Introduction
Breast cancer (BC) is a complex and heterogeneous
disease characterized by abnormal cell proliferation of the
epithelial cells lining the milk ducts (1). BC is the most common cancer and the
main cause of mortality due to cancer in women globally, with an
age-standardized incidence rate (world) of 46.8 per 100,000 and a
mortality rate of 12.7 per 100,000 for both sexes, as reported in
the most recent data (2). There is
a wide variety of drugs for the pharmacological treatment of BC.
However, most drugs act directly on DNA with severe adverse effects
(3). Histone deacetylase (HDAC)
inhibitors are a promising new class of anticancer drugs, which
indirectly modifies the expression of genes (4).
HDACs are zinc-dependent enzymes that remove acetyl
groups from lysine and arginine residues in histone proteins on DNA
leading to chromatin compaction and therefore transcription
repression (5). Overexpression of
HDACs has been reported in numerous cancer types and is directly
linked to accelerated cell proliferation and survival (6). There are 18 isoforms identified in
humans and the overexpression of isoform HDAC-6 has been correlated
with BC (7). HDAC inhibitors have
been successfully used in the treatment numerous types of cancer
inducing cell cycle arrest, activating extrinsic and intrinsic
apoptosis pathways, and autophagy in tumor cells, among other
mechanisms (8). Trichostatin A
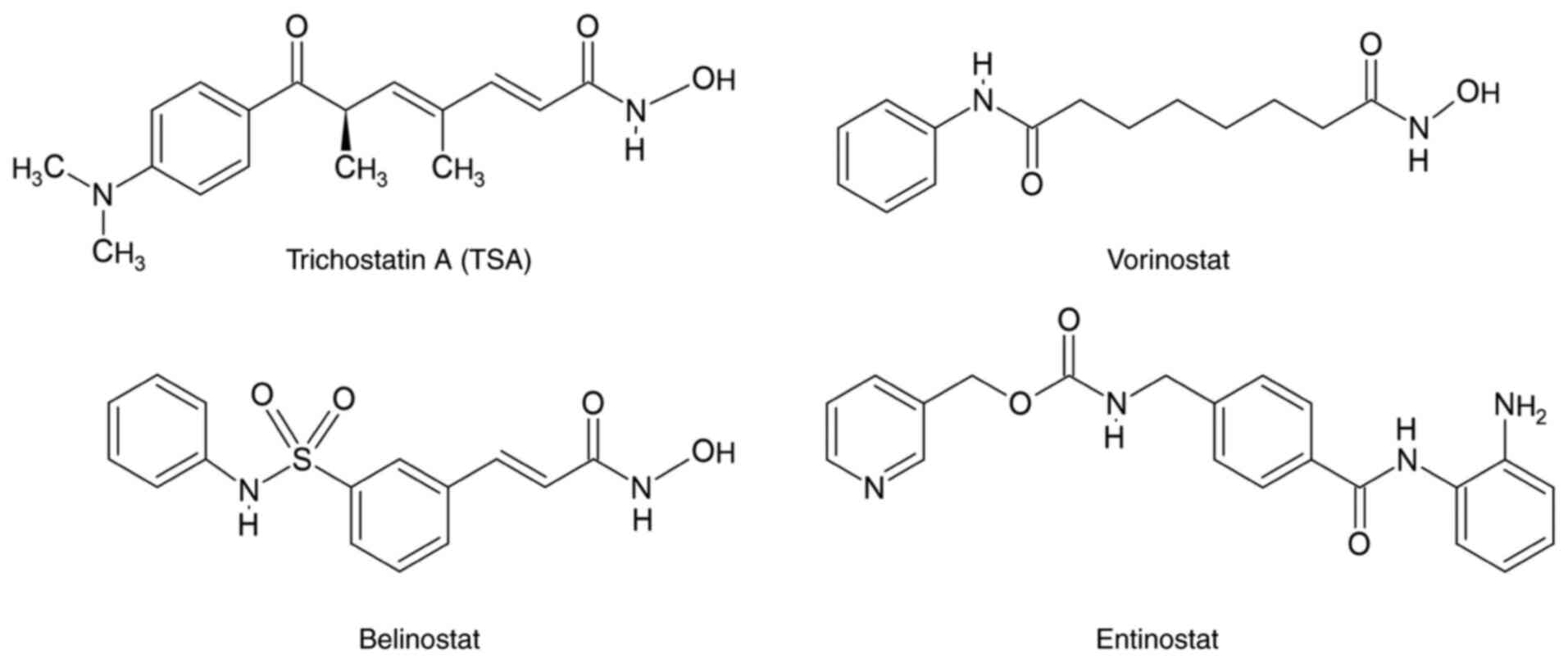
(TSA) vorinostat, entinostat and belinostat (Fig. 1) are some of the most known HDAC
inhibitors (9).
Belinostat was approved by the Food and Drug
Administration in 2014 for the treatment of peripheral T-cell
lymphoma (10). However,
effectiveness of belinostat over BC has been also reported
(11). Han et al (12) reported a significantly viability
inhibition of the triple-negative BC (TNBC) MDA-MB-231 cells,
suppressing its migration and invasion after belinostat treatment.
On the other hand, Tuncer et al (13,14)
demonstrated that belinostat inhibited the cell proliferation of
MCF-7 cells with IC50 at µM range; meanwhile Lu et
al (15) reported its
effectiveness over in vivo breast tumors.
Coumarin, a 2H-chromen-2-one heterocycle, is a
privileged scaffold with several medicinal properties (16). Coumarins have been reported to have
a wide variety of pharmacological activities, among them,
anticancer (17). A previous study
reported the anticancer behavior of coumarins by different
mechanisms including HDAC inhibition (18).
Following the authors' recent study on developing
anticancer compounds targeting HDAC6(19), in the present study, a set of six of
3-carboxy-coumarin sulfonamides, based in belinostat design, were
synthesized. Docking studies were performed to obtain the free
binding energies and to explore the interactions between the
designed compounds and HDAC6. The synthesized compounds were
evaluated for their cytotoxic activity against the BC cell lines
MCF-7 and MDA-MB-231 and the nonmalignant cells 3T3/NIH.
Materials and methods
Materials and equipment
The progress of all the reactions was monitored by
thin layer chromatography with ethyl acetate-hexane mixtures as
eluent. Salicylaldehyde, diethyl malonate, ethyl benzoylacetate,
piperidine, chlorosulfonic acid, aniline, benzylamine and
phenethylamine were purchased from MilliporeSigma and used as
received. The ACS grade solvents were purchased from CTR Scientific
(https://ctrscientific.com/) and used
without further purification. Melting points were measured on an
Electrothermal Mel-Temp 1201D apparatus. IR spectra were collected
using Varian 3100 FT-IR EXCALIBUR series spectrophotometer (Tecan
Group, Ltd.). All NMR spectra were recorded in a Bruker advance
TM-400 spectrometer (400 MHz 1H NMR, 100 MHz
13C NMR) in DMSO-d6 or
CDCl3 solutions using (CH3)4Si as
an internal reference compound; chemical shifts (δ) are in ppm and
coupling constants (nJ H-H) in Hz.
General procedure for synthesis of
coumarins 2, 6
Salicylaldehyde 1, diethyl malonate or ethyl
benzoylacetate, and piperidine were dissolved in 15 ml of ethanol
and placed under reflux at 78˚C for 24 h. After cooling, the
solvent was removed by vacuum filtration, and the resulting solid
was recrystallized from cold ethanol.
General procedure for synthesis of
sulfonyl chlorides 3, 7
One g of 1 or 2 was placed in a 25 ml ball flask and
15 equivalents of chloro-sulfonic acid were added dropwise with
stirring and cooling. Ice bath was removed, and the reaction
mixture was heated at 115˚C for 2 h. The cooled solution was added
dropwise to an ice-water mixture. The solid product was separated
by vacuum filtration, washed with cold water and dried at room
temperature.
General procedure for synthesis of
sulfonamides 4, 5, 8a-c
One gram of 3 or 7 and 2.2 equivalents of
triethylamine were dissolved in 20 ml of THF, dioxane or ethyl
acetate, then 2.2 equivalents of the amines a-c were added dropwise
with stirring at room temperature for 1 to 24 h. Sulfonamides were
purified by open column chromatography using a silica gel 60
(0.063-0.200 mm) column (Merck KGaA), with dimensions of 3 cm in
diameter and 50 cm in height. The mobile phase was passed through
the column at a flow rate of 2 ml/min, using ethyl acetate-hexane
mixtures as eluent. Spectroscopic data was presented in Table SI.
Ethyl 2-oxo-2H-chromen-3-carboxylate
(2)
White crystals, yield 72%, m.p. 88-90˚C. IR
(cm-1) v (C=O) 1760, v (C=O) 1672.
1H-NMR (CDCl3), δ (ppm): 8.53 (1H, s, H-4),
7.63 (1H, dd, 3J=7.3 Hz, H-7) 7.62 (1H, d,
3J=8.2 Hz, H-5), 7.35 (1H, d,
3J=7.8, H-8), 7.34 (1H, dd,
3J=7.6 Hz H-6), 4.40 (2H, q,
3J=7.2 Hz, H-12), 1.35 (3H, t,
3J=7.2 Hz, H-13). 13C-NMR
(CDCl3), δ (ppm): 163.0 (C-11), 156.6 (C-2), 155.1
(C-9), 148.4 (C-4), 134.3 (C-7), 129.5 (C-5), 124.8 (C-6), 118.3
(C-10), 117.8 (C-3), 116.6 (C-8), 61.8 (C-12), 14.1 (C-13). E. A.
C12H10O4 (%) Found: C (66.27), H
(4.76); Calculated: C (66.05), H (4.62).
6-(Chlorosulfonyl)-2-oxo-2H-chromen-3-carboxylic acid (3)
White solid, yield 72%, m.p. 96-99˚C. IR
(cm-1) v (C=O) 1731, v (C=O) 1692,
v (S=O) 1174. 1H-NMR (DMSO-d6):
δ (ppm): 8.77 (1H, s, H-4), 8.13 (1H, d, 4J=2.0
Hz, H-5), 7.91-7.88 (2H, m, H-7, H-8). 13C-NMR
(DMSO-d6), δ (ppm): 164.3 (C-11), 157.2 (C-2),
155.0 (C-9), 148.5 (C-4), 143.4 (C-6), 131.8 (C-7), 127.4 (C-5),
119.1 (C-10), 117.6 (C-3), 116.7 (C-8). E. A.
C10H5ClO6S. 10H20 (%)
Found: C (26.09), H (5.58); Calculated: C (41.61), H (1.75).
2-oxo-N-phenyl-6-(phenylsulfamoyl)-2H-chromen-3-car boxamide
(4a)
White solid, yield 20%, m.p. 233-235˚C. IR
(cm-1) v (N-H) 3200, v (C=O) 1703,
v (C=O) 1662, v (S=O) 1150. 1H-NMR
(DMSO-d6), δ (ppm): 8.93 (1H, s, H-4), 8.47 (1H,
d, 4J=2.3 Hz, H-5), 8.03 (1H, dd,
3J=7.1 Hz, 4J=2.3 Hz, H-7),
7.71 (1H, d, 3J=7.1 Hz, H-8), 7.72-7.02 (10H, m,
H-13-15, H-17-19). 13C-NMR (DMSO-d6),
δ (ppm): 160.1 (C-11), 159.8 (C-2), 156.36 (C-9), 146.3 (C-4),
138.7 (C-12), 137.9 (C-6), 136.7 (C-16), 131.7 (C-7), 129.7, 129.5
(C-14, C-18), 129.6 (C-5), 124.9, 124.8 (C-15, C-19), 122.7 (C-3),
120.8, 120.3 (C-13, C-17), 119.0 (C-10), 118.1 (C-8). E. A.
C22H16N2O5S (%) Found:
C (62.98), H (3.92), N (6.52); Calculated: C (62.85), H (3.84), N
(6.66).
2-oxo-N-(2-phenylethyl)-6-[(2-phenylethyl)sulfamoyl]-2H-chromene-3-carboxamide
(4c)
White solid, yield 22%, m.p. 192-194˚C. IR
(cm-1) v (N-H) 3331, v (C=O) 1710,
v (C=O) 1655, v (S=O) 1152. 1H-NMR
(CDCl3), δ (ppm): 8.89 (1H, s, H-4), 8.73 (1H, t, NH),
8.1 (1H, d, 4J=2.1 Hz, H-5), 8.01 (1H, dd,
3J=7.6 Hz, 4J=2.2 Hz, H-7),
7.47 (1H, d, 3J=7.6 Hz, H-8), 7.37-7.08 (10H, m,
H-15-17, H-21-23), 3.76 (2H, dd, 3J=6.6 Hz,
H-12), 3.31 (2H, dd, 3J=6.5 Hz, H-18), 2.97 (2H,
t, 3J=7.2 Hz, H-13), 2.82 (2H, t,
3J=6.7 Hz, H-19).13C-NMR
(CDCl3), δ (ppm): 160.5 (C-11), 160.2 (C-2), 156.2
(C-9), 147.1 (C-4), 138.5 (C-14), 137.4 (C-6), 137.3 (C-20), 131.7
(C-7), 128.9, 128.7, 128.6, 128.5 (C-15-16, C-21-22), 128.8 (C-5),
128.7 (C-17), 128.6 (C-23), 120.1 (C-3), 118.6 (C-10), 117.6 (C-8).
E. A. C26H24N2O5S (%)
Found: C (66.03), H (5.36), N (5.83); Calculated: C (65.53), H
(5.08), N (5.88).
N-benzyl-3-[(E)-(benzylimino)methyl]-4-hydroxybenzene-1-sulfonamide
(5)
Yellow crystals, yield 52%, m.p. 156-158˚C. IR
(cm-1) v (N-H) 3248, v (C=N) 1628,
v (S=O) 1153. 1H-NMR (CDCl3), δ (ppm):
8.39 (1H, s, H-4), 7.80 (1H, d, 4J=2.4 Hz, H-5),
7.77 (1H, dd, 3J=8.8 Hz, 4J=2.4
Hz, H-7) 7.42-7.20 (10H, m, H-13-15, H-18-20) 7.04 (1H, d,
3J=8.8 Hz, H-8), 4.83 (2H, s, H-11) 4.14 (2H, d,
3J=6.2 Hz, H-16).13C-NMR
(CDCl3), δ (ppm): 166.2 (C-9), 164.5 (C-4), 136.9
(C-17), 136.2 (C-12), 134.9 (C-6), 131.6 (C-5), 131.1 (C-7), 129.3,
128.9, 128.7, 128.0, 127.9, 127.8 (C-13-15, C-18-20), 118.5 (C-8),
117.9 (C-16). E. A.
C21H20N2O3S (%) Found:
C (66.56), H (5.68), N (7.22); Calculated: C (66.29), H (5.29), N
(7.36).
3-Benzoyl-2H-chromen-2-one (6)
White crystals, yield 71%, m.p. 134-136˚C. IR
(cm-1) v (C=O) 1717, v (C=O) 1656.
1H-NMR (CDCl3), δ (ppm): 8.11 (1H, s, H-4),
7.91 (2H, dd, 3J=7.1, 4J=1.6
Hz, H-13), 7.70-7.64 (2H, m, H-7, H-15), 7.63 (1H, d,
3J=7.6 H-5), 7.51 (2H, dd,
3J=7.8 Hz, H-14), 7.44 (1H, d,
3J=8.3 H-8), 7.38 (1H, dd,
3J=7.4, H-6). 13C-NMR
(CDCl3), δ (ppm): 191.6 (C-11), 158.3 (C-2), 154.8
(C-9), 145.3 (C-4), 136.2 (C-12), 133.8 (C-5), 133.6 (C-15), 129.5
(C-13), 129.1 (C-7), 128.6 (C-14), 127.1 (C-3), 124.9 (C-6), 118.2
(C-10), 116.9 (C-8). E. A.
C16H10O3 (%) Found: C (76.43), H
(4.11); Calculated: C (76.79), H (4.03).
3-[3-(Chlorosulfonyl)benzoyl]-2-oxo-2H-chromen-6-sulfonyl chloride
(7)
White solid, yield 85%, m.p. 75-78˚C. IR
(cm-1) v (C=O) 1743, v (C=O) 1665,
v (S=O) 1167. 1H-NMR (DMSO-d6),
δ (ppm): 8.53 (1H, s, H-4), 8.10 (1H, d, 4J=2.0
Hz, H-5), 8.08 (1H, dd, 4J=1.6 Hz, H-13), 7.92
(1H, dd, 3J=8.0 Hz, 4J=2.0 Hz,
H-15), 7.90 (2H, dd, 3J=7.7 Hz,
4J=1.6 Hz, H-7, H-15), 7.52 (1H, dd,
3J=7.8 Hz, H-16), 7.46 (1H, d,
3J=8.6 Hz, H-8). 13C-NMR
(DMSO-d6), δ (ppm): 191.1 (C-11), 158.4 (C-2),
154.4 (C-9), 149.0 (C-14), 146.0 (C-4), 145.4 (C-6), 136.1 (C-12),
131.4 (C-17), 131.2 (C15), 130.7 (C-7), 128.9 (C-16), 127.1 (C-13),
126.8 (C-3), 126.1 (C-5), 117.8 (C-10), 116.4 (C-8). E. A.
C16H8Cl2O7S2.
10H2O (%) Found: C (30.68), H (4.58); Calculated: C
(42.97), H (1.80).
2-oxo-N-phenyl-3-[3-(phenylsulfamoyl)benzoyl]-2H-chromene-6-sulfonamide
(8a)
Brown solid, yield 25%, m.p. 94-96˚C. IR
(cm-1) v (N-H) 3237, v (C=O) 1723,
v (C=O) 1669, v (S=O) 1151. 1H-NMR
(DMSO-d6), δ (ppm): 8.54 (1H, s H-4), 8.33 (1H,
d, 4J=2.3 Hz, H-5), 8.22 (1H, dd,
4J=1.6 Hz, H-13), 8.17 (1H, dd,
3J=7.8 Hz, 4J=1.6 Hz, H-17),
8.05 (1H, dd, 3J=7.3 Hz, 4J=2.3
Hz, H-7), 8.01 (1H, dd, 3J=7.8 Hz,
4J=1.7 Hz, H-15), 7.69 (1H, dd,
3J=7.8 Hz, H-16), 7.68 (1H, d,
3J=7.3 Hz, H-8), 7.26-7.0 (10H, m, H-19-21,
H-23-25). 13C-NMR (DMSO-d6), δ (ppm):
190.6 (C-11), 157.7 (C-2), 156.9 (C-9), 145.8 (C-4), 140.8 (C-12),
137.6, 137.6 (C-18, C-22), 137.0 (C-6), 136.4 (C-14), 134.2 (C-17),
131.8, 131.7 (C-7, C-15), 130.5 (C-16), 129.8, 129.7 (C-20, C-24),
129.4 (C-5),127.4 (C-13), 127.3 (C-3), 125.0, 124.9 (C-21, C-25),
121.1, 120.8 (C-19, C-23), 118.9 (C-10), 118.3 (C-8). E. A.
C28H20N2O7S2
(%) Found: C (60.14), H (3.93), N (5.11); Calculated: C (59.99), H
(3.59), N (4.99).
N-benzyl-3-[3-(benzylsulfamoyl)benzoyl]-2-oxo-2H-chromene-6-sulfonamide
(8b)
White crystals, yield 42%, m.p. 168-170˚C. IR
(cm-1) v (N-H) 3305, v (C=O) 1722,
v (C=O) 1676, v (S=O) 1153. 1H-NMR
(DMSO-d6), δ (ppm): 8.98 (1H, s H-4), 8.85 (1H,
dd, 4J=1.7 Hz, H-13), 8.79 (1H, d,
4J=2.2 Hz, H-5), 8.69 (1H, dd,
3J=7.8 Hz, 4J=2.3 Hz, H-17),
8.62-8.60 (2H, m, H-7, H-15), 8.22 (1H, dd,
3J=7.8 Hz, H-16), 8.05 (1H, d,
3J=8.7 Hz, H-8), 7.74-7.68 (10H, m, H-20-22,
H-25-27), 4.66 (4H, d, 3J=6.0 Hz, H-18, H-23).
13C-NMR (DMSO-d6), δ (ppm): 190.5
(C-11), 157.7 (C-2), 157.2 (C-9), 145.5 (C-4), 142.5 (C-12), 138.4
(C-6), 137.7, 137.7 (C-19, C-24), 136.6 (C-14), 133.4 (C-17),
132.0, 132.0 (C-7, C-15), 130.1 (C-16), 129.5 (C-5), 128.8, 128.4,
128.3, 127.9, 127.8 (C-20-22, C-25-27), 128.0 (C-3), 127.8 (C-13),
118.9 (C-10), 118.0 (C-8), 47.3 (C-18, C-23). E. A.
C30H24N2O7S2
(%) Found: C (61.58), H (4.29), N (4.85); Calculated: C (61.21), H
(4.11), N (4.76).
2-oxo-N-(2-phenylethyl)-3-{3-[(2-phenylethyl)sulfamoyl]benzoyl}-2H-chromene-6-sulfonamide
(8c)
Brown solid, yield 22%, m.p. 78-80˚C. IR
(cm-1) v (N-H) 3274, v (C=O) 1724,
v (C=O) 1667, v (S=O) 1148. 1H-NMR
(DMSO-d6): δ (ppm): 8.98 (1H, s H-4), 8.85 (1H,
s, H-13), 8.78 (1H, d, 4J=1.9 Hz, H-5), 8.68 (1H,
dd, 3J=7.8 Hz, 4J=1.6 Hz,
H-17), 8.60 (2H, dd, 3J=7.4 Hz,
4J=2.0 Hz, H-7, H-15), 8.21 (1H, dd,
3J=7.8 Hz, H-16), 8.05 (1H, d,
3J=7.4 Hz, H-8), 7.70-7.61 (10H, m, H-21-23,
H-27-29), 3.73-3.65 (4H, m, H-18, H-24) 3.29-3.24 (4H, m, H-19,
H-25). 13C-NMR (DMSO-d6), δ (ppm):
190.6 (C-11), 157.9 (C-2), 157.3 (C-9), 145.9 (C-4), 142.3 (C-12),
139.1, 139.0 (C-20, C-26), 138.1 (C-6), 137.7 (C-14), 133.5 (C-17),
131.9, 131.9 (C-7, C-15), 130.2 (C-16), 129.4 (C-5), 129.2, 129.2,
128.8, 126.8 (C-21-23, C-27-29), 128.0 (C-3), 127.7 (C-13), 119.1
(C-10), 118.0 (C-8), 45.0, 44.9 (C-18, C-24), 36.2 (C-19, C-25). E.
A.
C32H28N2O7S2
(%) Found: C (62.78), H (4.94), N (4.62); Calculated: C (62.32), H
(4.58), N (4.54).
Molecular docking
Molecular docking was performed using the AutoDock
Vina tool implemented in UCSF Chimera version 1.16 (www.cgl.ucsf.edu/chimera). 3D structures of the
test compounds were constructed using Maestro version 13.3
(Schrodinger, Inc.). The protein structure of HDAC was retrieved
from the Protein Data Bank (https://www.rcsb.org/) with the accession code 5EDU.
All water molecules and also the co-crystallized ligands were
removed from the crystallographic structure. The grid box was
defined surrounding the co-crystallized ligand trichostatin A (TSA)
within the HDAC6 active site. The grid box size was set at 20 Å, 20
Å, and 20 Å (x, y and z). In all simulations, the ligands were
flexible, and the protein remained static. 2D protein-ligand
interaction diagrams were generated through Maestro. Validation was
performed using AutoDock Vina tool in UCSF Chimera 1.16 by
redocking the co-crystallized ligand TSA. The 3D structure of TSA
was built through Maestro 13.3 and docked within the active site of
HDAC6 (5EDU). The grid box was centered at the crystallographic
coordinates of the co-crystallized ligand, and the grid box size
was set at 20 Å, 20 Å, and 20 Å. This validation was carried out
based on important interactions.
In vitro assays. Cell culture
A total of three cell lines were used to evaluate
the synthesized compounds: The human TNBC cell line MDA-MB-231
(cat. no. HTB-26), the human BC cell line MCF-7 (cat. no. HTB-22)
and the NIH/3T3 mouse fibroblast cell line. MCF-7 and MDA-MB-231 BC
cell lines were obtained from the American Type Culture Collection.
The MDA-MB-231 and MCF-7 BC cell lines were selected for the
present study due to their well-documented characteristics.
MDA-MB-231 is a TNBC cell line known for its aggressive and
invasive properties, representing a challenging subtype of BC. By
contrast, MCF-7 is an estrogen receptor-positive cell line that is
less aggressive and more responsive to hormone therapy.
The NIH/3T3 cell line was obtained from the
Parasitology Laboratory, Faculty of Medicine, Public Health, and
Nursing (FKKMK), University Gadjah Mada (Sleman, Indonesia). This
cell line is widely used to assess cellular mechanisms and was
chosen to provide a baseline for comparing the effects of the
synthesized compounds. Given that HDAC6 inhibition has been shown
to reverse metastatic traits and restore normal cellular
organization in cancer cells, NIH/3T3 cells, which do not exhibit
these malignancy-associated traits, offer a valuable control.
NIH/3T3 cells were included as non-malignant controls. All of them
were cultured in Dulbecco's modified Eagle's Medium (DMEM)
supplemented with 10% fetal bovine serum (FBS, Biowest; https://biowest.net/) and 1% antibiotic-antifungal
(penicillin G, sodium salt and 1% streptomycin sulfate). Cell
cultures were incubated at 37˚C under a 5% CO2 and 95%
air atmosphere; PBS-Trypsin-EDTA solution was used to detach the
cells when their confluency was up to 80%, and cells were seeded in
a 96-plate well with 10x103 cells in each well. After 24
h, the medium was replaced by compounds at concentrations of 6.25,
12.5, 25, 50 and 100 µM, previously dissolved in DMSO (0.1%). For
manipulation and visualization, a biosafety level 2 vertical
laminar flow cabinet (NUAIRE A2 NU-543-400) and an inverted
binocular microscope (MOTIC AE-20) were used, respectively.
Viability assay. Cells were seeded in a
tissue culture (TC)-treated 96-well flat-bottom microplate
(Corning, Inc.) at a density of 1x104 cells/well in
supplemented DMEM-HG and incubated for 24 h. Afterward, the medium
was removed, replaced with the treatments previous described in a
final volume of 100 µl/well, and the cells were incubated for 48 h
in a 5% CO2 atmosphere at 37˚C. At the end, the
treatments were removed and replaced with 100 µl/well 1X Alamar
Blue™ Cell Viability Assay Reagent (MilliporeSigma) in
phenol red-free medium for 4 h. The optical density was measured on
a microplate reader (iMark; Bio-Rad Laboratories, Inc.) at a
wavelength of 570-600 nm for excitation-emission. The percentage of
cytotoxicity was calculated as: 100-[(experimental OD value-blank
OD value)/(control OD value-blank OD value) x100%]. Cisplatin
(58.32 µM), doxorrubicin (2 µM) and belinostat (40 µM)
(MilliporeSigma) were used as positive controls. The compounds 4a,
c, 5, and8a-c were evaluated at a concentration of 40 µM.
Isolation of lymphocytes from human peripheral
blood and cell viability test. The methodology for isolation of
lymphocytes from human peripheral blood and cell viability test was
performed according to the methodology of Calderón-Segura et
al (20) and Hernández-Fuentes
et al (21) with slight
modifications. A total of 20 ml of heparinized venous blood from
three healthy volunteer donors was centrifuged at 500 x g for 20
min at room temperature (22-25˚C). The resulting cellular layer was
diluted 1:1 with HBSS, layered over Ficoll-Paque, and centrifuged
at 250 x g for 10 min at room temperature (22-25˚C). Lymphocytes
were then collected, washed twice in RPMI-1640 medium (Biowest,
Inc.) by centrifugation at 250 x g at room temperature (22-25˚C)
for 10 min, and resuspended in RPMI-1640 medium (37˚C) supplemented
with 1% penicillin/streptomycin. The lymphocyte pellet was
immediately assessed for cellular viability using a Neubauer
chamber. Cell viability was determined before and after treatments
using the trypan blue exclusion method, where trypan blue
penetrates the damaged membrane of dead cells and stains the
nucleus. A mixture of 10 µl of cell pellet and 10 µl of trypan blue
was incubated for 3 min, and then the number of dead cells out of
100 consecutive cells was counted in duplicate (20,21).
The compounds (4a, c, 5, 8a-c) and belinostat were evaluated at a
concentration of 40 µM. All experiments were performed in
triplicate in independent assays.
The present study was approved by the Ethics
Committee of the Clinical Research Center of the National Cancer
Institute (approval no. CEICANCL23062023-DISULFA-21; Colima,
Mexico). Cells were isolated from three healthy male volunteer
donors, all of whom provided oral informed consent for the
collection of their samples. The donors were aged between 26-27
years, with no history of drug use or medication intake 72 h prior
to the sample collection.
Statistical analysis
The results are presented as the mean ± standard
deviations (SD). In the viability assay, the data represent
the mean of three independent experiments with 10 replicates per
experiment. Group differences were evaluated using the
Kruskal-Wallis statistical test, followed by a Dunn's post hoc test
for multiple comparisons. All statistical analyses were conducted
using SPSS Statistics 20 software (IBM Corp.). Statistically
significant difference is denoted by the corresponding symbols in
the figures for P<0.05, were necessary.
Results and Discussion
Design features of 3-carboxy-coumarin
sulfonamides
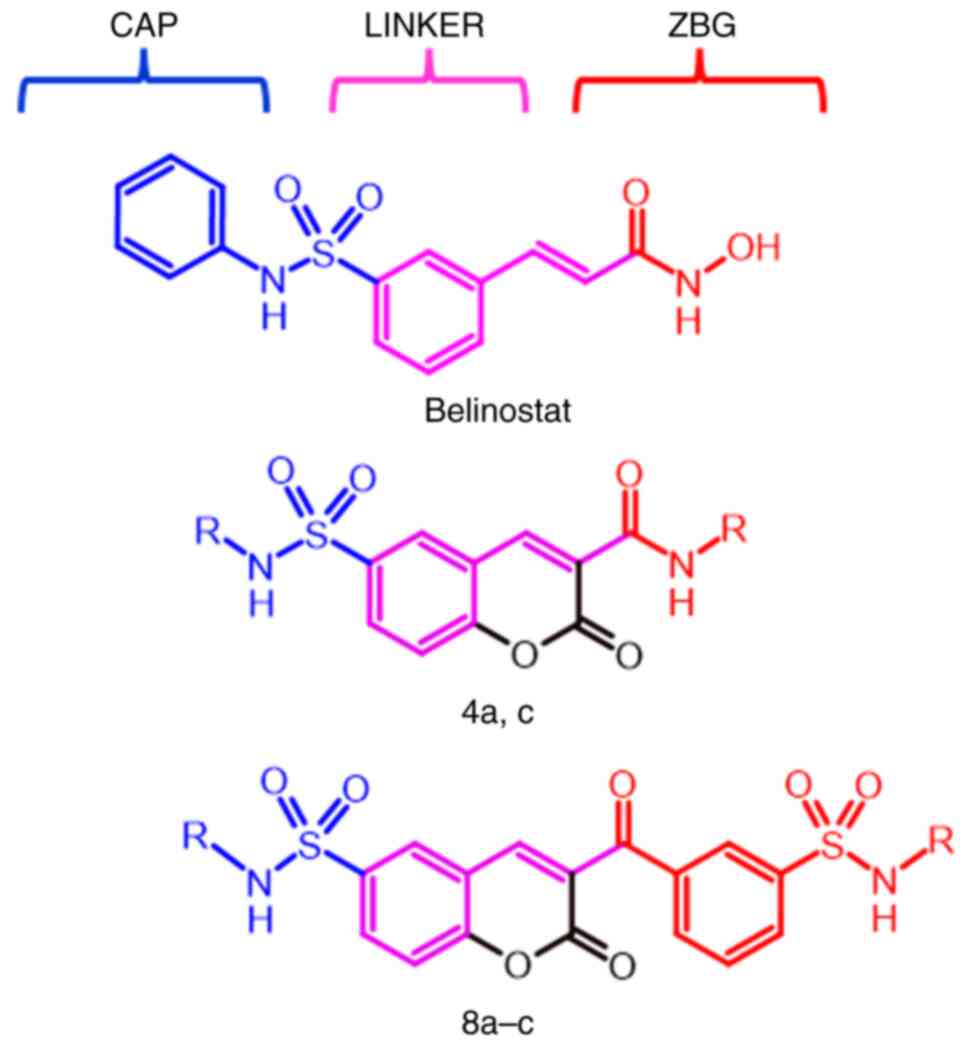
The general pharmacophore model for the HDAC
inhibitors essentially consists in three parts: i) A cap group, a
hydrophobic region that interacts with the external domain of the
enzyme; ii) a zinc binding group (ZBG) that coordinates the zinc
ion in the active site of the enzyme; and iii) a linker, a
semi-flexible chain (generally a six carbon unsaturated chain) that
connects the cap group and the ZBG and set them in within the
binding site for interactions (22-24).
Hydroxamate is the most common ZBG for its strong
binding affinity with metal ions; however, this feature leads to
lack of selectivity, binding to multiple metalloenzymes, generating
side effects, besides being metabolically unstable (25,26).
Additionally, numerous other studies have reported non-hydroxamate
inhibitors with significant in vitro activity over HDACs
(27-29).
On the other hand, numerous studies indicated that HDAC6 active
site cavity is wider and shallower than other isoforms, allowing to
accommodate bulkier molecules (30-32).
Moreover, aromatic moieties are useful for increasing affinity and
selectivity for HDAC6 (33,34). In this context, the coumarin
scaffold was incorporated on the linker region (Fig. 2), maintaining the sulfonamide
moiety, the aromatic central ring and the α,β-unsaturated system of
belinostat; and the hydroxamate moiety was replaced by the same
aromatic substituents used in the cap group.
Chemistry
The synthesis of the designed sulfonamides 4a, c, 5
and 8a-c is demonstrated in Fig. 3.
This approach includes a three-step reaction sequence. Coumarins
(2,6) were synthesized by the Knoevenagel
condensation of salicylaldehyde with the corresponding ketone in
accordance with previously described methods (35,36).
Chlorosulfonation of 2 and 6 was carried out without solvent, with
a large excess of chlorosulfonic acid and under reflux to provide
sulfonyl chlorides 3 and 7 in favorable yields. The treatment of
sulfonyl chloride 3 with aniline or/and phenethylamine provided
sulfonamides 4a, c meanwhile with benzylamine provided 5 probably
due to steric effects. On the other hand, treatment of 7 with the
corresponding amines provided sulfonamides 8a-c. All sulfonamides
were obtained at room temperature in the presence of triethylamine
as a base.
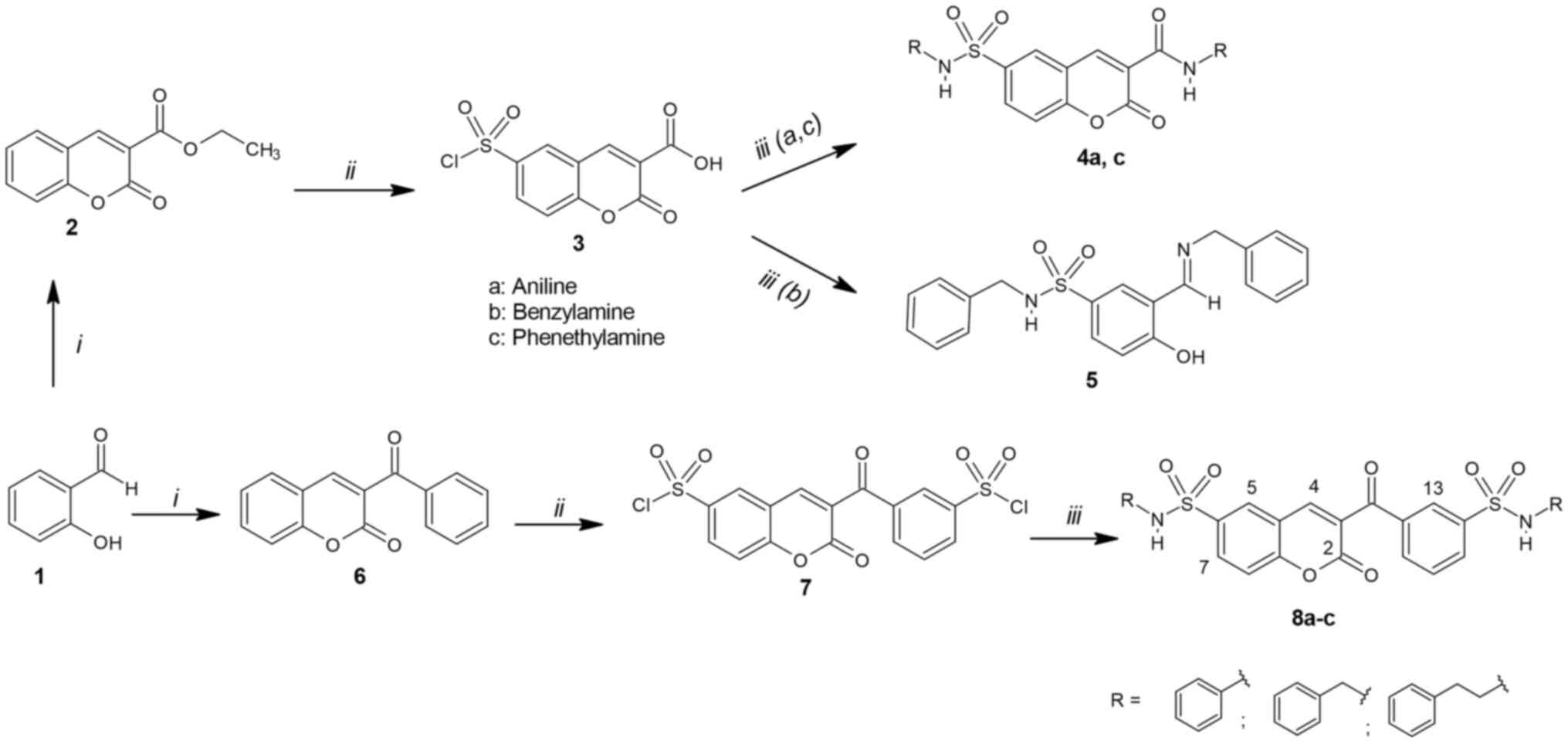 | Figure 3Scheme of synthesis of
3-carboxy-coumarin sulfonamides 4a, c, 5 and 8a-c. Reagents and
conditions: i) Diethyl malonate/ethyl benzoylacetate, piperidine,
EtOH, reflux 24 h; ii) HSO3Cl, reflux 2 h; iii) amines
(a, b, c), Et3N, THF, stirring 1-24 h, room
temperature. |
In the 1H NMR spectra the H-4 signal
indicated the formation of coumarins (2,6),
observed as the most de-shielded signal due to the intramolecular
hydrogen bond with the O-carbonyl as previously reported by
García-Báez et al (37). H-4
appeared in 8.53 ppm in compound 2 and shifted to higher
frequencies in the 8.89-8.93 ppm range due to amide formation in
4a, c; meanwhile it shifted from 8.11 ppm in compound 6 to
8.54-8.98 ppm in sulfonamides 8a-c (Table I). The chemical shifts of
sulfonamide N-H proton varied according to the amine residue; the
N-H signals were observed in the 4.60-7.62 ppm range with the
alkylamines benzylamine and phenylethylamine (compounds 4c, 5,
8b-c), whereas with aniline (compounds 4a, 8a) the N-H appeared in
the 10.40-10.68 ppm range. The amidic N-H proton appeared in
8.73-10.54 ppm range in compounds 4a, c. The aromatic protons
appeared as expected in the region 7.34-7.63 ppm for 2 and 6,
7.52-8.13 ppm for 3 and 7, and 7.69-8.79 ppm for 4a-8c (Table SI).
 | Table ISelected chemical shifts in ppm
(1H-NMR/13C-NMR) for compounds 2-8c in
CDCl3 or DMSO-d6 at 400 MHz. |
Table I
Selected chemical shifts in ppm
(1H-NMR/13C-NMR) for compounds 2-8c in
CDCl3 or DMSO-d6 at 400 MHz.
| Compound | H-4 | C-6 | NHA | NHB |
|---|
| 2a | 8.53 | 124.8 | - | |
| 6a | 8.11 | 124.9 | | |
| 3b | 8.77 | 143.4 | | |
| 7b | 8.53 | 145.4 | | |
| 4ab | 8.93 | 137.9 | 10.68 | 10.54 |
| 4ca | 8.89 | 137.4 | 4.60 | 8.73 |
| 5a | - | 134.9 | 4.83 | - |
| 8ab | 8.54 | 137.0 | 10.52 | 10.40 |
| 8bb | 8.98 | 138.4 | 7.62 | 7.62 |
| 8cb | 8.98 | 138.1 | 7.23 | 7.28 |
The 13C NMR spectra C-6 carbon was
de-shielded from 124.8-124.9 ppm range in compounds 2 and 7 to
143.4-145.4 ppm range in compounds 3 and 7 after chlorosulfonation
reaction occurred; meanwhile in the sulfonamides 4a, c, 5, 8a-c it
was observed in the 134.9-138.4 ppm range due to the electron-donor
effect of the amines. The IR spectra (Figs. S1 and S2) showed four characteristic stretching
absorption bands at 3331-3200, 1760-1703, 1692-1656 and 1174-1151
cm-1 corresponding to sulfonamide N-H, exocyclic C=O,
lactonic C=O and S=O, respectively.
Molecular docking
To determine the possible interaction mode between
the synthesized compounds (4a, c, 5 and 8a-c) and the HDAC6
catalytic site, molecular docking was performed using a validated
molecular program (Chimera 1.16). Validation of the method was
performed by redocking the co-crystallized ligand TSA where
coordination with zinc ion of HDAC6 in a bidentate fashion was
observed (Fig. S3). The molecular
docking studies showed that all ligands reached the catalytic
binding site of HDAC6.
Free binding energies ΔG (kcal/mol) are listed in
Table II. ΔG values are in the
-6.9 to -8.7 kcal/mol range, close to the value obtained for the
reference compound, belinostat (-8.3 kcal/mol). Compounds 8a-c,
having an additional aromatic ring, exhibit the most favorable ΔG
values, being close or even higher than belinostat in some cases.
This is consistent with previous studies suggesting that the HDAC6
catalytic cavity is wider than other isoforms, thus bulky aromatic
rings are well tolerated in the molecule design (38).
| Table IIBinding energies ΔG (kcal/mol) of
molecules docked into the active site of histone deacetylase 6. |
Table II
Binding energies ΔG (kcal/mol) of
molecules docked into the active site of histone deacetylase 6.
| Compounds | ΔG (Kcal/mol) |
|---|
| 4a | -7.4 |
| 4c | -6.9 |
| 5 | -7.2 |
| 8a | -8.7 |
| 8b | -8.3 |
| 8c | -7.9 |
| Belinostat | -8.3 |
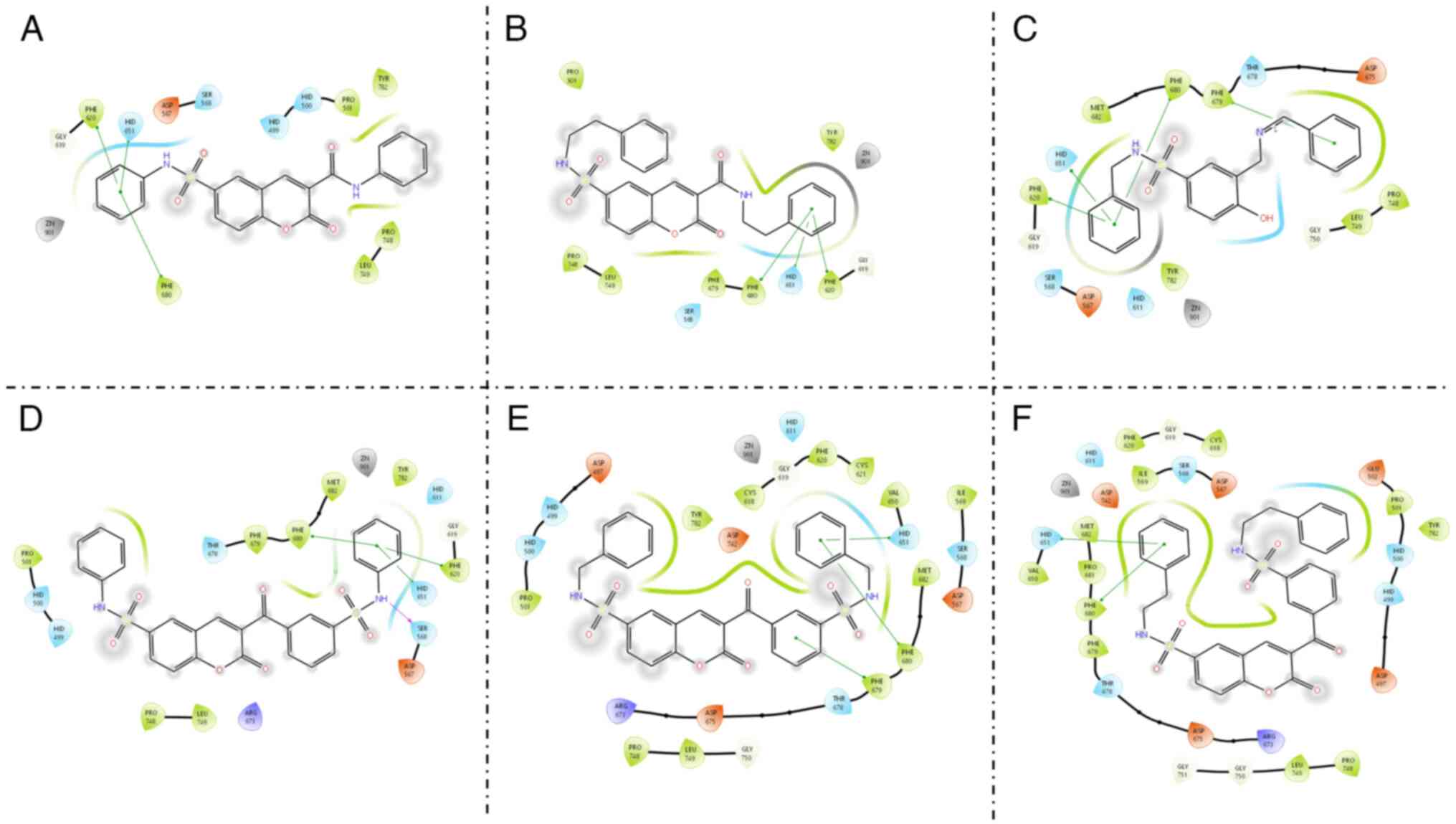
All ligands interact with Ser 568, Phe 620, Hid
651, Phe 679 and Phe 680 amino acid residues (Fig. 4), which are common interactions
among the HDAC6 inhibitors (39-42).
These interactions are similar to those found in belinostat (Gly
619, Hid 651, Phe 679 and Phe 680) where the main difference is
that in ligands 4a, c, 5, 8a-c no coordination with the zinc ion is
observed which can be explained by the absence of a traditional ZBG
as N-OH in these ligands. As can be observed, hydrogen bonding is
formed between sulfonamide N-H of compound 8a and Ser568;
meanwhile, the N-R-phenyl moiety of all ligands is involved in π-π
interactions with the aromatic residues surrounding the HDAC6
cavity.
In vitro evaluation
In a preliminary study, the cytotoxic efficacy of
six newly synthesized compounds was evaluated in the MDA-MB-231
cell line, alongside three control agents-belinostat, cisplatin and
doxorubicin. The compounds, which share structural similarities
with belinostat, were tested to determine their comparative
effectiveness. Belinostat, tested at a concentration of 40 µM,
served as a direct comparator given its structural relevance. The
well-established chemotherapy agents, cisplatin at 58.32 µM and
doxorubicin at 2 µM, known for their potent cytotoxic effects in
this cell line, served as benchmarks. The evaluation included
compounds labeled as 4a, 5, 8b, 8c, 4c and 8a.
The results, shown in Fig. 5, highlight the cytotoxic activities
(expressed as percentages) of the compounds tested. Compounds 4a,
5, 8c, 4c and 8a exhibited lower cytotoxic activity levels, though
comparable to that of doxorubicin. It is important to emphasize
that, although the mean inhibition percentages were slightly lower,
only compounds 4 and 4c showed statistically significant
differences when compared individually to doxorubicin (P<0.05,
for both comparisons). The other compounds (5, 8b, 8c and 8a) did
not present statistically significant differences (P>0.05, for
all comparisons) under the conditions of the analysis performed.
Regarding cisplatin, another chemotherapeutic agent used to treat
this type of cancer, a similar trend was observed to that with
doxorubicin. Compounds 4a, 5, 8c, 4c and 8a exhibited lower
cytotoxic activity levels compared with cisplatin. However, only
compounds 4 and 4c showed statistically significant differences
when individually compared with cisplatin (P<0.05, for both
comparisons). The other compounds (5, 8b, 8c and 8a) did not
present statistically significant differences (P>0.05, for all
comparisons) under the conditions of the analysis performed.
Notably, compound 8b exhibited a 1.12-fold greater potency than
cisplatin at comparable concentrations, demonstrating the highest
cytotoxic activity with an average of 61.99±8.82%. This was
followed by compound 8c, which showed 46.14±1.61% cytotoxicity, and
compound 8a, with 45.09±8.03%. These results highlight 8b, 8a and
8c as the most potent compounds, prompting further detailed
cytotoxicity testing in the MDA-MB-231 cell lines.
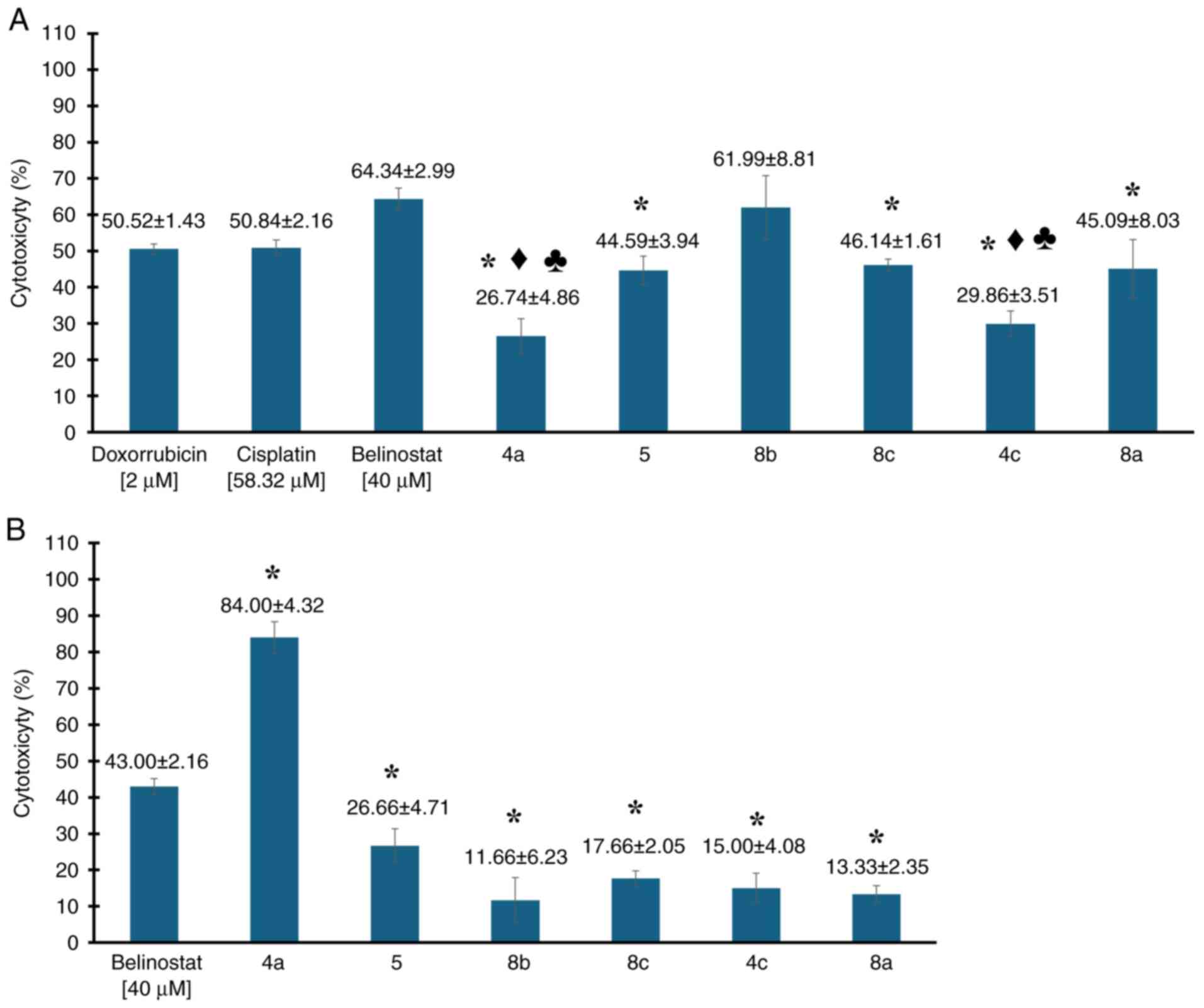 | Figure 5(A) Preliminary evaluation of
cytotoxic activity of 3-carboxy-coumarin sulfonamides (compounds
4a, 5, 8b, 8c, 4c and 8a) at 40 µM in MDA-MB-231 cell line,
compared with standard chemotherapeutic agents belinostat (40 µM),
doxorubicin (2 µM) and cisplatin (58.32 µM). (B) Evaluation of
leukocyte viability after exposure to compounds 4a, 5, 8b, 8c, 4c
and 8a and belinostat at 40 µM. Data are presented as the mean ±
SD. All assays were performed in triplicate. *P<0.05
vs. belinostat; ♦P<0.05 vs. doxorrubicin;
♣P<0.05 vs. cisplatin. |
On the other hand, under the tested conditions,
belinostat exhibited cytotoxic activity at 64.34±2.99%. Although
belinostat is primarily used to treat other cancers, such as
peripheral T-cell lymphoma, it serves as the core structure for the
compounds examined. However, in preliminary evaluations, compounds
4a, 5, 8c, 4c and 8a exhibited lower cytotoxicity percentages
compared with belinostat. Notably, compound 8b showed inhibition
percentages similar to those of belinostat, without statistically
significant differences (P=0.459). By contrast, the rest of the
compounds demonstrated statistically significant differences when
compared with belinostat (P<0.05, for all comparisons).
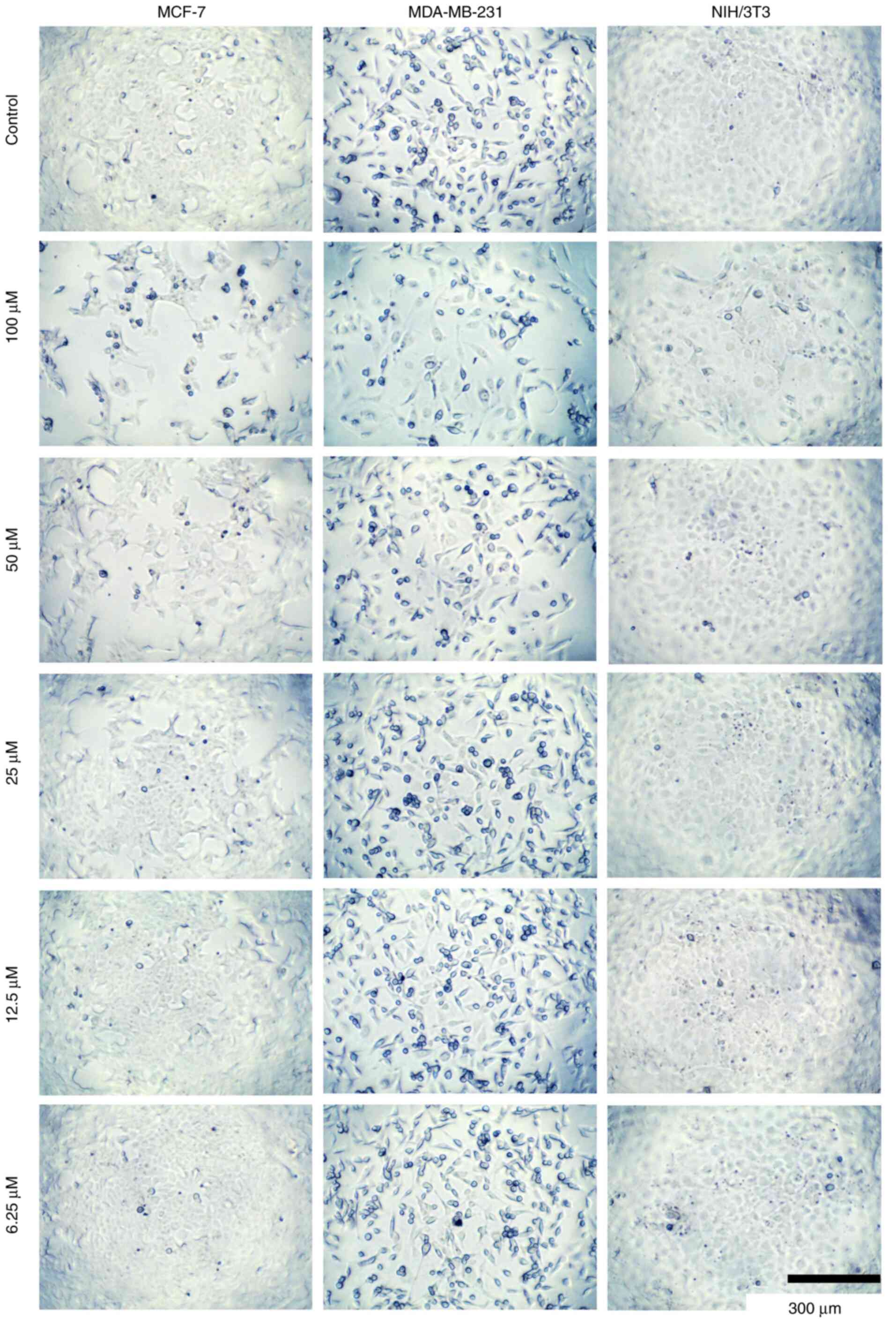
Additionally, morphological changes were observed in MCF-7,
MDA-MB-231 and NIH/3T3 cells incubated with different
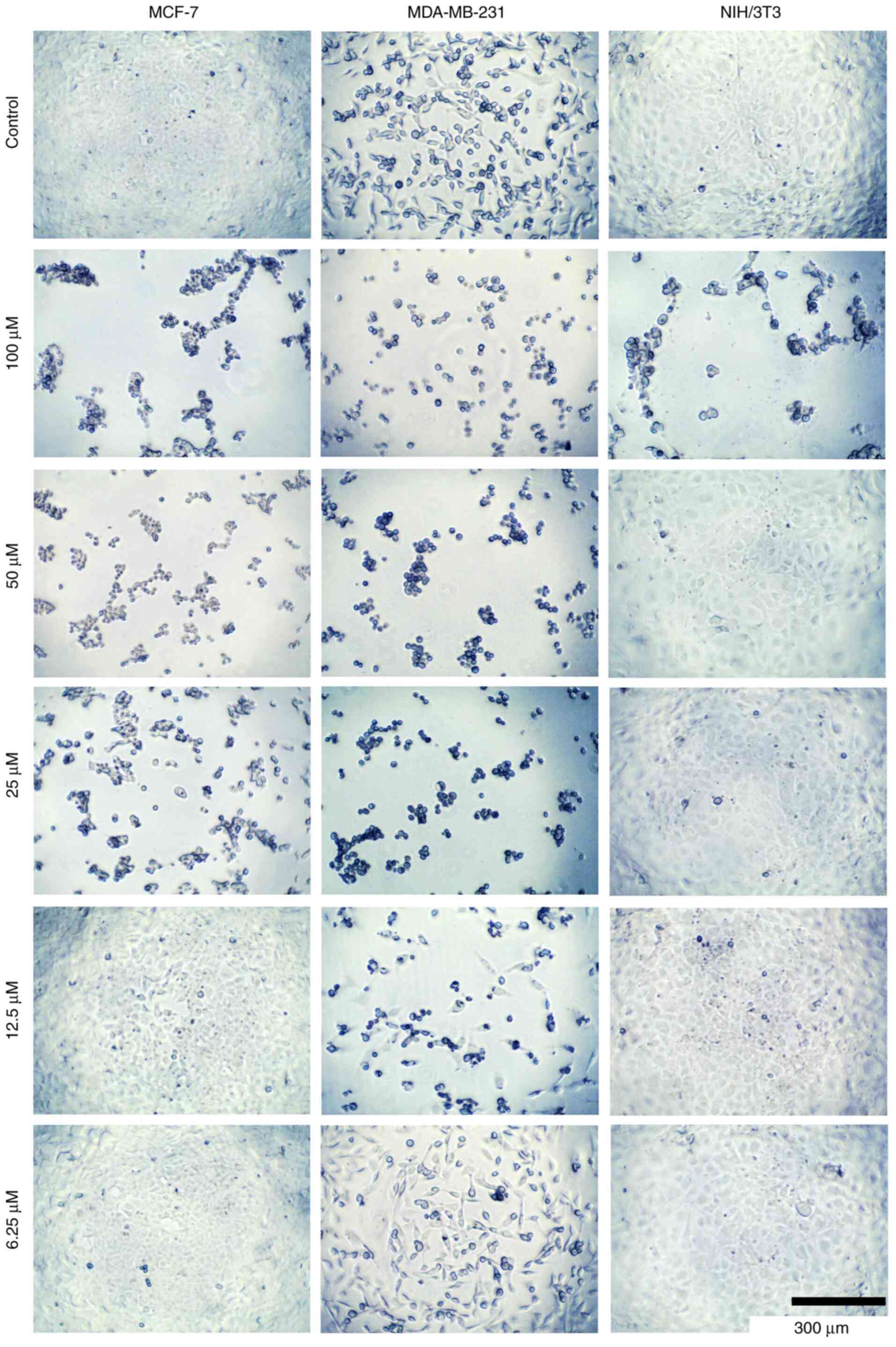
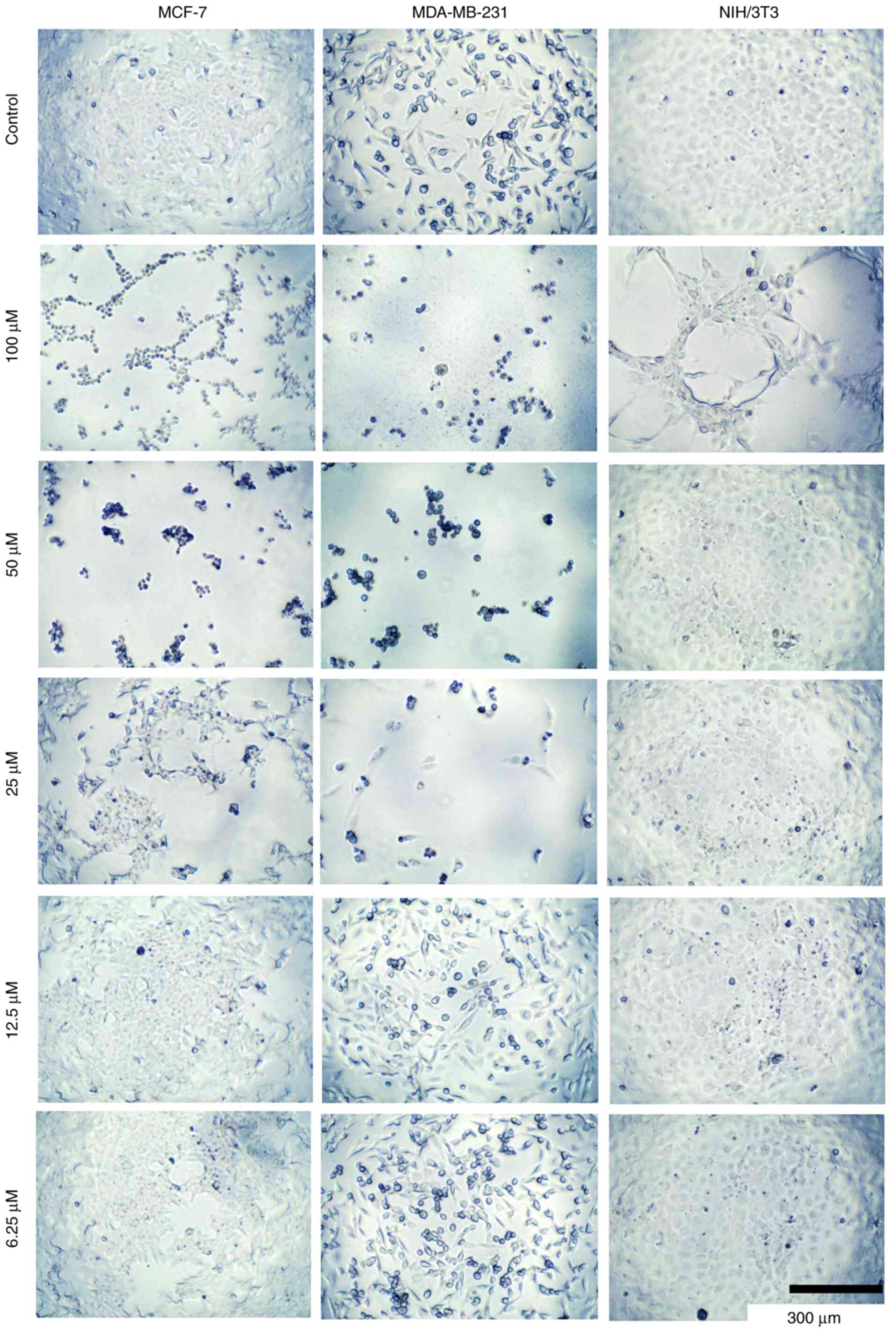
concentrations of compounds 8c, 8b, and 5 (Fig. 6, Fig.
7 and Fig. 8, respectively).
These changes potentially alter the cell structure to a more
spheroidal form, possibly affecting the cytoskeleton and related
proteins. However, further studies are required to confirm these
observations.
A brief structure-activity relationship analysis
revealed some structural differences that could potentially account
for the observed effects. For instance, compound 4a, which is
structurally similar to belinostat, demonstrated a cytotoxic
activity of 26.5%. This compound has a high structural analogy to
belinostat but features a coumarin system replacing the aromatic
ring (linker, Fig. 2) adjacent to
the sulfonamide group. At this point it is important to highlight
the fact that the sulfonamide group was proposed to be important to
maintain the activity of the molecules; this last point correlates
with the results obtained in the present study (24). Additionally, this the incorporation
of a coumarin heterocycle has also been reported to increase the
cytotoxicity activity against cancer cell lines such as
MDA-MB-231(43).
Compounds 5, 8a and 8c demonstrated cytotoxic
activities exceeding 50% at a concentration of 40 µM. Dose-response
curves were obtained to determine the IC50 values of
compounds 5 and 8b-c against MCF-7, MDA-MB-231 and 3T3/NIH cells.
All three compounds exhibited IC50 values in the
micromolar range (17-85 µM, Table
III). Compound 8b showed the best antiproliferative activity
over both MCF-7 and MDA-MB-231 cell lines. However, compound 8c
exhibited an improved safety profile, being less cytotoxic to
normal 3T3/NIH cells and with similar IC50 values over
MCF-7 and MDA-MB-231 to those of compound 8b.
| Table IIIIC50 values of compounds
5, 8b and 8c. |
Table III
IC50 values of compounds
5, 8b and 8c.
| Compound | MCF-7 | MDA-MB-231 | NIH/3T3 |
|---|
| 5 | 71±8 µM | 85±12 µM | 60±5 µM |
| 8b | 25±4 µM | 17±2 µM | 46±3 µM |
| 8c | 30±2 µM | 39±6 µM | 73±6 µM |
It is important to note that there are slight
structural differences among these compounds that could explain
their effects. Specifically, compounds 8a-c all incorporate a
coumarinic system within their structure. However, variations exist
in the spacers (methylene groups, CH2) between the
aromatic ring and the sulfonamide-coumarin system. Compound 8a
lacks methylene spacer, while compound 8b contains one methylene,
and compound 8c has two methylene spacers. These structural
variations result in cytotoxicity percentages of 45%, 62%, and 46%,
respectively, as represented in Fig.
5A. These findings suggested that the presence and number of
methylene spacers are critical factors influencing the cytotoxic
activity of these compounds. This is a significant point for
discussion in future research efforts to further elucidate the
mechanism of action and optimize the therapeutic potential of these
compounds.
Regarding compound 5, it structurally differs from
the others previously discussed as it lacks the coumarinic system;
however, it still exhibits a comparable level of cytotoxicity at
44%. A detailed analysis of its structure revealed a similarity to
belinostat, with a critical distinction: Compound 5 incorporates a
single methylene spacer between the aromatic ring and the sulfonyl
group. This observation is noteworthy because, as observed with
compounds 8a-c, the presence and number of methylene spacers have
had a substantial impact on cytotoxic activity. These facts have
been observed in other derivatives of compounds with HDAC activity,
such as belinostat, where an increase in the spacer length is
ultimately detrimental to cytotoxic activity (44). This similarity raises the
possibility that the phenyl ring, combined with a methylene spacer
and a sulfonamide group, could be a key structural motif for
modulating the biological activity in these kinds of molecules.
However, further research is required to delve deeper into this
structural feature, exploring how it influences efficacy and could
potentially be optimized for therapeutic use in cancer
treatment.
In the present study, it was observed that the
belinostat derivatives have significant effects on BC cell lines,
but further research is needed to fully elucidate their mechanisms
of action. HDAC6 plays a critical role in BC (45), as it is involved in the invasive
behavior of tumor cells and impacts the epithelial organization of
HER2-positive BC cells. HDAC6 also deacetylates HMGN2 to regulate
STAT5a activity and BC growth (46). Clinical trials have identified that
HDAC6 mRNA expression levels can be a prognostic factor and marker
of endocrine responsiveness; patients with HDAC6-positive BC have
longer progression-free survival and increased overall survival
(46). It is noteworthy that a
previous study found that HDAC4, 6, and 8 levels are higher in
MDA-MB-231 cells compared with MCF-7 cells (47). This finding aligns with the results
of the present study, where the MDA-MB-231 cell line revealed
different sensitivities to the compounds compared with MCF-7 cells
(Tables II and III). The observed variations in
IC50 values for the compounds across these cell lines
can be partially attributed to the differential expression levels
of HDACs. Further exploration of these differences, along with
comprehensive physiological, toxicological and morphophysiological
evaluations, will be crucial in understanding the full impact of
these belinostat derivatives and their potential as therapeutic
agents.
Additionally, an experiment was conducted where
leukocytes from healthy patients were exposed to the synthesized
compounds (4a, 5, 8b, 8c, 4c and 8a) at 40 µM, with belinostat used
as a reference (structural core of the compounds). The results
indicated that while these compounds demonstrated cytotoxic
activity against cancer cell lines, some exhibited slight cytotoxic
effects on healthy cells. One compound that demonstrated
particularly strong effects was 4a, which exhibited cytotoxic
activity of ~84%, making it at least 1.95-fold more cytotoxic than
belinostat, with a statistically significant difference
(P<0.05), indicating its non-selective cytotoxicity across both
cancerous and healthy cells. By contrast, compounds 5, 8b, 8c, 4c
and 8a exhibited higher viability in leukocytes, with percentages
ranging from 73-87%, demonstrating 3-4-fold lower potency than
belinostat against healthy cells. Despite this, these compounds
maintained strong cytotoxic effects against MDA-MB-231 cancer
cells, with statistically significant differences (P<0.05, for
all comparisons) compared with belinostat. Considering all the
aforementioned information, it could be possibly suggested that
these compounds may have similar mechanisms of action that allow
for selective cytotoxic effects, impacting cancer cells more than
healthy cells. The variations in their effects on leukocytes could
be related to differences in their chemical structures, such as the
presence of methylene spacers and the coumarin system, which might
influence their affinity for specific molecular targets. Compound
4a's broad cytotoxicity, while potent, may lack this selectivity,
making it less ideal for therapeutic applications where sparing
healthy cells is crucial. By contrast, the other compounds' ability
to reduce cancer cell viability while preserving a significant
proportion of healthy leukocytes suggests a more favorable
therapeutic profile, likely due to more selective interactions with
their targets. Although this compound appears to exert considerable
activity on leukocytes, it would be important in future studies to
evaluate its potential in hematological cancer cell lines, as well
as to explore other cell lines to fully understand its therapeutic
potential.
However, the broad cytotoxicity of compound 4a,
while potent, raises the question of whether its mechanism of
action could involve pathways beyond HDAC inhibition (45,46),
possibly affecting other cellular targets. To further understand
the potential therapeutic application of these compounds, it would
be interesting to explore whether the observed loss of viability is
driven by apoptotic or necrotic mechanisms, as this could
significantly influence their safety profile. Additionally,
investigating whether these compounds impact other molecular
pathways apart from HDACs could reveal broader implications for
their use in cancer treatment. These questions highlight the need
for more detailed studies to elucidate the precise mechanisms at
play and to assess the full therapeutic potential of these
compounds.
It is important to acknowledge the limitations of
the present study. While various BC cell lines were considered,
such as MDA-MB-231 and MCF-7, to enhance the generalizability of
the present findings across different BC subtypes, these specific
cell lines were chosen due to their distinct levels of HDAC6, a
critical factor in the activity of the compounds studied. Although
other BC cell lines could be relevant for similar research, the
comparison against HDAC6 levels in MDA-MB-231 and MCF-7 cells was
central to the focus of the current study. In future research, the
authors plan to evaluate additional BC cell lines, including
MCF-10A, which is widely used as a model in toxicity studies due to
its structural similarity to the normal human mammary epithelium
(48,49), to provide a more comprehensive
understanding of the compounds' effects.
Another point to discuss is the choice of
chemotherapeutics employed. Additionally, cell cycle flow
cytometric analysis will be also included in future experiments to
evaluate HDAC inhibitors, including the belinostat derivatives.
This additional analysis will help to understand the mechanisms by
which these compounds affect cell cycle regulation and include
detailed evaluations of physiological, toxicological and
morphophysiological parameters.
The primary goal of the present study was to assess
whether the belinostat derivatives could enhance biological
activity compared with existing chemotherapeutic agents. To this
end, belinostat, doxorubicin and cisplatin were used in our
experiments. Belinostat served as a reference for comparing the
biological effects of the derivatives, doxorubicin was included as
a standard compound commonly used in Mexico (50,51),
and cisplatin was selected due to its extensive use and
demonstrated efficacy in numerous studies (52,53),
particularly in the context of BC treatments in Mexico (50). Future experiments will also aim to
evaluate the effects of other chemotherapeutics, such as
carboplatin and oxaliplatin, among others, to provide a more
comprehensive overview of the effects of belinostat derivatives and
their potential to enhance biological activity. Additionally,
animal experiments will be also considered in future studies to
verify the effect of drug action in vivo, which will help
increase the reliability of the research results.
In conclusion, the synthesis, biological evaluation
and molecular docking studies of 3-carboxy-coumarin sulfonamides
have provided compelling evidence of their potential as effective
HDAC inhibitors, particularly targeting HDAC6. The present study
revealed that structural features, such as the presence and length
of methylene spacers and the incorporation of coumarin systems, are
crucial in modulating cytotoxic activity. Molecular docking results
showed strong interactions within the HDAC6 catalytic site, with
aromatic π-π interactions playing a significant role. These
interactions not only align with the observed cytotoxic profiles
but also offer insights into further optimization of these
compounds. By focusing on enhancing these structural elements,
there is substantial potential to develop more potent and selective
HDAC inhibitors, which could become valuable tools in cancer
therapy. This comprehensive approach, integrating synthesis,
biological testing and computational studies, paves the way for the
rational design of next-generation anticancer agents, ensuring both
effectiveness and specificity.
Supplementary Material
IR spectra (cm-1) of
compounds 2, 3, 4a, 4c and 5.
IR spectra (cm-1) of
compounds 6, 7, 8a, 8b and 8c.
2D representation of the interactions
of (A) TSA co-crystallized and (B) TSA docked within the histone
deacetylase 6 active site for validation purposes. TSA,
trichostatin A.
Chemical shifts in ppm
(1H-NMR) for compounds 2-8c in CDCl3 or
DMSO-d6.
Acknowledgements
The authors would like to thank Professor Julio V.
Barrios Nuñez from the University of Colima (Colima, Mexico) for
their assistance with English language editing.
Funding
Funding: The present study was supported by SIP-IPN (grant no.
20241669) and the University of Colima and CONACYT (grant no CVU
514536).
Availability of data and materials
The data generated in the present study may be
found in the Figshare platform under accession number 26999875.v2
or at the following URL: https://doi.org/10.6084/m9.figshare.26999875.v2.
Authors' contributions
FJMM, IIPM and JLMA conceptualized the study. JLMA,
GAHF, HPD, MOV and ACL developed methodology. ASEG performed
software analysis. IDE validated data. JLMA, ACL, HPD and FJM
conducted formal analysis. GAHF, IIPM and FJMM performed
investigation. FJMM, IDE, IIPM and HPD provided resources. FJMM
curated data, performed visualization and project administration.
JLMA, GAHF and ACL wrote the original draft. GAHF, FJMM and IIPM
wrote, reviewed and edited the manuscript. FJMM and IIPM supervised
the study. IDE, FJMM and IIPM confirm the authenticity of all the
raw data. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Clinical Research Center of the National Cancer
Institute (approval no. CEICANCL23062023-DISULFA-21; Colima,
Mexico). Cells were isolated from three healthy male volunteer
donors, all of whom provided oral informed consent for the
collection of their samples. The donors were aged between 26-27
years, with no history of drug use or medication intake 72 h prior
to the sample collection.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
JLMA, 0009-0002-9367-9069; GAHF,
0000-0003-4685-3095; HPD, 0000-0002-4375-6217; MOV,
0000-0003-2052-201X; IIPM, 0000-0002-9645-2049; ACL,
0000-0002-2549-4823; ASEG, 0009-0001-2487-7697; IDE,
0000-0001-9848-862X; FJMM, 0000-0001-6951-9837.
References
|
1
|
Lüönd F, Tiede S and Christofori G: Breast
cancer as an example of tumour heterogeneity and tumour cell
plasticity during malignant progression. Br J Cancer. 125:164–175.
2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Akram M, Iqbal M, Daniyal M and Khan AU:
Awareness and current knowledge of breast cancer. Biol Res.
50(33)2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
van den Boogaard WMC, Komninos DSJ and
Vermeij WP: Chemotherapy side-effects: Not All DNA damage is equal.
Cancers (Basel). 14(627)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Eckschlager T, Plch J, Stiborova M and
Hrabeta J: Histone deacetylase inhibitors as anticancer drugs. Int
J Mol Sci. 18(1414)2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ružić D, Đoković N, Nikolić K and Vujić Z:
Medicinal chemistry of histone deacetylase inhibitors. Arh Farm.
71:73–100. 2021.
|
|
6
|
Yang H, Salz T, Zajac-Kaye M, Liao D,
Huang S and Qiu Y: Overexpression of histone deacetylases in cancer
cells is controlled by interplay of transcription factors and
epigenetic modulators. FASEB J. 28:4265–4279. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sakamoto KM and Aldana-Masangkay GI: The
role of HDAC6 in cancer. J Biomed Biotechnol.
2011(875824)2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Matthews GM, Newbold A and Johnstone RW:
Intrinsic and extrinsic apoptotic pathway signaling as determinants
of histone deacetylase inhibitor antitumor activity. Adv Cancer
Res. 116:165–197. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bondarev AD, Attwood MM, Jonsson J,
Chubarev VN, Tarasov VV and Schiöth HB: Recent developments of HDAC
inhibitors: Emerging indications and novel molecules. Br J Clin
Pharmacol. 87:4577–4597. 2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Campbell P and Thomas CM: Belinostat for
the treatment of relapsed or refractory peripheral T-cell lymphoma.
J Oncol Pharm Pract. 23:143–147. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
El Omari N, Bakrim S, Khalid A, Albratty
M, Abdalla AN, Lee LH, Goh KW, Ming LC and Bouyahya A: Anticancer
clinical efficiency and stochastic mechanisms of belinostat. Biomed
Pharmacother. 165(115212)2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Han XL, Du J, Zheng YD, Dai JJ, Lin SW,
Zhang BY, Zhong FB, Lin ZG, Jiang SQ, Wei W and Fang ZY: CXCL1
clone evolution induced by the HDAC inhibitor belinostat might be a
favorable prognostic indicator in triple-negative breast cancer.
Biomed Res Int. 2021(5089371)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tuncer Z, Kurar E and Duran T:
Investigation of the effect of belinostat on MCF-7 breast cancer
stem cells via the Wnt, Notch, and Hedgehog signaling pathway.
Saudi Med J. 45:121–127. 2024.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Tuncer Z, Duran T, Gunes C and Kurar E:
Apoptotic effect of belinostat (PXD101) on MCF-7 cancer cells. Ann
Med Res. 28:941–945. 2021.
|
|
15
|
Lu P, Gu Y, Li L, Wang F, Yang X and Yang
Y: Belinostat suppresses cell proliferation by inactivating
Wnt/β-catenin pathway and promotes apoptosis through regulating PKC
pathway in breast cancer. Artif Cells Nanomed Biotechnol.
47:3955–3960. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Stefanachi A, Leonetti F, Pisani L, Catto
M and Carotti A: Coumarin: A natural, privileged and versatile
scaffold for bioactive compounds. Molecules. 23(250)2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Flores-Morales V, Villasana-Ruíz AP,
Garza-Veloz I, González-Delgado S and Martinez-Fierro ML:
Therapeutic effects of coumarins with different substitution
patterns. Molecules. 28(2413)2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yang F, Zhao N, Song J, Zhu K, Jiang CS,
Shan P and Zhang H: Design, synthesis and biological evaluation of
novel coumarin-based hydroxamate derivatives as histone deacetylase
(Hdac) inhibitors with antitumor activities. Molecules.
24(2569)2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Balbuena-Rebolledo I, Rivera-Antonio AM,
Sixto-López Y, Basurto J, Rosales-Hernández MC, Mendieta-Wejebe JE,
Martínez-Martínez FJ, Olivares-Corichi IM, García-Sánchez JR,
Guevara-Salazar JA, et al: Dihydropyrazole-carbohydrazide
derivatives with dual activity as antioxidant and
anti-proliferative drugs on breast cancer targeting the HDAC6.
Pharmaceuticals (Basel). 15(690)2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Calderón-Segura ME, Gómez-Arroyo S,
Villalobos-Pietrini R, Martínez-Valenzuela C, Carbajal-López Y,
Calderón-Ezquerro Mdel C, Cortés-Eslava J, García-Martínez R,
Flores-Ramírez D, Rodríguez-Romero MI, et al: Evaluation of
genotoxic and cytotoxic effects in human peripheral blood
lymphocytes exposed in vitro to neonicotinoid insecticides news. J
Toxicol. 2012(612647)2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hernández-Fuentes GA, Delgado-Enciso I,
Enríquez-Maldonado IG, Delgado-Machuca JJ, Zaizar-Fregoso S,
Hernandez-Rangel AE, Garcia-Casillas AC, Guzman-Esquivel J,
Rodriguez-Sanchez IP, Martinez-Fierro ML, et al: Antitumor effects
of annopurpuricin A, an acetogenin from the roots of annona
purpurea. Rev Bras Farmacogn. 34:111–121. 2024.
|
|
22
|
Haji Agha Bozorgi A and Zarghi A: Search
for the pharmacophore of histone deacetylase inhibitors using
pharmacophore query and docking study. Iran J Pharm Res.
13:1165–1172. 2014.PubMed/NCBI
|
|
23
|
Zagni C, Citarella A, Oussama M, Rescifina
A, Maugeri A, Navarra M, Scala A, Piperno A and Micale N:
Hydroxamic acid-based histone deacetylase (HDAC) inhibitors bearing
a pyrazole scaffold and a cinnamoyl linker. Int J Mol Sci.
20(945)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhang JH, Mottamal M, Jin HS, Guo S, Gu Y,
Wang G and Zhao LM: Design, synthesis and evaluation of belinostat
analogs as histone deacetylase inhibitors. Future Med Chem.
11:2765–2778. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Shen S and Kozikowski AP: Why hydroxamates
may not be the best histone deacetylase inhibitors-what some may
have forgotten or would rather forget? ChemMedChem. 11:15–21.
2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhang L, Zhang J, Jiang Q, Zhang L and
Song W: Zinc binding groups for histone deacetylase inhibitors. J
Enzyme Inhib Med Chem. 33:714–721. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Vickers CJ, Olsen CA, Leman LJ and Ghadiri
MR: Discovery of HDAC inhibitors that lack an active site
Zn(2+)-binding functional group. ACS Med Chem Lett. 3:505–508.
2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Traoré MDM, Zwick V, SimoÌes-Pires CA,
Nurisso A, Issa M, Cuendet M, Maynadier M, Wein S, Vial H, Jamet H
and Wong YS: Hydroxyl ketone-based histone deacetylase inhibitors
to gain insight into class I HDAC selectivity versus that of HDAC6.
ACS Omega. 2:1550–1562. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yu W, Liu J, Clausen D, Yu Y, Duffy JL,
Wang M, Xu S, Deng L, Suzuki T, Chung CC, et al: Discovery of zthyl
ketone-based highly selective HDACs 1, 2, 3 inhibitors for HIV
latency reactivation with minimum cellular potency serum shift and
reduced hERG activity. J Med Chem. 64:4709–4729. 2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Osko JD and Christianson DW: Structural
basis of catalysis and inhibition of HDAC6 CD1, the enigmatic
catalytic domain of histone deacetylase 6. Biochemistry.
58:4912–4924. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lobera M, Madauss KP, Pohlhaus DT, Wright
QG, Trocha M, Schmidt DR, Baloglu E, Trump RP, Head MS, Hofmann GA,
et al: Selective class IIa histone deacetylase inhibition via a
nonchelating zinc-binding group. Nat Chem Biol. 9:319–325.
2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Butler KV, Kalin J, Brochier C, Vistoli G,
Langley B and Kozikowski AP: Rational design and simple chemistry
yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J
Am Chem Soc. 132:10842–10846. 2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sixto-López Y, Gómez-Vidal JA, de Pedro N,
Bello M, Rosales-Hernández MC and Correa-Basurto J: Hydroxamic acid
derivatives as HDAC1, HDAC6 and HDAC8 inhibitors with
antiproliferative activity in cancer cell lines. Sci Rep.
10(10462)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Osko JD and Christianson DW: Structural
determinants of affinity and selectivity in the binding of
inhibitors to histone deacetylase 6. Bioorg Med Chem Lett.
30(127023)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Villa-Martínez CA, Magaña-Vergara NE,
Rodríguez M, Mojica-Sánchez JP, Ramos-Organillo ÁA, Barroso-Flores
J, Padilla-Martínez II and Martínez-Martínez FJ: Synthesis, optical
characterization in solution and solid-state, and DFT calculations
of 3-acetyl and
3-(1'-(2'-phenylhydrazono)ethyl)-coumarin-(7)-substituted
derivatives. Molecules. 27(3677)2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Martínez-Martínez FJ, Padilla-Martínez II
and Trujillo-Ferrara J: 1H and 13C NMR
assignments of 2-oxo-2H-1-benzopyran-3-acyl and -3-amide
derivatives. Magn Reson Chem. 39:765–767. 2001.
|
|
37
|
García-Báez EV, Martínez-Martínez FJ,
Höpfl H and Padilla-Martínez II: π-Stacking Interactions and CH···X
(X=O, Aryl) hydrogen bonding as directing features of the
supramolecular self-association in 3-carboxy and 3-amido coumarin
derivatives. Cryst Growth Des. 3:35–45. 2003.
|
|
38
|
Shen S and Kozikowski AP: A patent review
of histone deacetylase 6 inhibitors in neurodegenerative diseases
(2014-2019). Expert Opin Ther Pat. 30:121–136. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Vögerl K, Ong N, Senger J, Herp D,
Schmidtkunz K, Marek M, Müller M, Bartel K, Shaik TB, Porter NJ, et
al: Synthesis and biological investigation of phenothiazine-based
benzhydroxamic acids as selective histone deacetylase 6 inhibitors.
J Med Chem. 62:1138–1166. 2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Campiani G, Cavella C, Osko JD, Brindisi
M, Relitti N, Brogi S, Saraswati AP, Federico S, Chemi G, Maramai
S, et al: Harnessing the role of HDAC6 in idiopathic pulmonary
fibrosis: Design, synthesis, structural analysis, and biological
evaluation of potent inhibitors. J Med Chem. 64:9960–9988.
2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Saraswati AP, Relitti N, Brindisi M, Osko
JD, Chemi G, Federico S, Grillo A, Brogi S, McCabe NH, Turkington
RC, et al: Spiroindoline-capped selective HDAC6 inhibitors: Design,
synthesis, structural analysis, and biological evaluation. ACS Med
Chem Lett. 11:2268–2276. 2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Xu Y, Tang H, Xu Y, Guo J, Zhao X, Meng Q
and Xiao J: Design, synthesis, bioactivity evaluation, crystal
structures, and in silico studies of new α-amino amide derivatives
as potential histone deacetylase 6 inhibitors. Molecules.
27(3335)2022.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Takla FN, Bayoumi WA, El-Messery SM and
Nasr MNA: Developing multitarget coumarin based anti-breast cancer
agents: Synthesis and molecular modeling study. Sci Rep.
13(13370)2023.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Sodji QH, Kornacki JR, McDonald JF,
Mrksich M and Oyelere AK: Design and structure activity
relationship of tumor-homing histone deacetylase inhibitors
conjugated to folic and pteroic acids. Eur J Med Chem. 96:340–359.
2015.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zhang Z, Yamashita H, Toyama T, Sugiura H,
Omoto Y, Ando Y, Mita K, Hamaguchi M, Hayashi S and Iwase H: HDAC6
expression is correlated with better survival in breast cancer.
Clin Cancer Res. 10:6962–6968. 2004.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Rey M, Irondelle M, Waharte F, Lizarraga F
and Chavrier P: HDAC6 is required for invadopodia activity and
invasion by breast tumor cells. Eur J Cell Biol. 90:128–135.
2011.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Park SY, Jun JA, Jeong KJ, Heo HJ, Sohn
JS, Lee HY, Park CG and Kang J: Histone deacetylases 1, 6 and 8 are
critical for invasion in breast cancer. Oncol Rep. 25:1677–1681.
2011.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Vale N, Silva S, Duarte D, Crista DMA,
Pinto da Silva L and Esteves da Silva JCG: Normal breast epithelial
MCF-10A cells to evaluate the safety of carbon dots. RSC Med Chem.
12:245–253. 2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Paine TM, Soule HD, Pauley RJ and Dawson
PJ: Characterization of epithelial phenotypes in mortal and
immortal human breast cells. Int J Cancer. 50:463–473.
1992.PubMed/NCBI View Article : Google Scholar
|
|
50
|
United Mexican States and General Health
Council: 2018 Edition of the Basic Framework and Catalogue of
Medicines. Official Journal of the Federation, Mexico, 2018 (In
Spanish). https://www.dof.gob.mx/nota_detalle.php?codigo=5544613&fecha=23/11/2018#gsc.tab=0.
|
|
51
|
Jasso L, Lifshitz A, Arrieta O, Burgos R,
Campillo C, Celis MÁ, de la Llata M, Domínguez J, Halabe J, Islas
S, et al: Importance of the list of essential medicines in medical
prescription. Gac Med Mex. 156:598–599. 2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Romani AMP: Cisplatin in cancer treatment.
Biochem Pharmacol. 206(115323)2022.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Nauta IH, Klausch T, van de Ven PM,
Hoebers FJP, Licitra L, Poli T, Scheckenbach K, Brakenhoff RH,
Berkhof J and René Leemans C: The important role of cisplatin in
the treatment of HPV-positive oropharyngeal cancer assessed by
real-world data analysis. Oral Oncol. 121(105454)2021.PubMed/NCBI View Article : Google Scholar
|