Introduction
Chronic myeloid leukemia (CML) is caused by the
proliferation of abnormal blood cells due to hematopoietic stem
cell abnormalities. Global incidence of CML is 1.6-2 per 100,000
individuals, accounting for 15-20% of all leukemia cases. The
expected increase in the incidence of CML in the United States, as
estimated from 2010 to 2050, ranges from 70,000 cases to 180,000
cases (1-3).
The Philadelphia chromosome is detected in >90% of patients with
CML, and BCR::ABL1 tyrosine kinase encoded by this chromosome is a
major factor in CML pathogenesis (4).
Imatinib, initially introduced as a BCR::ABL1
tyrosine kinase inhibitor (TKI), has become a primary treatment
option (5). Prior to the
development of BCR::ABL1 inhibitors, CML was mainly treated using
interferon α, busulfan, and hydroxyurea therapy or allogeneic
hematopoietic stem cell transplantation, resulting in a median
survival of 10 years, and a 3-6 year survival rate of 10-20%, or a
5-year survival rate of 40-60%. Notably, development of BCR::ABL1
inhibitors, including imatinib, has significantly increased the
5-year survival rate to >90% (2,6).
However, 20-30% of patients with CML acquire resistance to
BCR::ABL1 TKIs, including imatinib (7). BCR::ABL1 TKI resistance can be
categorized into two types: BCR::ABL1-independent and
BCR::ABL1-dependent resistance. A major BCR::ABL1-dependent
resistance mechanism involves point mutations, accounting for
40-60% of BCR::ABL1 inhibitor resistance cases (8). The BCR::ABL1-independent resistance
mechanism includes the activation of bypass pathways, such as the
Janus tyrosine kinase/signal transducer and activator of
transcription, phosphatidylinositol-3 kinase (PI3K)/Akt,
WNT/β-catenin, and mitogen activated protein kinase
kinase/extracellular regulated protein kinase pathways (8-10).
However, the detailed mechanisms of BCR::ABL1 TKI resistance remain
unclear.
MDM2 is associated with p53 via its N-terminus to
prevent p53 transcriptional activity, and acts as an E3 ubiquitin
ligase for p53, promoting the cytoplasmic localization of p53 and
its degradation in proteasomes (11). MDM2 expression has been revealed to
increase with disease progression in CML and to facilitate
self-renewal of CML stem cells by reducing p53 expression (12,13).
MDM2 inhibitors have been demonstrated to enhance the sensitivity
to nilotinib and ABT-737, a BH-3 mimetic, in CD34-positive and
progenitor cells of patients with CML (14). JNJ-26854165, an MDM2 inhibitor, was
shown to trigger cell death in BCR::ABL1 T315I mutation-harboring
32D cells (15). However, the
specific effects of BCR::ABL1-independent resistance on
imatinib-resistant CML cells remain unclear.
The aim of the present study was to assess whether
MDM2 inhibitors can trigger cell death in BCR::ABL1-independent
imatinib-resistant CML cells.
Materials and methods
Reagents
Nutlin-3 (Selleck Chemicals), NSC-66811 (Calbiochem;
Merck KGaA), and pifithrin-α (Tokyo Kasei Kogyo Co., Ltd.) were
solubilized in dimethyl sulfoxide (DSMO; FUJIFILM Wako Pure
Chemical Corporation) and dispersed in phosphate-buffered saline
(0.05 M, pH 7.4). The maximum final concentration of DMSO used to
dissolve the reagents was 0.2, and 0.2% DMSO was added to the
control (0 µM) to which no MDM2 inhibitor was added.
Cell culture
The human CML cell line, K562 (cat. no. JCRB0019),
was obtained from the Japanese Cancer Research Resources Bank.
K562/IR and K562/DR cells were previously established at the
laboratory of the Division of Pharmacotherapy, Department of
Pharmacy, Kindai University (Higashi-Osaka, Japan) (9,10).
Both cell lines were maintained in the RPMI-1640 (Merck KGaA)
supplemented with 100 µg/ml penicillin, 100 U/ml streptomycin, and
10% fetal bovine serum (Thermo Fisher Scientific, Inc.) in a 5%
CO2 atmosphere.
Cell survival analysis
Effects of NSC-66811 and Nutlin-3 on cell viability
were assessed utilizing the trypan blue dye exclusion assay
(16,17). Briefly, cells were seeded in 96-well
plates at 2x103 cells/well and cultured for 24 h and
37˚C. NSC-66811 and Nutlin-3 were then added to the cells at 0.5,
1, 5, 10 and 25 µM. To evaluate cell viability with a combination
of 20 µM pifithrin-α and MDM2 inhibitors (NSC-66811 or Nutlin-3),
each MDM2 inhibitor was added 2 h after the addition of
pifithrin-α. Following 3 days of culture at 37˚C, cells were
suspended in 0.4% trypan blue solution at room temperature for 3
min and then analyzed utilizing a hematometer. Cell viability was
calculated by assessing the live and dead cells under a light
microscope.
Apoptosis analysis
Detection of apoptosis was assessed utilizing the
Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit
(cat. no. 15342-54; Nacalai Tesque, Inc.), following the
manufacturer's guidelines (18).
Briefly, cells were harvested, rinsed twice with PBS, and
resuspended in Annexin V binding buffer to achieve a concentration
of 1x106 cells/ml. The modified cell mixture was
combined with 5 µl of both Annexin V-FITC and PI solutions, then
allowed to incubate for a duration of 15 min at room temperature.
Following the incubation, 400 µl of Annexin V binding buffer was
introduced, and the sample was subsequently examined utilizing the
BD LSR Fortessa cell analyzer (Becton-Dickinson and Company) and
analyzed by Flow Jo software (Ver. 10; Flowjo LLC).
Caspase-3 activity analysis
Caspase-3 activity was assessed utilizing the
caspase-3/CPP32 fluorometric assay kit (cat. no. K105-25;
BioVision, Inc.), following the manufacturer's guidelines. Cells
were exposed to NSC-66811 (10 and 25 µM) and Nutlin-3 (10 and 25
µM) for 2 days at 37˚C, washed with PBS, and processed with the
lysis buffer included in the kit. The lysate was incubated with 1
mM Asp-Glu-Val-Asp-7-amino-4-trifluoromethylcoumarin (AFC; included
in the kit) at 37˚C for 1 h. The levels of AFC (excitation
wavelength, 400 nm; emission wavelength, 505 nm) emitted from the
substrate were analyzed utilizing a fluorescence spectrophotometer
(F-4500; Hitachi, Ltd.). Records were calibrated for lysate protein
concentration and represented as the change in proteolytic cleavage
(pM) of the substrate per hour per milligram of protein. The
protein concentrations in the lysates were measured utilizing a BCA
protein assay kit (cat. no. T9300A; Takara Bio, Inc.).
Western blotting
The extraction of cells was carried out utilizing
the ProteoExtract Subcellular Proteome Extraction Kit from
MilliporeSigma (cat. no. 539790), following the manufacturer's
guidelines, and the analysis was performed through western
blotting, as previously reported (10). Briefly, using the ProteoExtract
Subcellular Proteome Extraction Kit, the extracted cytoplasmic and
nuclear fractions (20 µg) were blotted to polyvinylidene difluoride
(PVDF) membranes (cat. no. IPVH00010; MilliporeSigma) after
electrophoresis on a 10% sodium dodecyl sulfate-polyacrylamide gel.
The PVDF membranes were then treated with 3% skim milk at room
temperature for 30 min, followed by overnight reaction at 4˚C with
the primary antibodies listed below. The membranes were then washed
three times with 0.1% TBS-T (cat. no. 207-18061; FUJIFILM Wako Pure
Chemical Corporation) for 5 min and reacted with horseradish
peroxidase (HRP)-conjugated secondary antibody for 1 h at room
temperature. Then, after washing three times with 0.1% TBS-T for 5
min, the antibody was reacted with ImmobilonForte (cat. no.
WBLUF0500; MilliporeSigma) for 5 min at room temperature, and
visualized by CS analyzer (Ver 3.0; ATTO Corporation). The protein
concentrations in the lysates were measured utilizing a BCA protein
assay kit (Takara Bio, Inc.). The following antibodies were
employed for the assay: Anti-lamin A/C (cat. no. sc-376248;
dilution 1:1,000), anti-MDM2 (cat. no. sc-965; dilution 1:1,000),
anti-p53 (cat. no. sc-126; dilution 1:1,000), anti-Puma (cat. no.
sc-377015; dilution 1:1,000), anti-Bax (cat. no. sc-7480; dilution
1:1,000), anti-Noxa (cat. no. sc-56169; dilution 1:1,000), anti-p21
(cat. no. sc-6246; dilution 1:1,000), anti-caspase-3 (cat. no.
sc-56053; dilution 1:1,000) (all from Santa Cruz Biotechnologies,
Inc.), anti-β-actin (cat. no. A2228; dilution 1:3,000;
MilliporeSigma), HRP-conjugated anti-mouse antibody (cat. no. 7076;
dilution 1:3,000) and anti-rabbit antibody (cat. no. 7074; dilution
1:3,000) (Cell Signaling Technology, Inc.).
Gene expression omnibus (GEO)
datasets
MDM2 expression was analyzed utilizing
GSE14671(19) and GSE33224(20) obtained from the GEO database of the
National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/geo/). In GSE14671,
MDM2 expression in each patient was detected on the Affymetrix
microarray platform (GPL570), and MDM2 expression in the 24
imatinib responders and 12 imatinib non-responders was evaluated.
In GSE33224, MDM2 expression in each patient was detected on the
Agilent microarray platform (GPL4133), and MDM2 expression was
assessed in 12 imatinib responders and 8 imatinib non-responders
(without the BCR::ABL1 mutation). To evaluate GSE14671 and GSE33224
collectively, MDM2 expression in imatinib non-responders was
detected when the mean number of imatinib responders was set at
one.
Statistical analysis
Statistical analysis was performed utilizing SPSS
version 21.0 software (IBM, Inc.). The results are expressed as the
mean ± standard deviation. Analysis was conducted utilizing one-way
analysis of variance (ANOVA), followed by Dunnett's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Downregulation of p53 and
overexpression of MDM2 expression in K562/IR cells
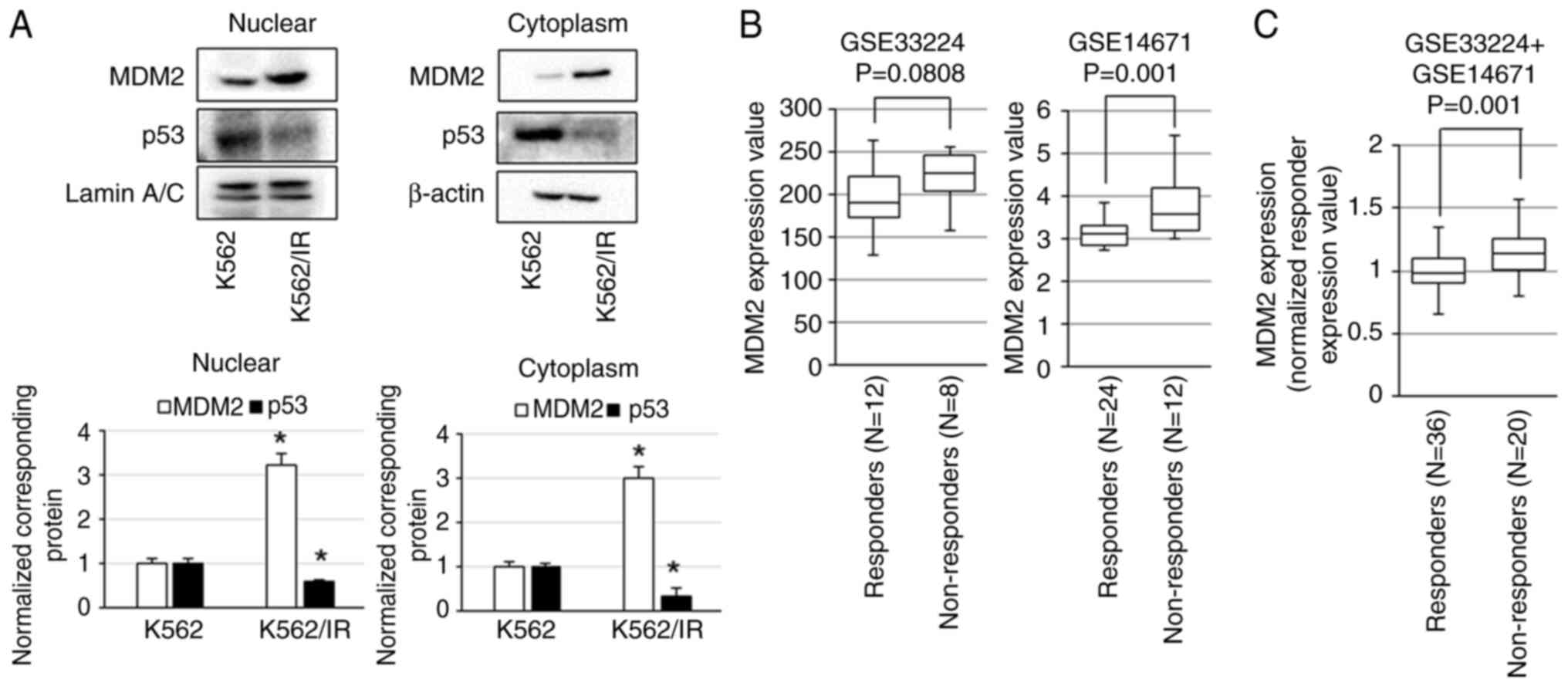
Protein levels of p53 and MDM2 in K562/IR and K562
cells were examined. The expression levels of MDM2 were elevated in
both nuclear and cytoplasmic fractions in K562/IR cells, whereas
the expression levels of p53 were decreased in K562/IR cells
compared with K562 cells (Fig. 1A).
Next, the GEO datasets, GSE14671 and GSE33224, were used to
determine the association between MDM2 expression and imatinib
resistance in patients with CML. In GSE33224, MDM2 expression
tended to be higher in imatinib non-responders than in imatinib
responders, and in GSE14671, MDM2 expression was found to be
significantly higher in the imatinib non-responders than in the
imatinib responders (Fig. 1B). In
the combined analysis of GSE14671 and GSE33224 datasets, MDM2
expression was significantly higher in imatinib non-responders than
in imatinib responders (Fig. 1C).
These results indicated that MDM2 is a target molecule for
BCR::ABL1-independent resistance.
Inhibition of MDM2 by NSC-66811 and
Nutlin-3 induces the apoptosis of K562/IR cells
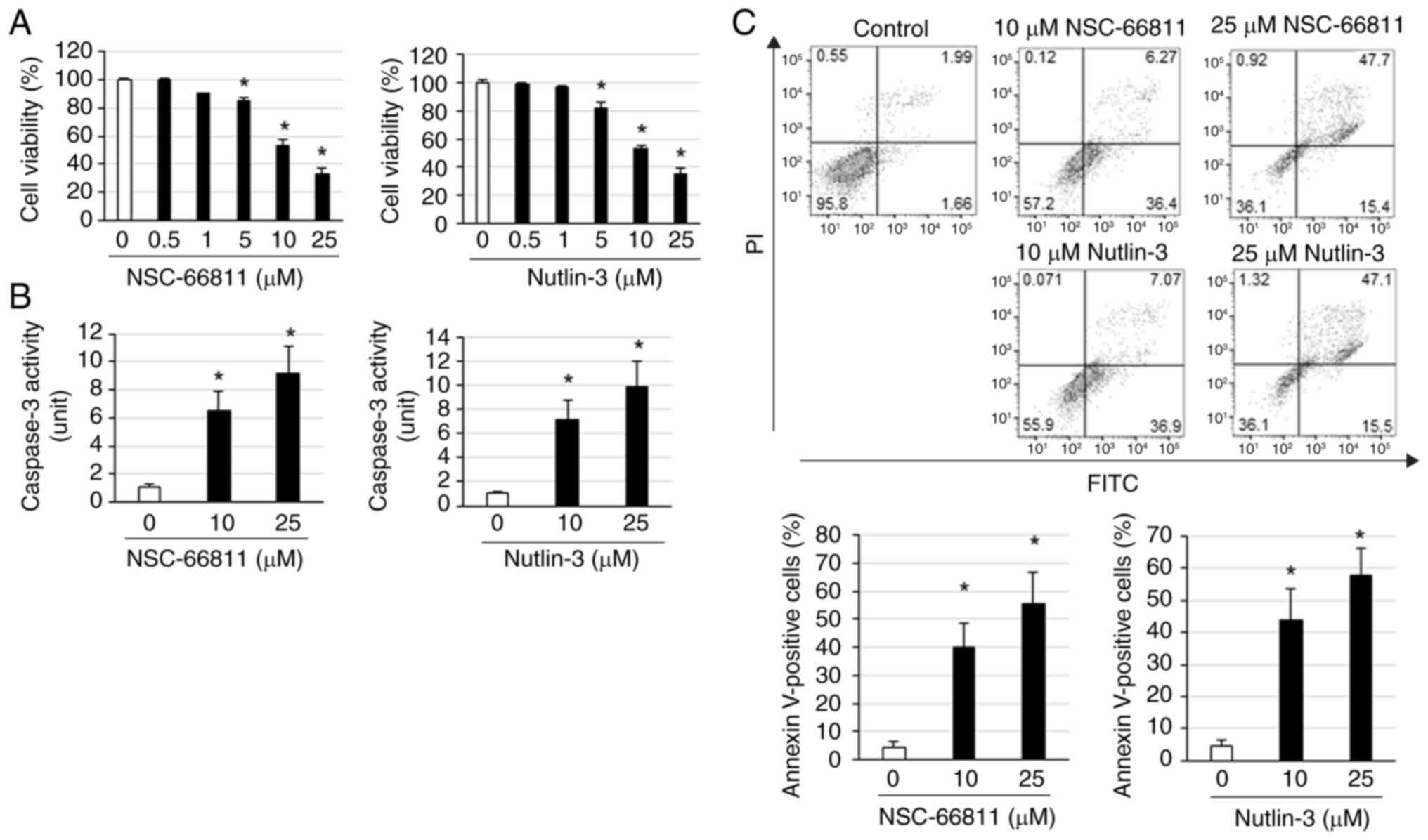
Next, it was assessed whether NSC-66811 and Nutlin-3
promote cell death in K562/IR cells. Both NSC-66811 and Nutlin-3
induced cell death in K562/IR cells in a concentration-dependent
manner (Fig. 2A). Additionally,
NSC-66811 and Nutlin-3 did not induce cell death in K562 cells at
concentrations that induced cell death in K562/IR cells (Fig. S1A). NSC-66811 and Nutlin-3
adequately potentiated the cell death-inducing effects of imatinib,
cytarabine, and busulfan (Fig.
S1B-D). NSC-66811 and Nutlin-3 increased the expression of
cleaved caspase-3 and activation of caspase-3, respectively
(Figs. 2B and 3B). Furthermore, the percentages of
early/late apoptotic or necrotic cells by treatment with NSC-66811
and Nutlin-3 were 36.4/6.27 and 0.12% (10 µM NSC-66811), 15.4/47.79
and 0.92% (25 µM NSC-66811), 36.9/7.07 and 0.071% (10 µM Nutlin-3),
15.5/47.1 and 1.32% (25 µM Nutlin-3), respectively (Fig. 2C) and administration with NSC-66811
and Nutlin-3 increased the number of Annexin V-positive cells in
K562/IR cells (Fig. 2C). These
results indicated that NSC-66811 and Nutlin-3 induced apoptosis in
K562/IR cells and potentiated the cell death-inducing effects of
imatinib, cytarabine, and busulfan.
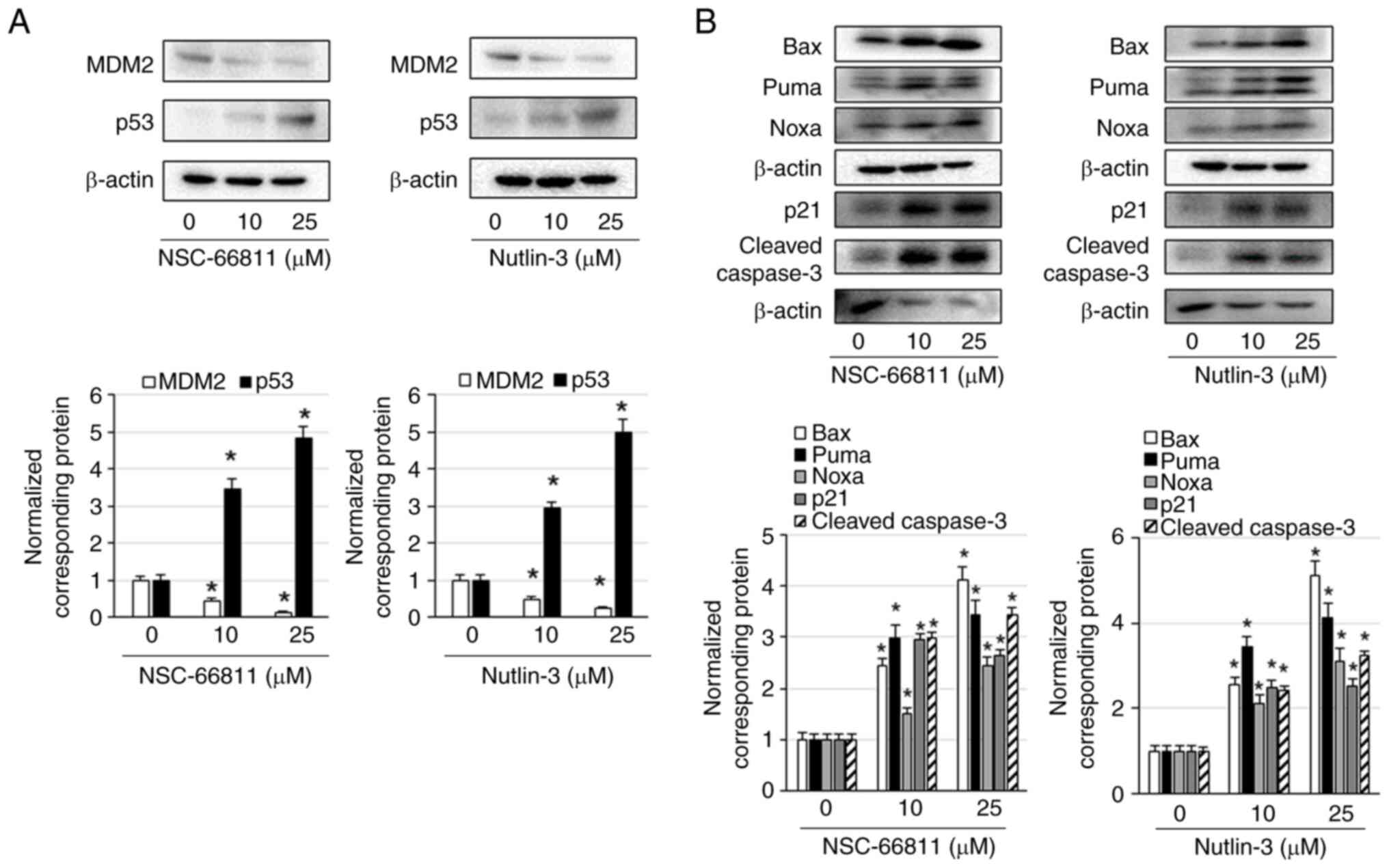 | Figure 3NSC-66811 and Nutlin-3 increase the
expression levels of p53 and decrease the expression levels of MDM2
in K562/IR cells. K562/IR cells were treated with the indicated
concentrations of NSC-66811 and Nutlin-3 for 3 days. (A) Cell
lysates were assessed via immunoblotting with antibodies against
MDM2, p53, and β-actin. MDM2 and p53 expression levels were
normalized to those of β-actin. Results are representative of three
independent experiments. *P<0.01 vs. untreated cells
(0.1% DMSO). (B) Cell lysates were assessed via immunoblotting with
antibodies against Bax, Puma, Noxa, p21, cleaved caspase-3, and
β-actin. The expression levels of Bax, Puma, Noxa, p21, and cleaved
caspase-3 were normalized to those of β-actin. Results are
representative of three independent experiments.
*P<0.01 vs. untreated cells (0.1% DMSO). MDM2, E3
ubiquitin-protein ligase Mdm2. |
A previous study demonstrated that the activation of
the MET pathway is involved in the BCR::ABL1 inhibitor resistance
mechanism in K562/IR cells (10).
To investigate whether MDM2 inhibitors also induce cell death in
cells with BCR::ABL1 inhibitor resistance mechanisms other than
this pathway, dasatinib-resistant K562/DR cells with BCR::ABL1
resistance mechanisms were analyzed by activating ERK1/2 via MOS
and TPL2 overexpression (9). First,
the protein levels of p53 and MDM2 in the K562/DR and K562 cells
were examined, and it was found that MDM2 expression was elevated
in K562/DR cells, both in the nuclear and cytoplasmic fractions,
whereas p53 expression levels were lower than those in K562 cells
(Fig. S2A). In addition, NSC-66811
and Nutlin-3 significantly induced the death of K562/DR cells and
potentiated the cell death-inducing effects of dasatinib (Fig. S2B). These results indicated that
MDM2 inhibitors induce cell death in CML cells with BCR::ABL1
resistance mechanisms, at least those involving MET pathway
activation and ERK1/2 activation through MOS and TPL2
overexpression.
NSC-66811 and Nutlin-3 increase Bax,
Puma, Noxa, and p21 levels by enhancing p53 expression and
decreasing MDM2 expression in K562/IR cells
To confirm the mechanisms underlying the
apoptosis-inducing effects of NSC-66811 and Nutlin-3, MDM2 and p53
levels in cells were examined. NSC-66811 and Nutlin-3 increased the
levels of p53 and decreased the levels of MDM2 in the cytoplasm of
K562/IR cells (Fig. 3A). Bax, Puma,
and Noxa, which are activated by p53, are BH-3-only proteins that
are involved in the intrinsic apoptotic pathway in the mitochondria
and p53 increases the expression of p21 via promoted transcription
of p21 (21,22). In the present study, the alterations
in these apoptosis-related factor levels after the treatment of
K562/IR cells with NSC-66811 and Nutlin-3 were examined. NSC-66811
and Nutlin-3 increased the levels of Bax, Puma, Noxa, and p21 in
K562/IR cells (Fig. 3B). These
results indicated that NSC-66811 and Nutlin-3 increase Bax, Puma,
Noxa, and p21 levels by promoting p53 expression and inhibiting
MDM2 expression in K562/IR cells.
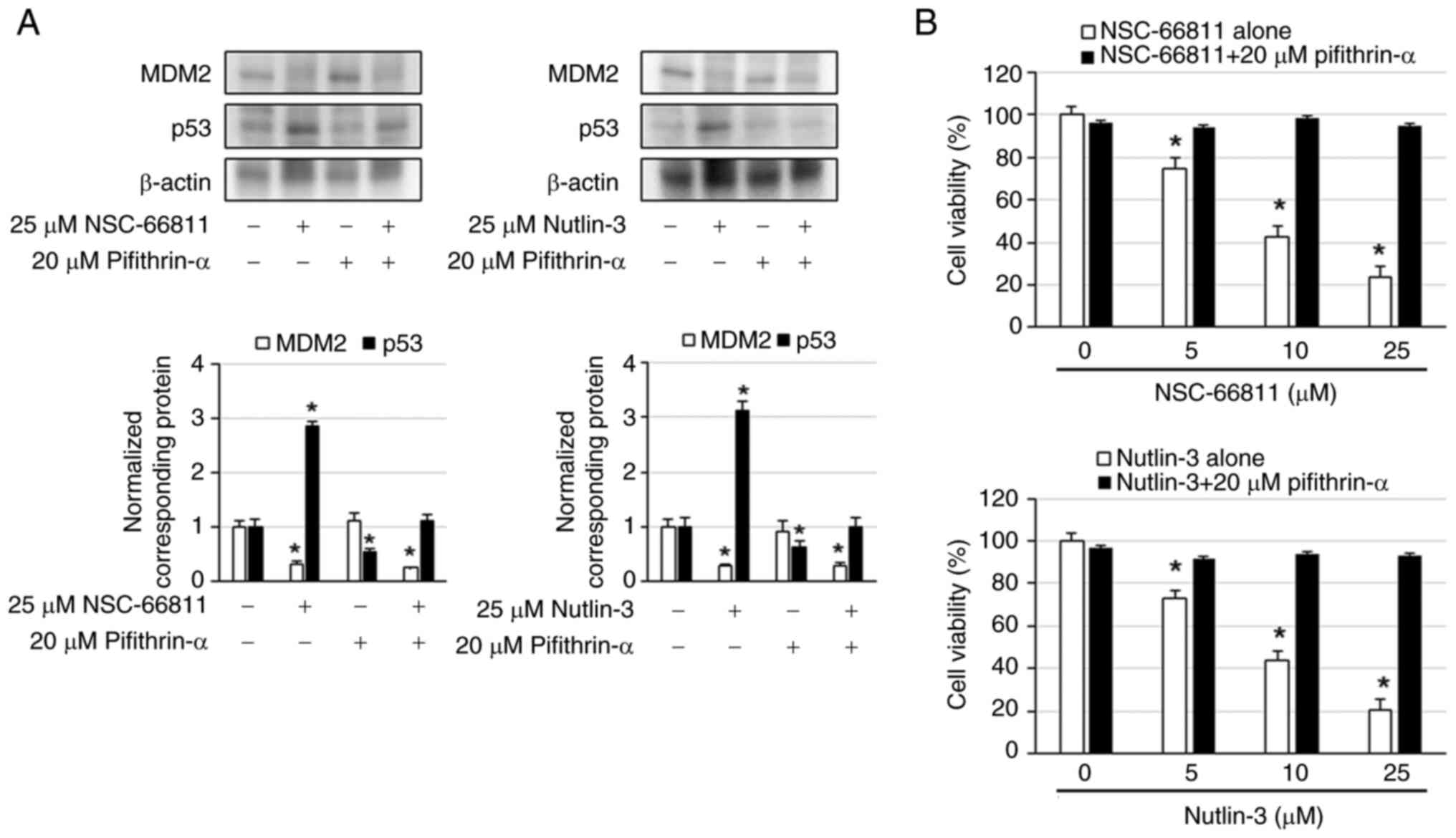
To investigate whether the increased expression of
p53 is involved in the induction of cell death of K562/IR cells by
MDM2 inhibitors, cell viability was examined when p53 inhibitor
pifithrin-α was combined with NSC-66811 or Nutlin-3. The results
revealed that NSC-66811 and Nutlin-3 decreased MDM2 expression and
increased p53 expression, whereas pifithrin-α did not alter MDM2
expression and decreased p53 expression (Fig. 4A). MDM2 expression in combination
with MDM2 inhibitor and pifithrin-α was comparable to that of MDM2
inhibitor alone, whereas p53 expression was reduced compared with
MDM2 inhibitor alone and comparable to that of no treatment
(Fig. 4A). Additionally,
pifithrin-α suppressed NSC-66811- and Nutlin-3-induced cell death
in K562/IR cells (Fig. 4B). These
results indicated that the MDM2 inhibitor promoted cell death by
decreasing the expression of MDM2 and increasing the expression of
p53.
Discussion
In present study, it was demonstrated that
BCR::ABL1-independent imatinib-resistant K562/IR cells and
dasatinib-resistant K562/DR cells exhibited higher MDM2 levels and
lower p53 levels in the cytoplasm and nucleus than
imatinib-sensitive and dasatinib-sensitive K562 cells. Moreover,
imatinib non-responders with CML had markedly higher MDM2 levels
than the imatinib responders with CML. Mutations in MDM2 promoter
induce MDM2 overexpression, accelerating the shift from chronic to
blast crisis phase, thereby worsening the prognosis of patients
with CML (23). Inactivation of p53
has been revealed to be correlated with low imatinib sensitivity
in vitro, in vivo, and in patients with CML (24). Therefore, the MDM2/p53 axis has been
demonstrated to contribute to BCR::ABL1 TKI resistance in CML,
suggesting the potential of agents modulating the MDM2/p53 axis for
CML therapy.
In the present study, NSC-66811 and Nutlin-3
enhanced the number of Annexin V-positive cells, caspase-3
activity, and cleaved caspase-3 expression, thereby inducing
apoptosis in K562/IR cells. Additionally, NSC-66811 and Nutlin-3
potentiated the cell death-inducing effects of imatinib,
cytarabine, and busulfan in K562/IR cells, and the cell
death-inducing effect of dasatinib in K562/DR cells. Moreover,
NSC-66811 and Nutlin-3 increased Bax, Puma, Noxa, and p21 levels by
decreasing MDM2 expression and increasing p53 expression in K562/IR
cells. Furthermore, pifithrin-α, a p53 inhibitor, attenuated the
NSC-66811- and Nutlin-3-inducing cell death in K562/IR cells.
Imatinib-resistant CML cells have been shown to exhibit lower p53
and Bax levels and higher MDM2 levels than imatinib-sensitive CML
cells (25). Nilotinib-resistant
acute lymphocytic leukemia cells were revealed to overexpress MDM2;
MDM2 inhibition using PI3K/mammalian target of rapamycin dual
inhibitors overcame nilotinib resistance in these cells (26). MDM2 inhibitors have been
demonstrated to induce apoptosis by increasing the levels of
BAX, PUMA, and NOXA, the target genes of p53,
in CML blast crisis cells with or without the BCR::ABL1
T315I mutation (27).
Moreover, MDM2 inhibitor and BCR::ABL1 TKI combination treatment
reduced the viability of CML stem cells (12). These findings indicate the
therapeutic effects of MDM2 inhibitors against BCR::ABL1 TKI
resistance.
It has been reported that mepacrine induces cell
death via increased p53 expression, and that an MDM2 inhibitor
potentiates BCR::ABL1 TKI-induced cell death in CML leukemic stem
cells (28). Additionally,
activation of BCR::ABL1 increased the expression of MDM2, thereby
abrogating p53 activation in CML cells (13). It has also been indicated that
BCR::ABL TKI enhances the expression of the TP53 gene in the
serum of patients with chronic phase CML, and drugs that activate
p53 may potentiate the efficacy of BCR::ABL1 TKIs (29). Mutations in MDM2 have also been
shown to be risk factors for developing CML (30). Clinical trials of RG7112, a
derivative of Nutlin-3, in CML, chronic lymphocytic leukemia, acute
lymphocytic leukemia, and acute myeloid leukemia have shown grade 3
and 4 neutropenia in 22% of the patients. Of the 30 patients, 5
achieved complete and partial responses, while RG7112 was not
effective as treatment in CML, chronic lymphocytic leukemia, acute
lymphocytic leukemia, or acute myeloid leukemia (31). Therefore, a combination therapy with
MDM2 inhibitors and other anticancer agents may be necessary.
Furthermore, it has been previously reported that Nutlin-3 enhanced
the apoptosis-inducing effect of nilotinib in CML stem/progenitor
cells, indicating that MDM2 inhibitors such as Nutlin-3 may be
effective against BCR::ABL1 TKI primary resistance in CML
stem/progenitor cells (14). In the
present study, MDM2 inhibitors potentiated the death-inducing
effects of imatinib, cytarabine, busulfan, and dasatinib in K562/IR
and K562/DR cells, demonstrating that MDM2 inhibitors are valuable
against BCR::ABL1 TKI-acquired resistant CML cells. These findings
indicate that MDM2 inhibitors that induce p53 activation may be
valuable as concomitant agents for BCR::ABL1 TKI, cytarabine, and
busulfan, and as agents to overcome acquired resistance to
BCR::ABL1 TKI.
It has been reported that K562 cells have a
single-base insertion mutation between codons 135 and 136,
indicating a lack of function (32). However, imatinib resistance was
revealed to be correlated with low p53 expression in K562/G cells
(25). Additionally, the mutant p53
harbored in K562 cells recovered wild-type p53 function in
12-O-tetradecanoylphorbol 13-acetate-resistant K562 cells
(33). In the present study, it was
observed that MDM2 inhibitors induced cell death in K562/IR and
K562/DR cells, but not in K562 cells, at the same concentrations.
These findings suggest that BCR::ABL1 TKI-resistant K562 cells may
convert mutant p53 into p53 with wild-type function, which needs to
be elucidated in subsequent studies.
In the present study, 5, 10 and 25 µM were used as
administered concentrations of Nutlin-3. Although Nutlin-3 has not
been clinically evaluated in humans, a derivative of Nutlin-3,
RG-7112, has been clinically studied in liposarcoma, with
steady-state plasma levels of ~12 µM, partial responses in 1 out of
20 patients, and stable disease in 14 patients (34). Nausea and vomiting, as well as
thrombocytopenia were also concerns in this clinical trial as
adverse events (34). Furthermore,
oral administration of 200 mg/kg of Nutlin-3 in mice has been shown
to produce a plasma Cmax of ~60 µM and to be well
tolerated in mice at this dose (35). Based on these findings, it is
possible that Nutlin-3 may be well tolerated in humans at a high
concentration (25 µM), but a lower concentration than 10 µM may be
better considering the clinical trial of RG-7112(34). Therefore, if the plasma
concentration of Nutlin-3 can be maintained at ~5 µM, it may be
viable in combination with BCR::ABL1 TKI and conventional
anticancer agents for the treatment of CML. However, whether 5 µM
of Nutlin-3 can actually be maintained in human plasma needs to be
investigated in the future.
Although it was demonstrated that MDM2 inhibitors
induce apoptosis via p53 activation in imatinib-resistant CML
cells, the present study has several limitations. First, as
aforementioned, the p53 gene status in K562/IR cells could not be
confirmed nor could the plasma concentrations in humans or mice
treated with Nutlin-3 be measured. Second, the effects of MDM2
inhibitors on tumor growth in K562/IR cell-bearing mice were not
examined. Third, the cell death-inducing effects of MDM2 inhibitors
in CML cells from patients with imatinib non-responder CML were not
confirmed. These are crucial factors for the clinical application
of MDM2 inhibitors and should be investigated in future
studies.
In conclusion, the expression levels of MDM2 were
increased in patients with CML who were non-responders to imatinib
and in imatinib-resistant CML cells. However, MDM2 inhibition
induced the apoptosis of imatinib-resistant CML cells by increasing
the expression levels of Bax, Puma, and Noxa via p53 activation. It
was previously found that activation of the MET pathway and
enhancing expression of TPL2 and MOS are associated with BCR::ABL1
inhibitor resistance in CML cells, and that HIF-1α inhibition
promotes cell death in BCR::ABL1 inhibitor-sensitive and -resistant
CML cells (9,10,36).
Overall, these findings, along with previous research by the
authors, highlight MDM2 as a therapeutic target to treat
imatinib-resistant CML.
Supplementary Material
NSC-66811 and Nutlin-3 potentiate the
cell death-inducing effects of imatinib, cytarabine, and busulfan
in K562/IR cells. (A) Following treatment of K562 and K562/IR cells
with NSC-66811 and Nutlin-3, cell viability was determined via
trypan blue dye exclusion assay. The cells were treated with the
indicated concentrations of NSC-66811 and Nutlin-3 for 3 days.
Results are representative of five independent experiments.
*P<0.01 vs. untreated cells (0.1% DMSO). (B-D)
K562/IR cells were treated with NSC-66811 or Nutlin-3 in
combination with (B) imatinib, (C) cytarabine, and (D) busulfan,
and cell viability was determined via trypan blue dye exclusion
assay. Each drug was administered at the same time. Cells were
treated with the indicated concentrations of NSC-66811, Nutlin-3,
imatinib, cytarabine, or busulfan for 3 days. Results are
representative of five independent experiments.
*P<0.01 vs. each concentration of MDM2 inhibitor
(NSC-66811 or Nutlin-3) alone.
Increased expression levels of MDM2
and decreased expression levels of p53 in K562/DR cells. (A)
Following incubation for 2 days, cell lysates were assessed via
immunoblotting with antibodies against MDM2, p53, β-actin, and
lamin A/C. The expression levels of MDM2 and p53 were normalized to
those of β-actin or lamin A/C. Results are representative of three
independent experiments. *P<0.01 vs. K562 cells. (B)
K562/DR cells were treated with NSC-66811 or Nutlin-3 in
combination with dasatinib, and cell viability was determined via
trypan blue dye exclusion assay. Each drug was administered at the
same time. Cells were treated with the indicated concentrations of
NSC-66811, Nutlin-3, and dasatinib for 3 days. Results are
representative of five independent experiments.
*P<0.01 vs. untreated cells (0.2% DMSO);
#P<0.01 vs. each concentration of MDM2 inhibitor
(NSC-66811 or Nutlin-3) alone. MDM2, E3 ubiquitin-protein ligase
Mdm2.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported in part by a
Grant-in-Aid for Scientific Research (C) (grant no. 23K06270) from
the Japan Society for the Promotion of Science (JSPS).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
AK and MT performed the experiments, acquired and
analyzed the data, and wrote the manuscript. TO, TM, RK, NN and TY
performed the experiments, and acquired and analyzed the data. AK
and MT confirm the authenticity of all the raw data. MT and SN
designed the experiments and revised the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Romero-Morelos P, González-Yebra AL,
Muñoz-López D, Lara-Lona E and González-Yebra B: Frequencies of
BCR::ABL1 transcripts in patients with chronic myeloid leukemia: A
meta-analysis. Genes (Basel). 15(232)2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Sun J, Hu R, Han M, Tan Y, Xie M, Gao S
and Hu JF: Mechanisms underlying therapeutic resistance of tyrosine
kinase inhibitors in chronic myeloid leukemia. Int J Biol Sci.
20:175–181. 2024.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Held N and Atallah EL: Real-world
management of CML: Outcomes and treatment patterns. Curr Hematol
Malig Rep. 18:167–175. 2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Marzocchi G, Castagnetti F, Luatti S,
Baldazzi C, Stacchini M, Gugliotta G, Amabile M, Specchia G,
Sessarego M, Giussani U, et al: Variant Philadelphia
translocations: Molecular-cytogenetic characterization and
prognostic influence on frontline imatinib therapy, a GIMEMA
working party on CML analysis. Blood. 117:6793–6800.
2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
El-Tanani M, Nsairat H, Matalka II, Lee
YF, Rizzo M, Aljabali AA, Mishra V, Mishra Y, Hromić-Jahjefendić A
and Tambuwala MM: The impact of the BCR-ABL oncogene in the
pathology and treatment of chronic myeloid leukemia. Pathol Res
Pract. 254(155161)2024.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kantarjian HM, Jain N, Garcia-Manero G,
Welch MA, Ravandi F, Wierda WG and Jabbour EJ: The cure of leukemia
through the optimist's prism. Cancer. 128:240–259. 2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hochhaus A, Baccarani M, Silver RT,
Schiffer C, Apperley JF, Cervantes F, Clark RE, Cortes JE,
Deininger MW, Guilhot F, et al: European LeukemiaNet 2020
recommendations for treating chronic myeloid leukemia. Leukemia.
34:966–984. 2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Poudel G, Tolland MG, Hughes TP and Pagani
IS: Mechanisms of resistance and implications for treatment
strategies in chronic myeloid leukaemia. Cancers (Basel).
14(3300)2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tsubaki M, Takeda T, Koumoto Y, Usami T,
Matsuda T, Seki S, Sakai K, Nishio K and Nishida S: Activation of
ERK1/2 by MOS and TPL2 leads to dasatinib resistance in chronic
myeloid leukaemia cells. Cell Prolif. 56(e13420)2023.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Tsubaki M, Takeda T, Kino T, Sakai K, Itoh
T, Imano M, Nakayama T, Nishio K, Satou T and Nishida S:
Contributions of MET activation to BCR-ABL1 tyrosine kinase
inhibitor resistance in chronic myeloid leukemia cells. Oncotarget.
8:38717–38730. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Tuval A, Strandgren C, Heldin A,
Palomar-Siles M and Wiman KG: Pharmacological reactivation of p53
in the era of precision anticancer medicine. Nat Rev Clin Oncol.
21:106–120. 2024.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Scott MT, Liu W, Mitchell R, Clarke CJ,
Kinstrie R, Warren F, Almasoudi H, Stevens T, Dunn K, Pritchard J,
et al: Activating p53 abolishes self-renewal of quiescent leukaemic
stem cells in residual CML disease. Nat Commun.
15(651)2024.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Trotta R, Vignudelli T, Candini O, Intine
RV, Pecorari L, Guerzoni C, Santilli G, Byrom MW, Goldoni S, Ford
LP, et al: BCR/ABL activates mdm2 mRNA translation via the La
antigen. Cancer Cell. 3:145–160. 2003.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Carter BZ, Mak PY, Mak DH, Ruvolo VR,
Schober W, McQueen T, Cortes J, Kantarjian HM, Champlin RE,
Konopleva M and Andreeff M: Synergistic effects of p53 activation
via MDM2 inhibition in combination with inhibition of Bcl-2 or
Bcr-Abl in CD34+ proliferating and quiescent chronic myeloid
leukemia blast crisis cells. Oncotarget. 6:30487–30499.
2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
You L, Liu H, Huang J, Xie W, Wei J, Ye X
and Qian W: The novel anticancer agent JNJ-26854165 is active in
chronic myeloid leukemic cells with unmutated BCR/ABL and T315I
mutant BCR/ABL through promoting proteosomal degradation of BCR/ABL
proteins. Oncotarget. 8:7777–7790. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tsubaki M, Takeda T, Noguchi M, Jinushi M,
Seki S, Morii Y, Shimomura K, Imano M, Satou T and Nishida S:
Overactivation of Akt contributes to MEK inhibitor primary and
acquired resistance in colorectal cancer cells. Cancers (Basel).
11(1866)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tsubaki M, Takeda T, Tomonari Y, Koumoto
YI, Imano M, Satou T and Nishida S: Overexpression of HIF-1α
contributes to melphalan resistance in multiple myeloma cells by
activation of ERK1/2, Akt, and NF-κB. Lab Invest. 99:72–84.
2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Morii Y, Tsubaki M, Takeda T, Otubo R,
Seki S, Yamatomo Y, Imano M, Satou T, Shimomura K and Nishida S:
Perifosine enhances the potential antitumor effect of
5-fluorourasil and oxaliplatin in colon cancer cells harboring the
PIK3CA mutation. Eur J Pharmacol. 898(173957)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
McWeeney SK, Pemberton LC, Loriaux MM,
Vartanian K, Willis SG, Yochum G, Wilmot B, Turpaz Y, Pillai R,
Druker BJ, et al: A gene expression signature of CD34+ cells to
predict major cytogenetic response in chronic-phase chronic myeloid
leukemia patients treated with imatinib. Blood. 115:315–325.
2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Silveira RA, Fachel AA, Moreira YB, De
Souza CA, Costa FF, Verjovski-Almeida S and Pagnano KBB:
Protein-coding genes and long noncoding RNAs are differentially
expressed in dasatinib-treated chronic myeloid leukemia patients
with resistance to imatinib. Hematology. 19:31–41. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hao Q, Chen J, Lu H and Zhou X: The ARTS
of p53-dependent mitochondrial apoptosis. J Mol Cell Biol.
14(mjac074)2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Aravindhan S, Younus LA, Hadi Lafta M,
Markov A, Ivanovna Enina Y, Yushchenkо NA, Thangavelu L, Mostafavi
SM, Pokrovskii MV and Ahmadi M: P53 long noncoding RNA regulatory
network in cancer development. Cell Biol Int. 45:1583–1598.
2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liu YC, Hsiao HH, Yang WC, Liu TC, Chang
CS, Yang MY, Lin PM, Hsu JF, Lee CP and Lin SF: MDM2 promoter
polymorphism and p53 codon 72 polymorphism in chronic myeloid
leukemia: The association between MDM2 promoter genotype and
disease susceptibility, age of onset, and blast-free survival in
chronic phase patients receiving imatinib. Mol Carcinog.
53:951–959. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Wendel HG, de Stanchina E, Cepero E, Ray
S, Emig M, Fridman JS, Veach DR, Bornmann WG, Clarkson B, McCombie
WR, et al: Loss of p53 impedes the antileukemic response to BCR-ABL
inhibition. Proc Natl Acad Sci USA. 103:7444–7449. 2006.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Cheng Y, Hao Y, Zhang A, Hu C, Jiang X, Wu
Q and Xu X: Persistent STAT5-mediated ROS production and
involvement of aberrant p53 apoptotic signaling in the resistance
of chronic myeloid leukemia to imatinib. Int J Mol Med. 41:455–463.
2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ding J, Romani J, Zaborski M, MacLeod RA,
Nagel S, Drexler HG and Quentmeier H: Inhibition of PI3K/mTOR
overcomes nilotinib resistance in BCR-ABL1 positive leukemia cells
through translational down-regulation of MDM2. PLoS One.
8(e83510)2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Peterson LF, Mitrikeska E, Giannola D, Lui
Y, Sun H, Bixby D, Malek SN, Donato NJ, Wang S and Talpaz M: p53
stabilization induces apoptosis in chronic myeloid leukemia blast
crisis cells. Leukemia. 25:761–769. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Adnan Awad S, Dufva O, Klievink J,
Karjalainen E, Ianevski A, Pietarinen P, Kim D, Potdar S, Wolf M,
Lotfi K, et al: Integrated drug profiling and CRISPR screening
identify BCR::ABL1-independent vulnerabilities in chronic myeloid
leukemia. Cell Rep Med. 5(101521)2024.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Al-Kuraishy HM, Al-Gareeb AI and
Al-Buhadilly AK: p53 gene (NY-CO-13) levels in patients with
chronic myeloid leukemia: The role of imatinib and nilotinib.
Diseases. 6(13)2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhao D and Liu T: The relationship between
MDM2 T309G polymorphism and leukemia in the Chinese population:
Evidence from a meta-analysis. Clin Lab. 63:1639–1645.
2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Burgess A, Chia KM, Haupt S, Thomas D,
Haupt Y and Lim E: Clinical overview of MDM2/X-targeted therapies.
Front Oncol. 6(7)2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Law JC, Ritke MK, Yalowich JC, Leder GH
and Ferrell RE: Mutational inactivation of the p53 gene in the
human erythroid leukemic K562 cell line. Leuk Res. 17:1045–1050.
1993.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Usuda J, Inomata M, Fukumoto H, Iwamoto Y,
Suzuki T, Kuh HJ, Fukuoka K, Kato H, Saijo N and Nishio K:
Restoration of p53 gene function in 12-O-tetradecanoylphorbor
13-acetate-resistant human leukemia K562/TPA cells. Int J Oncol.
22:81–86. 2003.PubMed/NCBI
|
|
34
|
Ray-Coquard I, Blay JY, Italiano A, Le
Cesne A, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, et al:
Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients
with MDM2-amplified, well-differentiated or dedifferentiated
liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol.
13:1133–1140. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Brennan RC, Federico S, Bradley C, Zhang
J, Flores-Otero J, Wilson M, Stewart C, Zhu F, Guy K and Dyer MA:
Targeting the p53 pathway in retinoblastoma with subconjunctival
Nutlin-3a. Cancer Res. 71:4205–4213. 2011.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Tsubaki M, Takeda T, Matsuda T, Kimura A,
Tanaka R, Nagayoshi S, Hoshida T, Tanabe K and Nishida S:
Hypoxia-inducible factor 1α inhibitor induces cell death via
suppression of BCR-ABL1 and Met expression in BCR-ABL1 tyrosine
kinase inhibitor sensitive and resistant chronic myeloid leukemia
cells. BMB Rep. 56:78–83. 2023.PubMed/NCBI View Article : Google Scholar
|