Introduction
Langerhans cell histiocytosis (LCH) is an uncommon
hematological disease with an average incidence of 2-5 cases per
million children and 1-2 cases per million adults annually
worldwide (1). The precise etiology
and pathogenesis of this disorder remain to be understood. Early
hypotheses suggested that LCH may be a reactive process, but recent
evidence identifying oncogenic BRAF or MAP2K1 mutations in the
majority of LCH cases has prompted a reassessment (1,2). This
evidence points to LCH as a clonal neoplasm originating from
unchecked proliferation and accumulation of immature myeloid
dendritic cells in bone marrow (1-3).
The clinical presentations of LCH include granulomatous lesions
consisting of clonal pathologic histiocytes and are linked to a
mortality rate of 10 to 20% (4).
Under the updated revised classification system of
histiocytoses, LCH has been split into three categories:
Single-system LCH, lung LCH and multi-system LCH with or without
risk organ involvement (spleen, liver and bone marrow) (5). The most common form of single-system
LCH is bone involvement, which can manifest as uni- or multi-focal
(5). In the case of multi-system
LCH, the bones are usually the most affected site, followed by the
pituitary gland and lung (6). Liver
involvement is less frequent, with incidence rates in adults of
16-27% of all LCH cases, while it is more frequent in children with
multisystem LHC, with an incidence reported from 19-60% (7).
The current study presents a rare case involving
bone, skin, lung and liver involvement in an adult multi-system
LCH, with the aim of contributing to reviewing the current
literature on this disease and providing guidance for medical
practitioners who encounter similar cases in their practice.
Case report
A 49-year-old man presented to the First People's
Hospital of Foshan, Foshan, China with a 1-week history of itchy
skin over his entire body on October, 2023. At first, food
allergies were suspected given that the patient presented with red
rashes scattered across his back and face. The vital signs were
stable after admission and the findings of physical examination
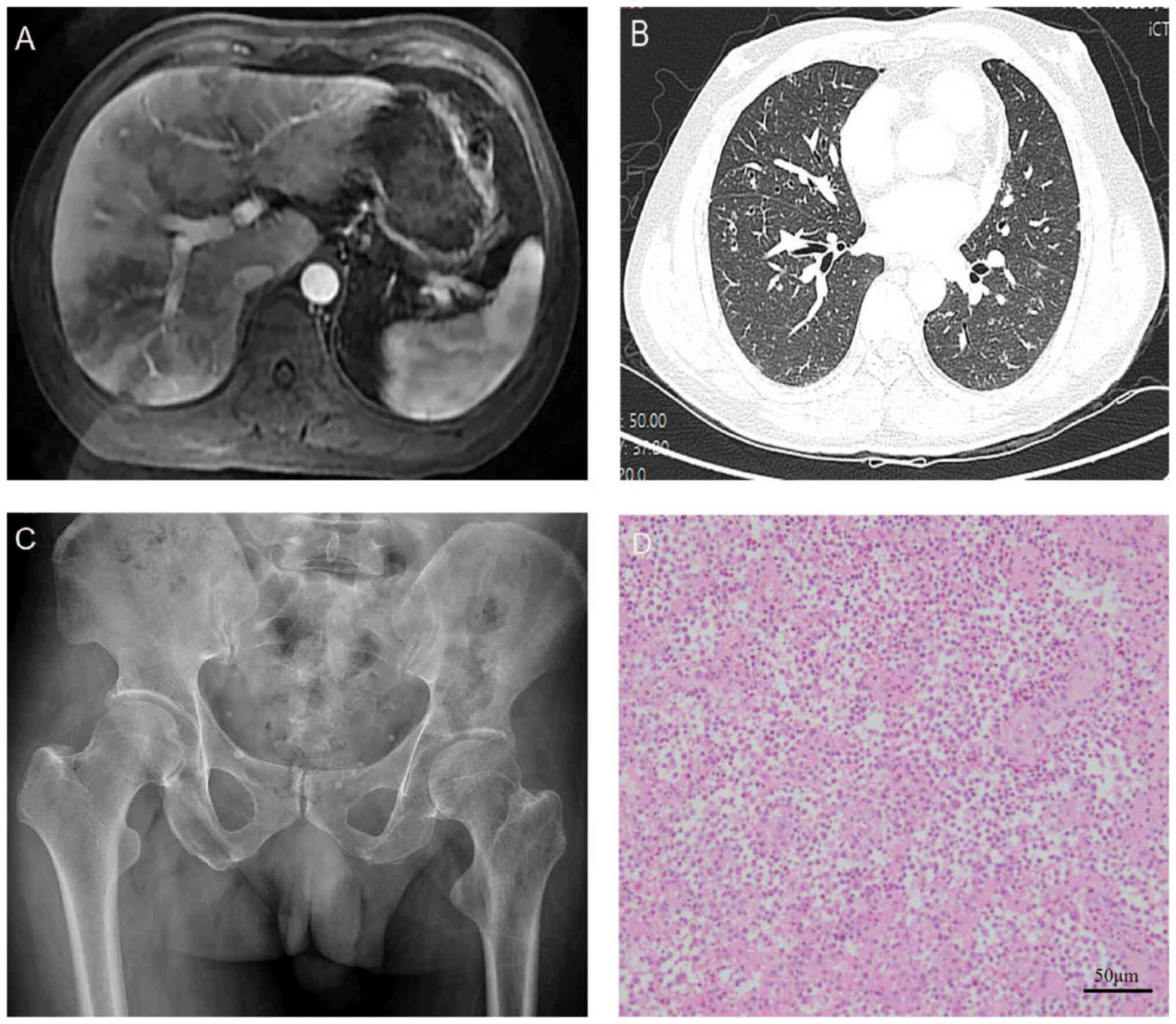
were all normal. CT imaging revealed multiple nodules and spots in
the chest and liver, indicating the possibility of infectious
lesions (Fig. 1A and B). However, conventional laboratory tests
(such as routine blood tests, condensation profile, procalcitonin,
protein C, α-fetoprotein and bone marrow examination) did not find
any abnormalities. The medical history of the patient revealed that
the patient had previously undergone resection of left iliac bone
lesions (Fig. 1C and D), and the histopathology examination
confirmed that the diagnosis was Langerhans cell histiocytosis
(LCH). Immunohistochemistry analysis showed positive staining for
S-100 protein, CD163, CD68, CD1a, lysozyme and langerin (CD207)
antigen. Therefore, the final diagnosis was determined to be
multi-system LCH with bone, skin, lung and liver involvement. The
patient was initiated on the front line drug, cytarabine, and after
three rounds of treatment, his condition did not improve
significantly. At the 2-year follow-up from the initial diagnosis,
the patient was still alive at the last follow-up on April,
2024.
Discussion
LCH, a rare inflammatory myeloid neoplasm that
affects both children and adults, may present with the accumulation
of mononuclear phagocytes in a wide range of tissues and organs,
including bone, oral cavity, skin, anogenital regions, liver,
lungs, spleen and lymph nodes (8).
Children tend to be more affected than adults. The pathogenesis of
LCH is not fully understood (9).
Whether LCH is reactive inflammation or neoplastic remains a matter
of debate. Nowadays, the etiology is generally considered to be a
BRAFV600E mutation, while activated MAP2K1 is the most
prevalent mutation in non-BRAFV600E mutation cases
(3). It is reported that
BRAFV600E, BRAF deletion and MAP2K1 mutation were
detected in 38.8, 25.4 and 19.4% of patients, respectively
(6). BRAF deletion has been
revealed to be associated with multisystem LHC in adult patients,
specifically those with liver involvement (6). Pathologically, LCH is distinguished by
the presence of abnormal Langerhans cell-like cells mixed with
inflammatory cells (10). The
pathogenesis of LCH may be related to cytokine storm in serum and
lesions. Ismail et al (11)
confirmed the finding of higher levels of plasma inflammatory
factor IL-17A in patients with LCH compared with controls. The
disease may manifest as a single-system disorder or as a
multi-system disorder, and acute-phase protein ITIH4 levels may
distinguish multi-system from single-system LCH (10). In the present study, although only
one enhancement lesion in the left iliac bone was pathologically
confirmed, typical imaging and findings of LCH were observed in the
skin, lung and liver. Therefore, the LCH in the current report was
considered to be multi-system.
Bone is the most commonly affected organ in patients
with LCH, involving single or multiple osseous sites. The head and
neck region is particularly susceptible, while iliac bone
infringement is relatively uncommon (12). Hashimoto et al (9) reported that all pediatric cases
involved the bones of the extremities, and all adult cases involved
the bones of the trunk. Patients with osseous LCH typically present
with symptoms such as swelling or pain. Skin involvement of LCH may
mirror a skin-limited disease that may resolve spontaneously or
with brief chemotherapy (13). It
is reported that 53% of individuals with multi-system LCH developed
skin lesions (13), which appear to
be a clinical indicator of a potentially multi-system LCH. Lung
involvement is not a common manifestation of LCH, and is associated
with smoking (14). Pulmonary LCH
is typically identified on CT as the presence of multiple nodules
and cysts.
In the present case, the predominant radiological
finding was that of multiple cystic lesions. Liver involvement is a
poor prognostic factor that indicates poor overall survival (OS)
and event-free survival (EFS) in adult LCH (6). Furthermore, simultaneous involvement
of the lung, skin, liver, and bone is uncommon, making multi-system
LCH a high-risk condition. In multi-system LCH with risk organ
involvement, survival rates decline to 70%, and roughly one-third
of patients witness disease reactivation post-treatment (3). Clinical manifestations are very
heterogeneous and the type and number of organs involved will
determine the symptoms that occur. Pathologic diagnosis is
characterized by infiltration of oval shaped cells with coffee
bean-like nuclei that express CD1a, S100 and Langerin (CD207)
(3,15). The current frontline treatment for
multi-system LCH remains empirically derived chemotherapy,
consisting of agents such as vinblastine, prednisone, cytarabine
and nucleoside analogues (such as cladribine) (15).
In a recent cohort, it was found that patients
receiving cytarabine-based first-line treatment had improved
event-free survival and overall survivals compared with patients
receiving other treatments (6).
However, the symptoms of the patient in the current study did not
improve when using cytarabine. Promising targeted agents (such as
BRAF and MEK inhibitors) alone or in combination with chemotherapy
are also under investigation.
Awada et al (16) described a 41-year-old female LCH
patient with an activated BRAFV600E mutation who had a
remarkable response to dabrafenib (BRAF inhibitor) 150 mg twice
daily and trametinib (MEK inhibitor) 2 mg once daily within days.
In addition, the patient was PET/CT negative after 2 months and
plasma BRAFV600E-mutant ctDNA became undetectable during
treatment (16). As described by
Awada et al (16), to the
best of our knowledge, this is the first report of successful
treatment of BRAFV600E mutant LCH with combined BRAF MEK
inhibition, and it also shows that detection of
BRAFV600E mutant circulating tumor DNA in plasma is a
promising biomarker for this disease. Messinger et al
reported that the MEK inhibitor trametinib results in a strong
response in patients with LCH (17). In that report a neonatal LCH with
MAP2K1p.K57_G61del mutation did seem to have a complete response
lasting 22 months, and two adolescent male patients with LCH with
BRAF p.N486_P490del mutation received trametinib for >1 year
with no reactivation in one and partial response in another
(including stable lung disease). Experience with patients with LCH
suggests that future prospective clinical trials will most likely
involve a combination of targeted drugs or targeted drugs combined
with chemotherapy to achieve the optimal therapeutic benefits and
safety profile of this promising new therapeutic strategy.
LCH is a rare and complex disease that can affect
multiple organ systems. It is often under-diagnosed or delayed in
diagnosis. The current study presents the case of a patient with
LCH affecting multiple organs including the bone, skin, lung and
liver. The aim of the present study is to raise awareness of this
disease and to help healthcare professionals identify, diagnose and
treat similar cases more quickly.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Key Cultivation
Project of ‘14th Five-Year Plan’ in Foshan (grant no. FSPY145202)
and the Foshan Science and Technology Program Project (grant no.
2220001005255).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
QL and HZ wrote the manuscript. QL, XH and JY
contributed to initial patient assessment and follow up. QL and HZ
obtained and analyzed patient clinical data. QL and FD contributed
to conception and have reviewed the manuscript and consistently
improved its content. FD and HZ confirm the authenticity of all the
raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent was provided by the patient
for publication of data and images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Reisi N, Raeissi P, Harati Khalilabad T
and Moafi A: Unusual sites of bone involvement in Langerhans cell
histiocytosis: A systematic review of the literature. Orphanet J
Rare Dis. 16(1)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Sconocchia T, Foßelteder J, Sconocchia G
and Reinisch A: Langerhans cell histiocytosis: Current advances in
molecular pathogenesis. Front Immunol. 14(1275085)2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Harmon CM and Brown N: Langerhans cell
histiocytosis: A clinicopathologic review and molecular
pathogenetic update. Arch Pathol Lab Med. 139:1211–1214.
2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zinn DJ, Chakraborty R and Allen CE:
Langerhans cell histiocytosis: Emerging insights and clinical
implications. Oncology (Williston Park). 30:122–139.
2016.PubMed/NCBI
|
|
5
|
Emile JF, Abla O, Fraitag S, Horne A,
Haroche J, Donadieu J, Requena-Caballero L, Jordan MB, Abdel-Wahab
O, Allen CE, et al: Revised classification of histiocytoses and
neoplasms of the macrophage-dendritic cell lineages. Blood.
127:2672–2681. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Cao XX, Duan MH, Zhao AL, Cai H, Chen J,
Gao XM, Liu T, Cai HC, Zhang L, Sun J, et al: Treatment outcomes
and prognostic factors of patients with adult Langerhans cell
histiocytosis. Am J Hematol. 97:203–208. 2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Reginelli A, Pignatiello M, Urraro F,
Belfiore MP, Toni G, Vacca G and Cappabianca S: Langerhans cell
histiocytosis with uncommon liver involvement: A case report. Am J
Case Rep. 21(e923505)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Sato A, Kobayashi M, Yusa N, Ogawa M,
Shimizu E, Kawamata T, Yokoyama K, Ota Y, Ichinohe T, Ohno H, et
al: Clinical and prognostic features of Langerhans cell
histiocytosis in adults. Cancer Sci. 114:3687–97. 2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hashimoto K, Nishimura S, Sakata N, Inoue
M, Sawada A and Akagi M: Treatment outcomes of langerhans cell
histiocytosis: A retrospective study. Medicina (Kaunas).
57(356)2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Murakami I, Oh Y, Morimoto A, Sano H,
Kanzaki S, Matsushita M, Iwasaki T, Kuwamoto S, Kato M, Nagata K,
et al: Acute-phase ITIH4 levels distinguish multi-system from
single-system Langerhans cell histiocytosis via plasma peptidomics.
Clin Proteomics. 12(16)2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ismail MB, Åkefeldt SO, Lourda M, Gavhed
D, Aricò M, Henter JI, Delprat C and Valentin H: High levels of
plasma interleukin-17A are associated with severe neurological
sequelae in Langerhans cell histiocytosis. Cytokine.
126(154877)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Lian C, Lu Y and Shen S: Langerhans cell
histiocytosis in adults: A case report and review of the
literature. Oncotarget. 7:18678–18683. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Simko SJ, Garmezy B, Abhyankar H, Lupo PJ,
Chakraborty R, Lim KP, Shih A, Hicks MJ, Wright TS, Levy ML, et al:
Differentiating skin-limited and multisystem Langerhans cell
histiocytosis. J Pediatr. 165:990–996. 2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Vassallo R, Ryu JH, Schroeder DR, Decker
PA and Limper AH: Clinical outcomes of pulmonary Langerhans' cell
histiocytosis in adults. N Engl J Med. 346:484–490. 2002.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Allen CE, Merad M and McClain KL:
Langerhans-cell histiocytosis. N Engl J Med. 379:856–868.
2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Awada G, Seremet T, Fostier K, Everaert H
and Neyns B: Long-term disease control of Langerhans cell
histiocytosis using combined BRAF and MEK inhibition. Blood Adv.
2:2156–2158. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Messinger YH, Bostrom BC, Olson DR, Gossai
NP, Miller LH and Richards MK: Langerhans cell histiocytosis with
BRAF p.N486_P490del or MAP2K1 p.K57_G61del treated by the MEK
inhibitor trametinib. Pediatr Blood Cancer.
67(e28712)2020.PubMed/NCBI View Article : Google Scholar
|