Introduction
The term epidermolysis bullosa (EB) encompasses an
inherited clinically heterogenous group of skin conditions with a
frequency of 1 in 17,000 live births (1). At least 20 different genes are
associated with the diagnosis of EB, which displays profound
genetic heterogeneity, further emphasized by the phenomenon of one
gene leading to both autosomal-dominant and autosomal-recessive
pattern inheritance with different phenotypes (2,3). The
involved genes encode structural proteins, essential for
intraepidermal adhesion, dermo-epidermal anchorage and skin
integrity (3,4). Thus, the main clinical symptom is
mechanical skin fragility presenting as mucocutaneous blistering,
erosions and ulceration caused by minimal trauma or friction
(5). Extracutaneous manifestation
(oral, oesophageal, tracheal, genitourinary and ocular mucous
membranes) has also been documented. According to the most recent
consensus reclassification from 2020, the 4 main types of EB based
on the lesions' location are EB simplex, junctional EB, dystrophic
EB (DEB) and Kindler's EB (4).
Currently, the most effective diagnostic approach for EB includes
transmission electron microscopy and immunofluorescence antigen
mapping of affected skin, along with DNA mutational analysis. The
genetic test is a particularly important component of the
diagnostic process for diagnostic confirmation (with a subtype), as
it also provides the basis for proper genetic counseling and allows
the application of prenatal diagnosis. At present, next-generation
sequencing (NGS) is considered the preferable strategy, as it
offers the ability to screen for multiple sequence variations and
is less expensive (5).
The current study presents a case report of a female
patient with a clinical diagnosis of dystrophic epidermolysis
bullosa, identifying a novel variant in the type VII collagen α1
chain (COL7A1) gene.
Materials and methods
Patient
A 17-year-old female patient was referred by a
dermatology specialist to the Genomic Laboratory of the Center of
Competence, Medical University Pleven (Pleven, Bulgaria), for
genetic testing and genetic counseling in June 2022. After
obtaining informed consent from the patient's parents, blood
samples were derived (in EDTA plastic tubes) from the patient and
the patient's parents.
Germline pathogenic variant
detection
Following the manufacturer's protocol, genomic DNA
was extracted from each sample with the MagCore Genomic DNA Whole
blood kit (MagCore®). Genetic testing of the patient and
the patient's parents was performed by NGS. The TruSight One
Expanded panel (Illumina. Inc.) was employed to prepare a library
according to the manufacturer's protocol. The panel contained oligo
probes for exons and exon-intron boundaries of 6,699 genes
associated with single-gene disorders. The procedures were
conducted following the manufacturer's instructions. Qualified
libraries were sequenced using the Illumina NextSeq 550 platform
with a 2x150 bp configuration (Illumina. Inc.), aligning the reads
to the hg19 reference human genome (6). Data output files in gVCF format were
entered into BaseSpace Variant Interpreter (Illumina. Inc.). Custom
filters (minimum read depth of 20x per variant and excluded silent
variants) were created and applied to improve variant annotation
and interpretation. The five-tier terminology system of the
American College of Medical Genetics and Genomics (7) was used for variant classification as
follows: Pathogenic, likely pathogenic, variant of unknown clinical
significance, likely benign and benign. The variants automatically
annotated by the software were manually checked in the primary
human genome databases ClinVar (www.ncbi.nlm.noh.gov/clinvar), dbSNP (www.ncbi.nlm.noh.gov/projrct/SNP) and
Ensembl (http://www.ensembl.org).
Results
Clinical data
The patient had been diagnosed with EB at birth.
According to the patient's history, during the first days after
birth, the following lesions had appeared: Vesicular rash on the
feet's dorsal surface, large bulla on the third and fourth finger
of the left hand, and vesicles on the lower lip and tongue. A skin
biopsy was performed on the third day after delivery for
histopathological evaluation. The results from the latter were as
follows: Subepidermal peeling of the epidermis with severe
hyperkeratosis, the granular layer was composed of by one cell row;
periodic acid-Schiff staining showed parts of the basement
membrane. The condition displayed a chronic manifestation with a
daily appearance of new vesicles, which, after bursting,
transformed into erosions that tended to heal for 7-10 days and
eventually evolved into white scars. The predominantly affected
areas were the palms, feet, elbows and knees. Due to the formation
of vesicles under the nails, the latter had fallen off completely
on all of the toes and partially on the fingers, including the
right thumb and index finger and left index and middle finger. The
patient self-reported the occasional appearance of erosions in the
oral cavity (tongue, gingival mucosa), which does not interfere
with proper eating habits. The patient complained of difficulties
swallowing, which had started a few months ago. X-ray examination
indicated no oesophageal stenosis. The last dermatology examination
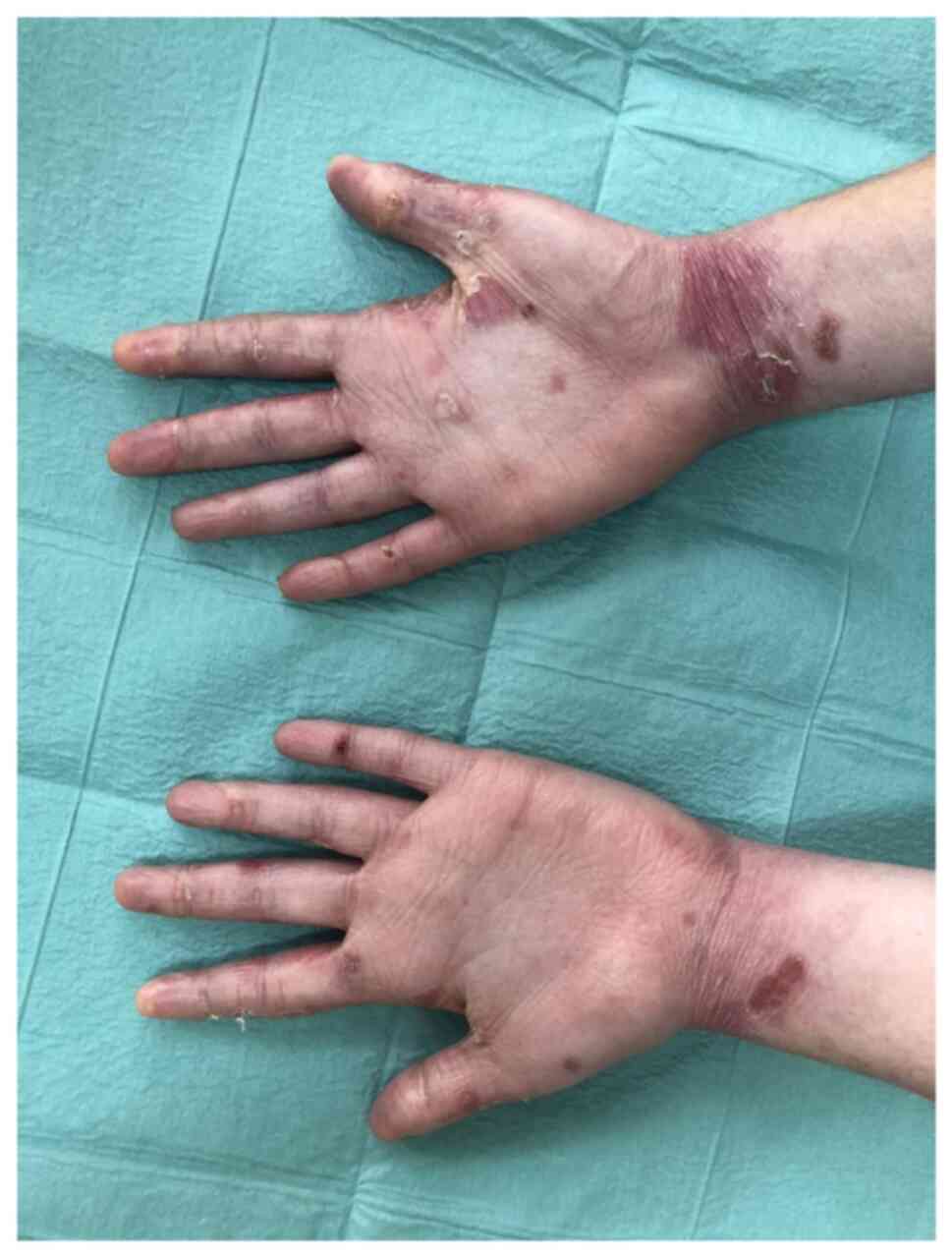
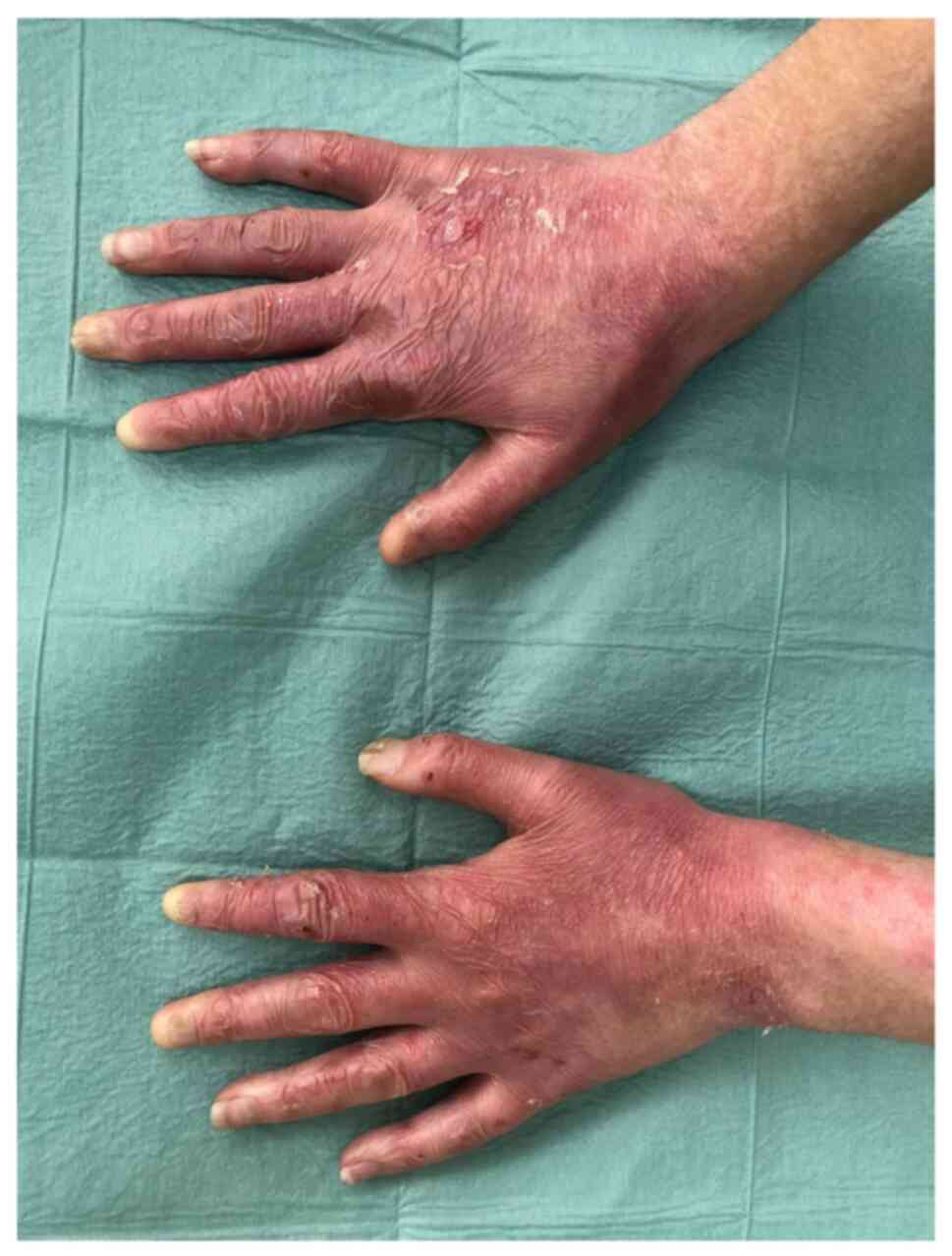
revealed blisters with clear content involving the dorsal and
palmar surfaces of the fingers, palms, feet, toes, elbows and knees
(Fig. 1). There were single
erosions on the gums and back of the tongue. The fingernails were
dystrophic (Fig. 2) and both toes
of both feet were with anonychia. There were no pathological
changes involving the scalp.
Genetic testing results
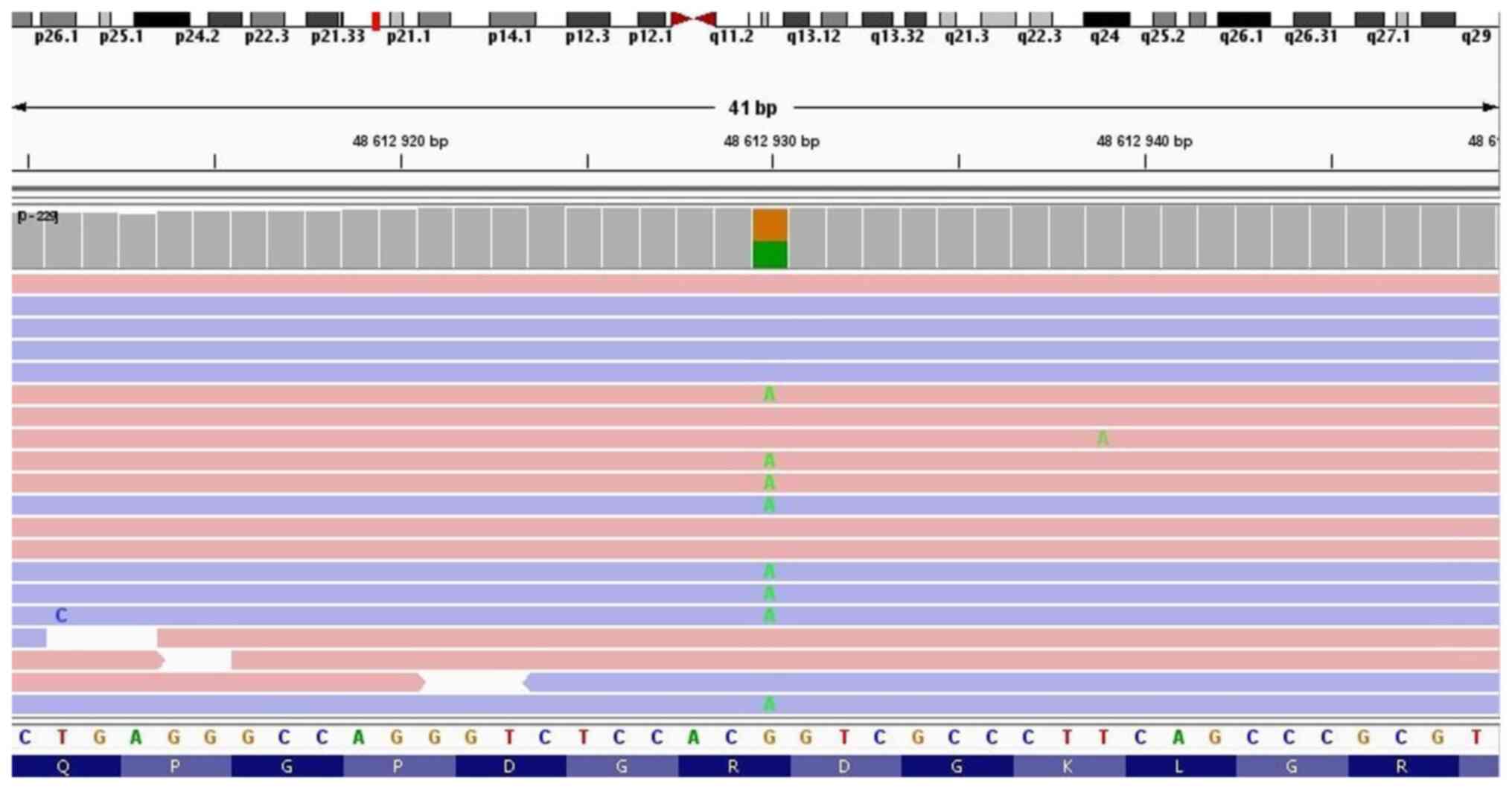
NGS testing of the patient detected two clinically
significant variants in the COL7A1 gene in a compound heterozygous
state: c.6022C>T p.(Arg2008Cys) (NM_000094.3) (Fig. 3) and c.3474del p.(Val1160Ter)
(NM_000094.3) (Fig. 4). The first
one c.6022C>T is a missense variant in which the amino acid
arginine (basic and polar) is substituted by cysteine (neutral and
slightly polar) in codon 6,022. According to the ClinVar database,
it is observed in individuals with dystrophic epidermolysis bullosa
and is classified as pathogenic. The second variant, c.3474del, is
a single base deletion of thymine in 3,474th position of the
nucleotide sequence. Although expected to result in a frameshift,
the amino acids Pro1158 and Gly1159 are not altered. The variant
leads to the formation of a premature translational stop signal
situated on exon 26. It has not been reported yet in the ClinVar
database and is classified as likely pathogenic. Segregation
analysis of the patient's parents revealed the c.6022C>T
p.(Arg2008Cys) variant (COL7A1 gene) in a heterozygous state in the
father and the c.3474del p.(Val1160Ter) variant in a heterozygous
state in the mother. Thus, the parental origin of each
COL7A1 variant in the patient was identified.
Discussion
DEB is the second most common type of EB, accounting
for 30% of all cases (8). It can be
inherited in an autosomal-dominant and autosomal-recessive manner.
Both modes of inheritance are attributed to mutations in the
COL7A1 gene, which encodes type VII collagen. The latter is
an essential component of the anchoring fibrils and is responsible
for epidermal basement membrane attachment to the dermal
extracellular matrix proteins (9).
Thus, the anchoring fibrils in patients with DEB are
morphologically altered and lead to sub-lamina densa plane of
tissue separation (10). The
clinical manifestation exhibits great variability-from isolated
nail dystrophy through to mild localized skin blistering to
generalized blistering with extracutaneous involvement (11). The dominant subtype of DEB (DDEB)
tends to exhibit less severe symptoms. Typical manifestations
include blistering limited to hands, feet, knees, elbows associated
with milia, atrophic scarring and nail dystrophy. Еxtracutaneous
involvement is rare (12).
Recessive DEB (RDEB) is less prevalent (1). The most common RDEB subtypes are the
severe generalized type (formerly known as Hallopeau-Siemens) and
intermediate RDEB (formerly known as non-Hallopeau-Siemens)
(13). Severe RDEB manifests as
congenital widespread skin blistering resulting in extensive
scarring with pseudosyndactyly, alopecia, extracutaneous
involvement such as corneal abrasions, mucous membrane blistering,
oesophageal strictures, kidney problems and cardiomyopathy
(13,14). Intermediate RDEB exhibits less
severe blistering with oral, dental, hair and nail changes
(14,15). Extracutaneous complications and
pseudosyndactyly are less common (16). Both subtypes are associated with an
increased risk of squamous cell carcinoma (15). The COL7A1 gene displays great
allelic heterogeneity, as >800 genetic variants have been
documented (17). It is difficult
to define clear genotype-phenotype correlations, given the massive
number of listed pathogenic variants and the clinical overlapping
among DEB subtypes.
However, certain patterns stand out. Multiple
mutations occur in exons 73-75, where 75% of DDEB pathogenic
variants reside. DDEB is predominantly a result of glycine
substitutions within the triple helix of the COL7A1 gene,
which affects the stability of fibrils involved in dermo-epidermal
anchorage (10,18). Severe RDEB is generally associated
with premature termination codon (PTC) in both COL7A1
alleles due to nonsense, frameshift and splice-site mutations,
leading to an insufficiency or a total lack of production of type
VII collagen. Intermediate RDEB results from compound
heterozygosity of the PTC variant on one allele and missense
variant on the other or simultaneous presence of two missense
variants on both alleles, i.e. at least one missense variant in the
COL7A1 gene is always present. The expected outcome is the
production of somewhat functional but abnormal type VII collagen
(10,14). Clinical manifestation of DDEB and
intermediate RDEB tends to be indistinguishable, posing diagnostic
difficulties, particularly if there is no family history of EB
(19).
In the presented case, the patient was assigned a
diagnosis of DEB. There was no family history of similar skin
conditions. The presented manifestation was relatively mild and
classifying it preliminarily as dominant or recessive was
difficult. Therefore, a segregation approach was considered most
suitable for diagnostic precision-the patient and the patient's
parents were simultaneously tested via NGS. The sequence data
analysis revealed a compound heterozygous state in the patient's
COL7A1 gene associated with the recessive form of DEB.
Segregation analysis of the patient's parents further revealed
their heterozygous carrier status and thus established the parental
origin of both COL7A1 variants. The paternal variant was
found to be c.6022C>T p.(Arg2008Cys). It is a missense located
in exon 73 that leads to substitution of the amino acid arginine
(basic and polar) by cysteine (neutral and slightly polar) at codon
2,008 of the COL7A1 protein. The Arg2008 amino acid is
considered to be clinically significant. Reported variants that
disrupt it have been classified as pathogenic. In silico
analysis shows that the variant has a deleterious effect on the
protein encoded by the COL7A1 gene. It has been reported in
individuals affected by DEB (https://www.ncbi.nlm.nih.gov/clinvar/variation/522791/).
According to the Genome Aggregation Database (GnomAD; https://gnomad.broadinstitute.org/), the
frequency of the c.6022C>T p.(Arg2008Cys) variant regarding the
non-Finnish European population is 0.000009. Based on the American
College of Medical Genetics and Genomics (ACMG) guidelines
(7), it was classified as
pathogenic. The variant of maternal origin was found to be
c.3474del p.(Val1160Ter). It is a novel one that had not been
identified before the analysis. Its frequency is unknown as it is
absent from the GnomAD database. The variant is a deletion of a
single nucleotide that leads to the formation of a PTC in exon 26,
out of 118 exons. It is expected to result in an absent or
truncated protein product. According to the ACMG guidelines, it was
predicted to be likely pathogenic.
In agreement with the literature data, the case
presents as an RDEB with a milder manifestation associated with a
missense and a PTC variant in a compound heterozygous state. The
missense mutation is situated in exon 73, even though the type of
DEB is not dominant. The inconclusive phenotype and lack of family
history necessitated DNA testing to establish the genetic origin of
DEB in the patient along with the right pattern of inheritance. Not
only did it provide an exact genetic diagnosis, but it also allowed
for informed reproductive choices to be undertaken within the
family. Establishing the carrier status of patients' parents for
variants in the COL7A1 gene determines a 25% risk for each
of their subsequent pregnancies to end up with an affected
offspring. Concerning the optimization of patient reproduction, it
is recommended that the patient's future partner is tested for
their COL7A1 gene carrier status. The latter is necessary to
estimate the risk for an affected offspring. If no pathogenic
variants in the COL7A1 gene of the partner are detected, the
risk is virtually zero. Detection of carrier status determines a
50% risk for an affected offspring.
In conclusion, the presented case with detected
variants in the COL7A1 gene may contribute to the
establishment of more certain genotype-phenotype correlations. The
overlapping of symptoms in different subtypes of DEB requires
genetic testing via NGS to determine the exact inheritance pattern.
The latter is essential information for accurate genetic counseling
regarding the subsequent reproduction of the patients' parents and
the patients' own reproduction.
Acknowledgements
The authors would like to thank Mrs. Yanka
Tzvetanova, a philologist and a senior teacher of English for
Medical Purposes and General English, who provided assistance in
editing the language of the current paper.
Funding
Funding: This study was supported by the European Regional
Development Fund with the leading organization Medical University
Pleven (grant no. BG05M2OP001-1.002-0010-C01).
Availability of data and materials
The NGS datasets generated and/or analyzed during
the current study are available in the ClinVar repository
(https://www.ncbi.nlm.nih.gov/clinvar/variation/3257728/).
The other data generated in the present study are included in the
figures and/or tables of this article.
Authors' contributions
IAY recruited the patient. SEN, ZBK, IAY and PPV
collected clinical and biological data. SEN and ZBK performed the
molecular analysis. IAY was responsible for the diagnosis and
treatment of the patient. ZBK analyzed the data. SEN, ZBK and KSK
were involved in the writing and revision of the manuscript. SEN
and PPV reviewed and revised the manuscript. All authors have read
and approved the final manuscript. SEN and ZBK confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
This study was approved by the Ethics Commission of
the Medical University Pleven (Pleven, Bulgaria). The patient's
parents provided written informed consent regarding their own
participation in the study along with the participation of the
patient (their daughter), who was <18 years of age at the time
of the analysis.
Patient consent for publication
Written informed consent for the publication of
their data was obtained from the patient's parents.
Competing interests
The authors have no competing interests to
declare.
References
|
1
|
Siañez-González C, Pezoa-Jares R and
Salas-Alanis JC: Congenital epidermolysis bullosa: A review. Actas
Dermosifiliogr. 100:842–856. 2009.PubMed/NCBI(In Spanish).
|
|
2
|
Prodinger C, Reichelt J, Bauer JW and
Laimer M: Epidermolysis bullosa: Advances in research and
treatment. Exp Dermatol. 28:1176–1189. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Sait H, Srivastava S and Saxena D:
Integrated management strategies for epidermolysis bullosa: Current
insights. Int J Gen Med. 15:5133–5144. 2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mariath LM, Santin JT, Schuler-Faccini L
and Kiszewski AE: Inherited epidermolysis bullosa: Update on the
clinical and genetic aspects. An Bras Dermatol. 95:551–569.
2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bardhan A, Bruckner-Tuderman L, Chapple
ILC, Fine JD, Harper N, Has C, Magin TM, Marinkovich MP, Marshall
JF, McGrath JA, et al: Epidermolysis bullosa. Nat Rev Dis Primers.
6(78)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Homo sapiens genome assembly GRCh37.
(n.d.). NCBI. https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001405.13/.
|
|
7
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hou PC, Del Agua N, Lwin SM, Hsu CK and
McGrath JA: Innovations in the treatment of dystrophic
epidermolysis bullosa (DEB): Current landscape and prospects. Ther
Clin Risk Manag. 19:455–473. 2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Nyström A, Bruckner-Tuderman L and Kiritsi
D: Dystrophic epidermolysis bullosa: Secondary disease mechanisms
and disease modifiers. Front Genet. 12(737272)2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chung HJ and Uitto J: Type VII collagen:
the anchoring fibril protein at fault in dystrophic epidermolysis
bullosa. Dermatol Clin. 28:93–105. 2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Dang N and Murrell DF: Mutation analysis
and characterization of COL7A1 mutations in dystrophic
epidermolysis bullosa. Exp Dermatol. 17:553–568. 2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sawka E and Funk T: Dominant dystrophic
epidermolysis bullosa with congenital absence of skin and
brachydactyly of the great toes. Pediatr Dermatol. 38:1251–1254.
2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Shinkuma S: Dystrophic epidermolysis
bullosa: A review. Clin Cosmet Investig Dermatol. 8:275–284.
2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Eichstadt S, Tang JY, Solis DC,
Siprashvili Z, Marinkovich MP, Whitehead N, Schu M, Fang F,
Erickson SW, Ritchey ME, et al: From clinical phenotype to
genotypic modelling: Incidence and prevalence of recessive
dystrophic epidermolysis bullosa (RDEB). Clin Cosmet Investig
Dermatol. 12:933–942. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Laimer M, Prodinger C and Bauer JW:
Hereditary epidermolysis bullosa. J Dtsch Dermatol Ges.
13:1125–1133. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Fine JD: Inherited epidermolysis bullosa.
Orphanet J Rare Dis. 5(12)2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ma THT, Luong TLA, Hoang TL, Nguyen TTH,
Vu TH, Tran VK, Nguyen DB, Trieu TS, Nguyen HH, Nong VH and Nguyen
DT: Novel and very rare causative variants in the COL7A1 gene of
Vietnamese patients with recessive dystrophic epidermolysis bullosa
revealed by whole-exome sequencing. Mol Genet Genomic Med.
9(e1748)2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yan Y, Meng Z, Hao S, Wang F, Jin X, Sun
D, Gao H and Ma X: Five novel COL7A1 gene mutations in three
Chinese patients with recessive dystrophic epidermolysis bullosa.
Ann Clin Lab Sci. 48:100–105. 2018.PubMed/NCBI
|
|
19
|
Varki R, Sadowski S, Uitto J and Pfendner
E: Epidermolysis bullosa. II. Type VII collagen mutations and
phenotype-genotype correlations in the dystrophic subtypes. J Med
Genet. 44:181–192. 2007.PubMed/NCBI View Article : Google Scholar
|