14740)2023.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zhang J, Liu X, Meng Y, Wu H, Wu Y, Yang B and Wang L: Autoimmune disease associated IFIH1 single nucleotide polymorphism related with IL-18 serum levels in Chinese systemic lupus erythematosus patients. Sci Rep. 8(9442)2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Xiao W, Feng J, Long H, Xiao B and Luo ZH: Case report: Aicardi-Goutières syndrome and singleton-merten syndrome caused by a gain-of-function mutation in IFIH1. Front Genet. 12(660953)2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Bursztejn AC, Briggs TA, del Toro Duany Y, Anderson BH, O'Sullivan J, Williams SG, Bodemer C, Fraitag S, Gebhard F, Leheup B, et al: Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: Overlap between Aicardi-Goutières and Singleton-Merten syndromes. Br J Dermatol. 173:1505–1513. 2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Asgari S, Schlapbach LJ, Anchisi S, Hammer C, Bartha I, Junier T, Mottet-Osman G, Posfay-Barbe KM, Longchamp D, Stocker M, et al: Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc Natl Acad Sci USA. 114:8342–8347. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Cananzi M, Wohler E, Marzollo A, Colavito D, You J, Jing H, Bresolin S, Gaio P, Martin R, Mescoli C, et al: IFIH1 loss-of-function variants contribute to very early-onset inflammatory bowel disease. Hum Genet. 140:1299–1312. 2021.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Shen M, Tang L, Shi X, Zeng X and Yao Q: NLRP12 autoinflammatory disease: A Chinese case series and literature review. Clin Rheum. 36:1661–1667. 2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Gruber C and Bogunovic D: Incomplete penetrance in primary immunodeficiency: A skeleton in the closet. Hum Genet. 139:745–757. 2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Erzurumluoglu AM, Shihab HA, Rodriguez S, Gaunt TR and Day IN: Importance of genetic studies in consanguineous populations for the characterization of novel human gene functions. Ann Hum Genet. 80:187–196. 2016.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Rodríguez-Ubreva J, Calvillo CL, Satter LR and Ballestar E: Interplay between epigenetic and genetic alterations in inborn errors of immunity. Trends Immunol. 44:902–916. 2023.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Peng XP, Al-Ddafari MS, Caballero-Oteyza A, El Mezouar C, Mrovecova P, Dib SE, Massen Z, Smahi MC, Faiza A, Hassaïne RT, et al: Next generation sequencing (NGS)-based approach to diagnosing Algerian patients with suspected inborn errors of immunity (IEIs). Clin Immunol. 256(109758)2023.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Romano R, Cillo F, Moracas C, Pignata L, Nannola C, Toriello E, De Rosa A, Cirillo E, Coppola E, Giardino G, et al: Epigenetic alterations in inborn errors of immunity. J Clin Med. 11(1261)2022.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Isailovic N, Daigo K, Mantovani A and Selmi C: Interleukin-17 and innate immunity in infections and chronic inflammation. J Autoimmun. 60:1–11. 2015.PubMed/NCBI View Article : Google Scholar
|














